
Current Understanding of the Role of Senescent Melanocytes in Skin Ageing


{kind=link}
{kind=link}
Abstract
:1. Introduction
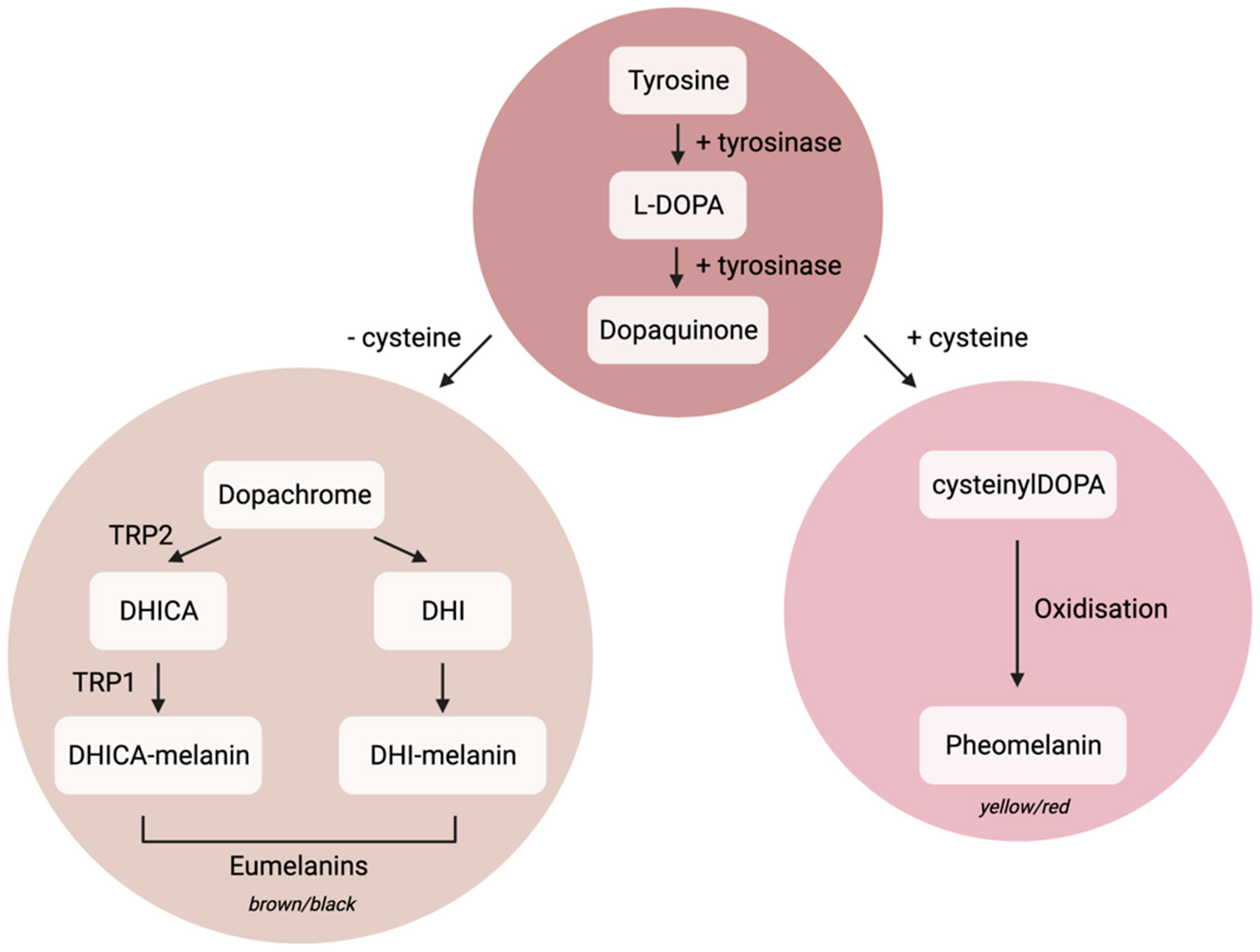
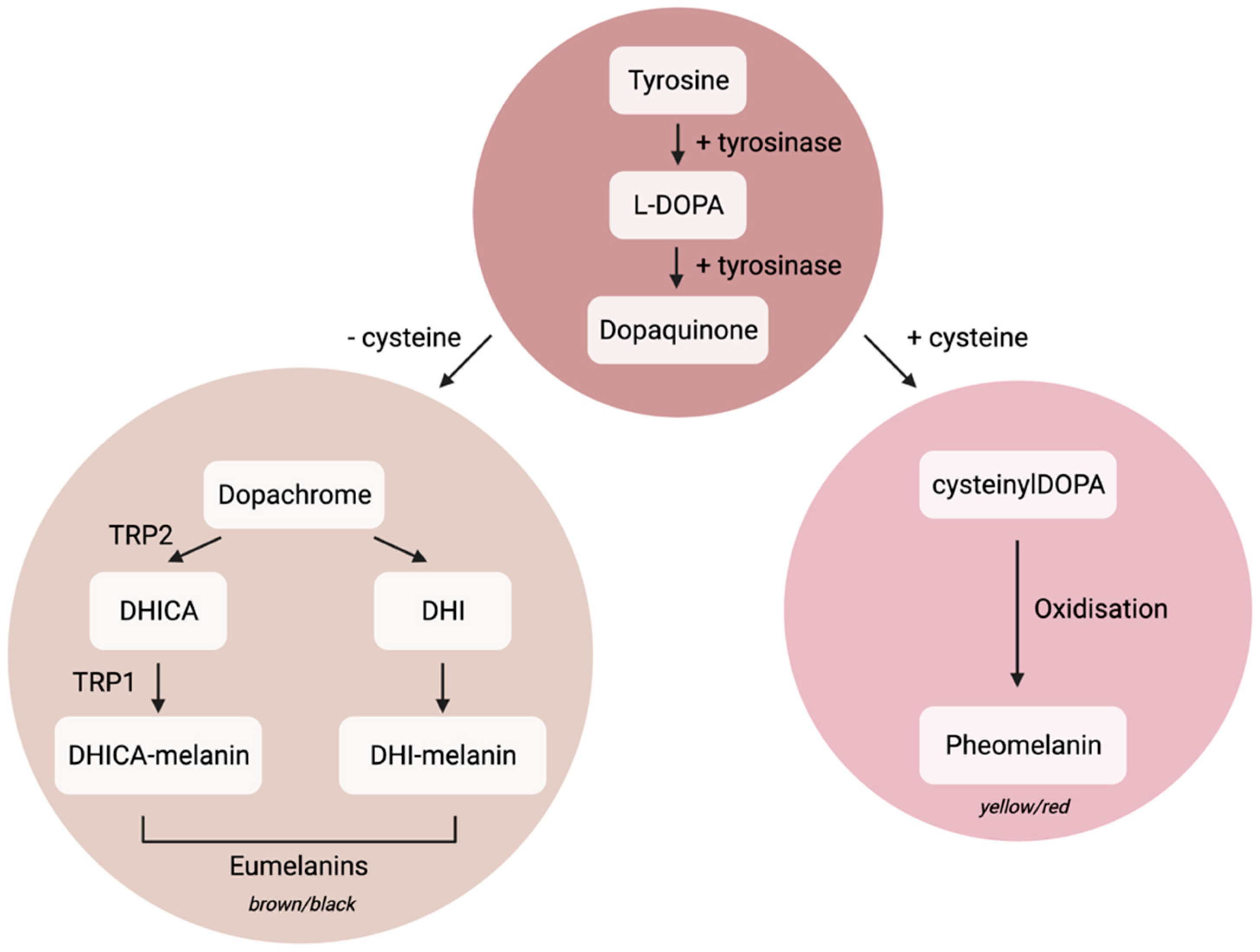
1.1. Melanocyte Cell Biology
1.2. Senescence
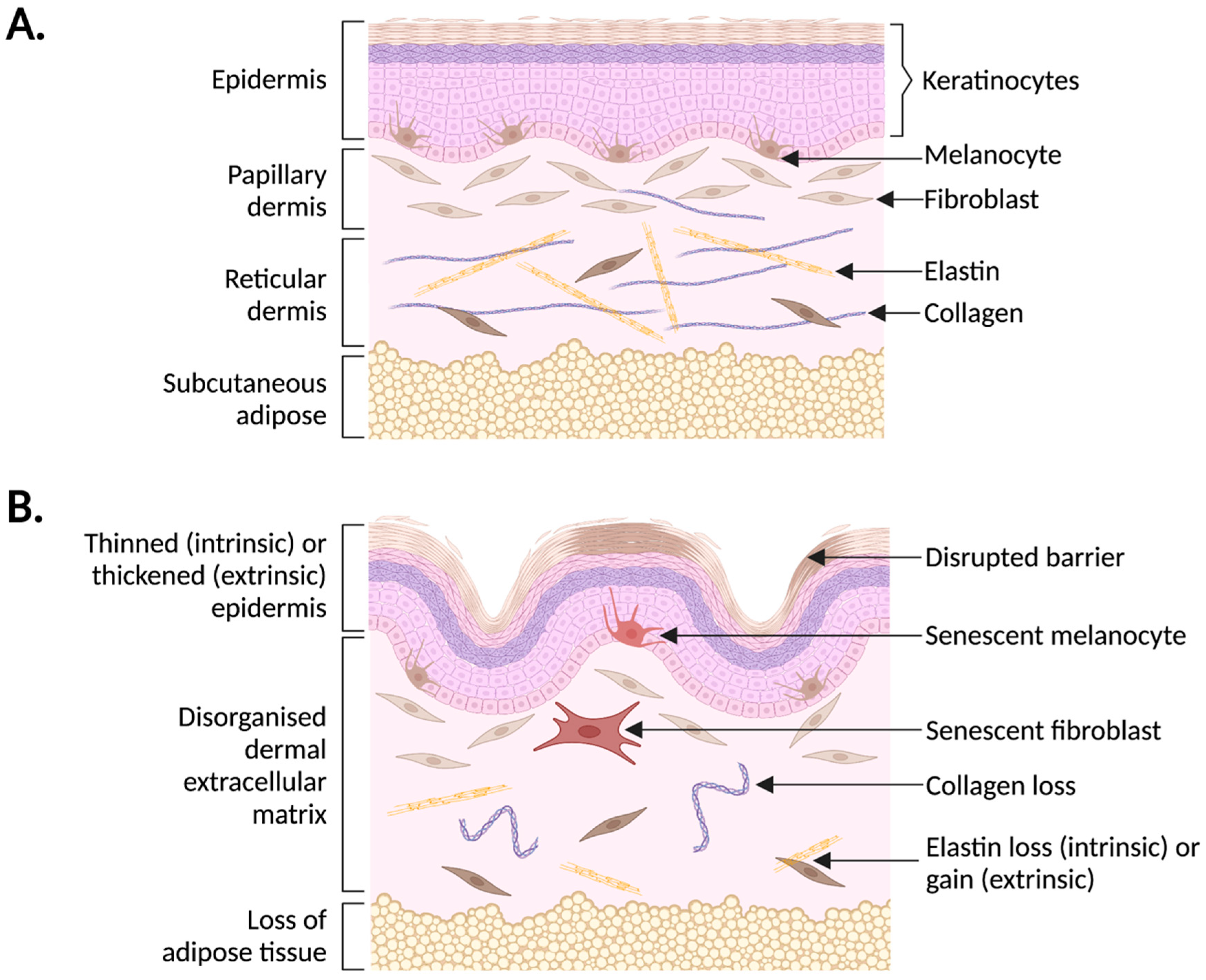
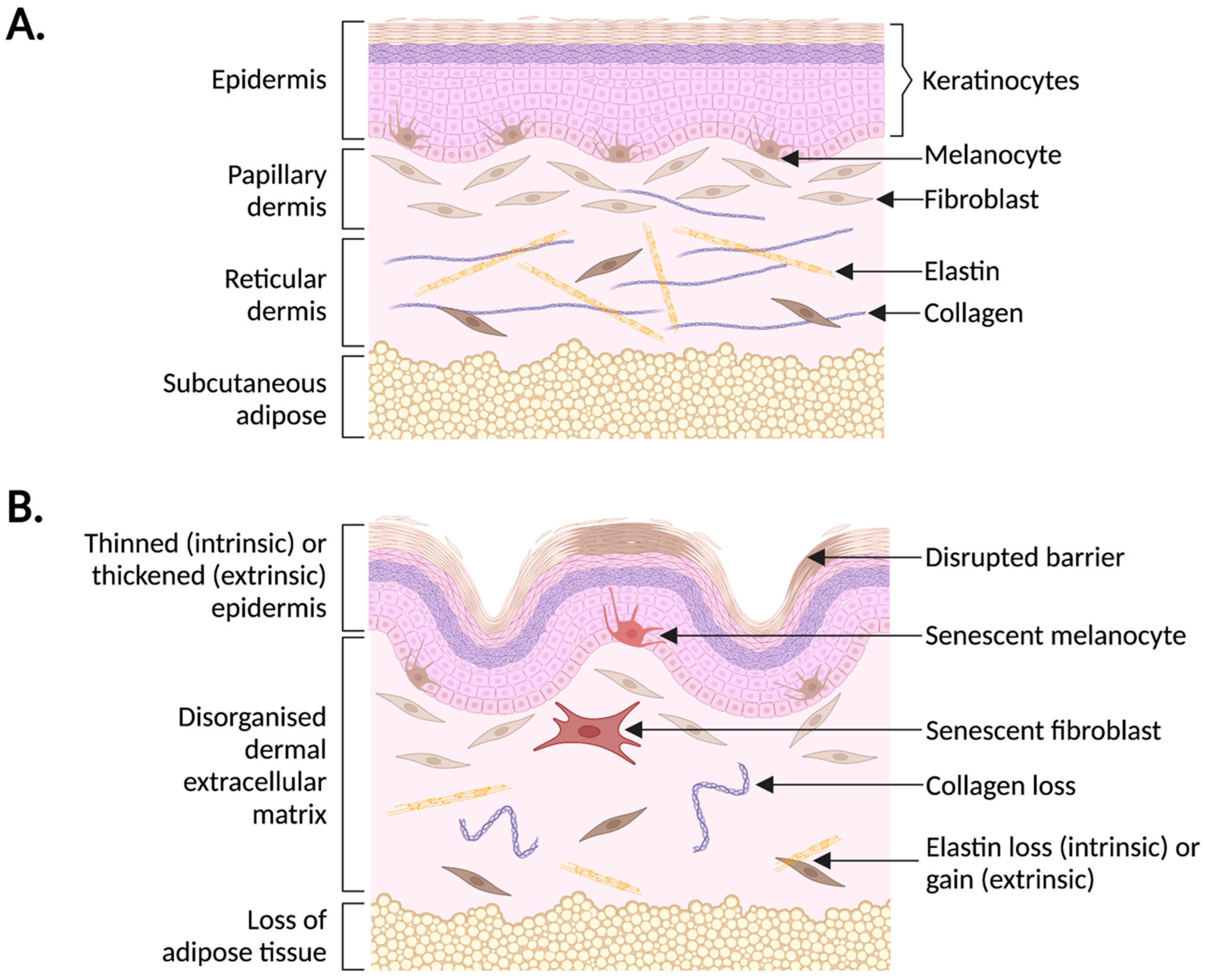
2. Human Skin Ageing
3. Senescence and Human Skin Ageing
4. Melanocyte Senescence
4.1. Mechanisms
4.2. UV-Induced Melanocyte Senescence
4.3. Evidence That Senescent Melanocytes Impact Human Skin Ageing In Vivo
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Handel, A.C.; Miot, L.D.B.; Miot, H.A. Melasma: A clinical and epidemiological review. An. Bras. Dermatol. 2014, 89, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Hearing, V.J. Melanocytes and their diseases. Cold Spring Harb. Perspect. Med. 2014, 4, a017046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, N.K.; Smalley, K.S.M.; Li, L.; Herlyn, M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. 2005, 18, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymińska, A. Skin melanocytes: Biology and development. Adv. Dermatol. Allergol./Postȩpy Dermatol. I Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef]
- Seiberg, M. Keratinocyte–melanocyte interactions during melanosome transfer. Pigment Cell Res. 2001, 14, 236–242. [Google Scholar] [CrossRef]
- Jiang, S.; Liao, Z.-K.; Jia, H.-Y.; Liu, X.-M.; Wan, J.; Lei, T.-C. The regional distribution of melanosomes in the epidermis affords a localized intensive photoprotection for basal keratinocyte stem cells. J. Dermatol. Sci. 2021, 103, 130–134. [Google Scholar] [CrossRef]
- Wasmeier, C.; Hume, A.N.; Bolasco, G.; Seabra, M.C. Melanosomes at a glance. J. Cell Sci. 2008, 121, 3995–3999. [Google Scholar] [CrossRef] [Green Version]
- Archambault, M.; Yaar, M.; Gilchrest, B.A. Keratinocytes and fibroblasts in a human skin equivalent model enhance melanocyte survival and melanin synthesis after ultraviolet irradiation. J. Investig. Dermatol. 1995, 104, 859–867. [Google Scholar] [CrossRef]
- D’Alba, L.; Shawkey, M.D. Melanosomes: Biogenesis, properties, and evolution of an ancient organelle. Physiol. Rev. 2019, 99, 1–19. [Google Scholar] [CrossRef]
- Kim, Y.; Kang, B.; Kim, J.C.; Park, T.J.; Kang, H.Y. Senescent Fibroblast–Derived GDF15 Induces Skin Pigmentation. J. Investig. Dermatol. 2020, 140, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Berson, J.F.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 2001, 12, 3451–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, M. Rab GTPases: Key players in melanosome biogenesis, transport, and transfer. Pigment Cell Melanoma Res. 2021, 34, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Moreiras, H.; Seabra, M.C.; Barral, D.C. Melanin Transfer in the Epidermis: The Pursuit of Skin Pigmentation Control Mechanisms. Int. J. Mol. Sci. 2021, 22, 4466. [Google Scholar] [CrossRef]
- Taylor, K.L.; Lister, J.A.; Zeng, Z.; Ishizaki, H.; Anderson, C.; Kelsh, R.; Jackson, I.; Patton, E.E. Differentiated melanocyte cell division occurs in vivo and is promoted by mutations in Mitf. Development 2011, 138, 3579–3589. [Google Scholar] [CrossRef] [Green Version]
- Halaban, R.; Langdon, R.; Birchall, N.; Ccuono, C.; Baird, A.; Scott, G.; Moellomann, G.; Mcguire, J. Paracrine stimulation of melanocytes by keratinocytes through basic fibroblast growth factor. Ann. N. Y. Acad. Sci. 1988, 548, 180–190. [Google Scholar] [CrossRef]
- Saldana-Caboverde, A.; Kos, L. Roles of endothelin signaling in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010, 23, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Medrano, E.E.; Yang, F.; Boissy, R.; Farooqui, J.; Shah, V.; Matsumoto, K.; Nordlund, J.J.; Park, H.Y. Terminal differentiation and senescence in the human melanocyte: Repression of tyrosine-phosphorylation of the extracellular signal-regulated kinase 2 selectively defines the two phenotypes. Mol. Biol. Cell 1994, 5, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.-X.; Zhang, R.-Z.; Xiao, L.; Wang, L. Effects of keratinocyte-derived and fibroblast-derived exosomes on human epidermal melanocytes. Indian J. Dermatol. Venereol. Leprol. 2022, 88, 322–331. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of melanoma. Medical. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Chow, J.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular senescence in naevi and immortalisation in melanoma: A role for p16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2003, 436, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, D.C. Human melanocyte senescence and melanoma susceptibility genes. Oncogene 2003, 22, 3063–3069. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Campisi, J.; di Fagagna, F.D. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Lawless, C.; Wang, C.; Jurk, D.; Merz, A.; von Zglinicki, T.; Passos, J.F. Quantitative assessment of markers for cell senescence. Exp. Gerontol. 2010, 45, 772–778. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Wallis, R.; Milligan, D.; Hughes, B.; Mizen, H.; López-Domínguez, J.A.; Eduputa, U.; Tyler, E.J.; Serrano, M.; Bishop, C.L. Senescence-associated morphological profiles (SAMPs): An image-based phenotypic profiling method for evaluating the inter and intra model heterogeneity of senescence. Aging 2022, 14, 4220. [Google Scholar] [CrossRef]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.-P.; Rodier, F.; Campisi, J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CTuttle, S.L.; Waaijer, M.E.C.; Slee-Valentijn, M.S.; Stijnen, T.; Westendorp, R.; Maier, A.B. Cellular senescence and chronological age in various human tissues: A systematic review and meta-analysis. Aging Cell 2020, 19, e13083. [Google Scholar]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Sellheyer, K. Pathogenesis of solar elastosis: Synthesis or degradation? J. Cutan. Pathol. 2003, 30, 123–127. [Google Scholar] [CrossRef]
- Giangreco, A.; Qin, M.; Pintar, J.E.; Watt, F.M. Epidermal stem cells are retained in vivo throughout skin aging. Aging Cell 2008, 7, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Krutmann, J.; Bouloc, A.; Sore, G.; Bernard, B.A.; Passeron, T. The skin aging exposome. J. Dermatol. Sci. 2017, 85, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Tigges, J.; Krutmann, J.; Fritsche, E.; Haendeler, J.; Schaal, H.; Fischer, J.W.; Kalfalah, F.; Reinke, H.; Reifenberger, G.; Stühler, K.; et al. The hallmarks of fibroblast ageing. Mech. Ageing Dev. 2014, 138, 26–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hong, Y.; Kim, M. Structural and Functional Changes and Possible Molecular Mechanisms in Aged Skin. Int. J. Mol. Sci. 2021, 22, 12489. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G. Molecular mechanisms of skin ageing. Mech. Ageing Dev. 2002, 123, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.E.; Kim, Y.; Kwon, S.; Kim, M.; Kim, Y.H.; Kim, J.-H.; Park, T.J.; Kang, H.Y. Senescent fibroblasts drive ageing pigmentation: A potential therapeutic target for senile lentigo. Theranostics 2018, 8, 4620. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, S.M.; Kwon, S.; Park, T.J.; Kang, H.Y. Senescent fibroblasts in melasma pathophysiology. Exp. Dermatol. 2019, 28, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Yoon, J.E.; Kim, Y.H.; Kim, Y.; Park, T.J.; Kang, H.Y. The potential skin lightening candidate, senolytic drug ABT263, for photoageing pigmentation. Br. J. Dermatol. 2021, 186, 740–742. [Google Scholar] [CrossRef]
- Kim, J.C.; Park, T.J.; Kang, H.Y. Skin-Aging Pigmentation: Who Is the Real Enemy? Cells 2022, 11, 2541. [Google Scholar] [CrossRef]
- Low, E.; Alimohammadiha, G.; Smith, L.A.; Costello, L.F.; Przyborski, S.A.; von Zglinicki, T.; Miwa, S. How good is the evidence that cellular senescence causes skin ageing? Ageing Res. Rev. 2021, 71, 101456. [Google Scholar] [CrossRef]
- Boer, M.; Duchnik, E.; Maleszka, R.; Marchlewicz, M. Structural and biophysical characteristics of human skin in maintaining proper epidermal barrier function. Adv. Dermatol. Allergol./Postȩpy Dermatol. I Alergol. 2016, 33, 1–5. [Google Scholar] [CrossRef]
- Watt, F.M.; Fujiwara, H. Cell-extracellular matrix interactions in normal and diseased skin. Cold Spring Harb. Perspect. Biol. 2011, 3, a005124. [Google Scholar] [CrossRef] [Green Version]
- Marcos-Garcés, V.; Molina Aguilar, P.; Bea Serrano, C.; García Bustos, V.; Benavent Seguí, J.; Ferrández Izquierdo, A.; Ruiz-Saurí, A. Age-related dermal collagen changes during development, maturation and ageing–a morphometric and comparative study. J. Anat. 2014, 225, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Quan, T.; Purohit, T.; Shao, Y.; Cho, M.K.; He, T.; Varani, J.; Kang, S.; Voorhees, J.J. Collagen fragmentation promotes oxidative stress and elevates matrix metalloproteinase-1 in fibroblasts in aged human skin. Am. J. Pathol. 2009, 174, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varani, J.; Spearman, D.; Perone, P.; Fligiel, S.E.; Datta, S.C.; Wang, Z.Q.; Shao, Y.; Kang, S.; Fisher, G.J.; Voorhees, J.J. Inhibition of type I procollagen synthesis by damaged collagen in photoaged skin and by collagenase-degrad ed collagen in vitro. Am. J. Pathol. 2001, 158, 931–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, R.A.; Grove, G. Human skin fibroblasts derived from papillary and reticular dermis: Differences in growth potential in vitro. Science 1979, 204, 526–527. [Google Scholar] [CrossRef]
- Kabashima, K.; Honda, T.; Ginhoux, F.; Egawa, G. The immunological anatomy of the skin. Nat. Rev. Immunol. 2019, 19, 19–30. [Google Scholar] [CrossRef]
- Bikle, D.D.; Xie, Z.; Tu, C.-L. Calcium regulation of keratinocyte differentiation. Expert Rev. Endocrinol. Metab. 2012, 7, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, H. Skin aging and dry skin. J. Dermatol. 2004, 31, 603–609. [Google Scholar] [CrossRef]
- Grove, G.L.; Kligman, A.M. Age-associated changes in human epidermal cell renewal. J. Gerontol. 1983, 38, 137–142. [Google Scholar] [CrossRef]
- Gilchrest, B.A.; Blog, F.B.; Szabo, G. Effects of aging and chronic sun exposure on melanocytes in human skin. J. Investig. Dermatol. 1979, 73, 141–143. [Google Scholar] [CrossRef] [Green Version]
- Debacq-Chainiaux, F.; Leduc, C.; Verbeke, A.; Toussaint, O. UV, stress and aging. Dermatoendocrinol 2012, 4, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. UV-induced skin damage. Toxicology 2003, 189, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, C.; Jiang, G. Research progress on skin photoaging and oxidative stress. Adv. Dermatol. Allergol./Postępy Dermatol. I Alergol. 2021, 38, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Igaki, T. Dissecting cellular senescence and SASP in Drosophila. Inflamm. Regen. 2016, 36, 25. [Google Scholar] [CrossRef] [Green Version]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16INK4a in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Durr, P.; Wlaschek, M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Fitsiou, E.; Pulido, T.; Campisi, J.; Alimirah, F.; Demaria, M. Cellular senescence and the senescence-associated secretory phenotype as drivers of skin photoaging. J. Investig. Dermatol. 2021, 141, 1119–1126. [Google Scholar] [CrossRef]
- Ogata, Y.; Yamada, T.; Hasegawa, S.; Sanada, A.; Iwata, Y.; Arima, M.; Nakata, S.; Sugiura, K.; Akamatsu, H. SASP-induced macrophage dysfunction may contribute to accelerated senescent fibroblast accumulation in the dermis. Exp. Dermatol. 2021, 30, 84–91. [Google Scholar] [CrossRef]
- Lupa, D.M.W.; Kalfalah, F.; Safferling, K.; Boukamp, P.; Poschmann, G.; Volpi, E.; Götz-Rösch, C.; Bernerd, F.; Haag, L.; Huebenthal, U.; et al. Characterization of skin aging–associated secreted proteins (SAASP) produced by dermal fibroblasts isolated from intrinsically aged human skin. J. Investig. Dermatol. 2015, 135, 1954–1968. [Google Scholar] [CrossRef]
- Özcan, S.; Alessio, N.; Acar, M.B.; Mert, E.; Omerli, F.; Peluso, G.; Galderisi, U. Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging 2016, 8, 1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, Y.V.; Rattanavirotkul, N.; Olova, N.; Salzano, A.; Quintanilla, A.; Tarrats, N.; Kiourtis, C.; Müller, M.; Green, T.; Adams, P.D.; et al. Notch signaling mediates secondary senescence. Cell Rep. 2019, 27, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinmüllner, R.; Zbiral, B.; Becirovic, A.; Stelzer, E.M.; Nagelreiter, F.; Schosserer, M.; Lämmermann, I.; Liendl, L.; Lang, M.; Terlecki-Zaniewicz, L.; et al. Organotypic human skin culture models constructed with senescent fibroblasts show hallmarks of skin aging. NPJ Aging Mech. Dis. 2020, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waaijer, M.E.C.; Parish, W.E.; Strongitharm, B.H.; Van Heemst, D.; Slagboom, P.E.; De Craen, A.J.M.; Sedivy, J.M.; Westendorp, R.G.J.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef]
- Schoenmaker, M.; De Craen, A.J.M.; Meijer, P.H.E.M.D.; Beekman, M.; Blauw, G.J.; Slagboom, P.; Westendorp, R.G.J. Evidence of genetic enrichment for exceptional survival using a family approach: The Leiden Longevity Study. Eur. J. Hum. Genet. 2006, 14, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Waaijer, M.E.C.; Gunn, D.A.; Adams, P.D.; Pawlikowski, J.S.; Griffiths, C.E.M.; van Heemst, D.; Slagboom, P.E.; Westendorp, R.G.J.; Maier, A.B. P16INK4a positive cells in human skin are indicative of local elastic fiber morphology, facial wrinkling, and perceived age. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2016, 71, 1022–1028. [Google Scholar] [CrossRef] [Green Version]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef]
- Bennett, D.C.; Medrano, E.E. Molecular regulation of melanocyte senescence. Pigment Cell Res. 2022, 15, 242–250. [Google Scholar] [CrossRef]
- Zimmermann, T.; Pommer, M.; Kluge, V.; Chiheb, C.; Muehlich, S.; Bosserhoff, A.-K. Detection of Cellular Senescence in Human Primary Melanocytes and Malignant Melanoma Cells In Vitro. Cells 2022, 11, 1489. [Google Scholar] [CrossRef] [PubMed]
- Lämmerhirt, L.; Kappelmann-Fenzl, M.; Fischer, S.; Pommer, M.; Zimmermann, T.; Kluge, V.; Matthies, A.; Kuphal, S.; Bosserhoff, A.K. Knockdown of Lamin B1 and the Corresponding Lamin B Receptor Leads to Changes in Heterochromatin State and Senescence Induction in Malignant Melanoma. Cells 2022, 11, 2154. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, L.; Lamm, S.; Henz, I.; Kappelmann-Fenzl, M.; Haferkamp, S.; Meierjohann, S.; Hellerbrand, C.; Kuphal, S.; Bosserhoff, A.K. Role of melanoma inhibitory activity in melanocyte senescence. Pigment Cell Melanoma Res. 2019, 32, 777–791. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 1995, 217, 65–77. [Google Scholar] [CrossRef]
- Ouelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.; Han, S.Y.; Song, J. Dynamics of ARF regulation that control senescence and cancer. BMB Rep. 2016, 49, 598–606. [Google Scholar] [CrossRef] [Green Version]
- Ha, L.; Ichikawa, T.; Anver, M.; Dickins, R.; Lowe, S.; Sharpless, N.E.; Krimpenfort, P.; DePinho, R.A.; Bennett, D.C.; Sviderskaya, E.V.; et al. ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc. Natl. Acad. Sci. USA 2007, 104, 10968–10973. [Google Scholar] [CrossRef] [Green Version]
- Sviderskaya, E.V. p16Ink4a in melanocyte senescence and differentiation. J. Natl. Cancer Inst. 2002, 94, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.D.M.; Cook, M.G.; Chong, H.; Hossain, M.; Pandha, H.S.; Bennett, D.C. Senescence evasion in melanoma progression: Uncoupling of DNA-damage signaling from p53 activation and p21 expression. Pigment Cell Melanoma Res. 2013, 26, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Shi, J.; Sun, L.; Song, L.; Zhao, W.; Xiong, X.; Lu, Y. The involvement of ERK1/2 and p38 MAPK in the premature senescence of melanocytes induced by H2O2 through a p53-independent p21 pathway. J. Dermatol. Sci. 2022, 105, 88–97. [Google Scholar] [CrossRef]
- Jenkins, N.C.; Grossman, D. Role of melanin in melanocyte dysregulation of reactive oxygen species. Biomed. Res. Int. 2013, 2013, 9087972. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Hearing, V.J. The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 2008, 84, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San Juan, L.; Cagigal, M.L.; Fernandez-Flores, A.; Mayorga, M.; Gandarillas, A. Protooncogene MYC drives human melanocyte melanogenesis and senescence. Cancer Gene Ther. 2022, 29, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Medrano, E.E. Melanin accumulation accelerates melanocyte senescence by a mechanism involving p16INK4a/CDK4/pRB and E2F1. Ann. N. Y. Acad. Sci. 2000, 908, 71–84. [Google Scholar] [CrossRef]
- Martic, I.; Wedel, S.; Jansen-Dürr, P.; Cavinato, M. A new model to investigate UVB-induced cellular senescence and pigmentation in melanocytes. Mech. Ageing Dev. 2020, 190, 111322. [Google Scholar] [CrossRef]
- Barker, D.; Dixon, K.; Medrano, E.E.; Smalara, D.; Im, S.; Mitchell, D.; Babcock, G.; Abdel-Malek, Z.A. Comparison of the responses of human melanocytes with different melanin contents to ultraviolet B irradiation. Cancer Res. 1995, 55, 4041–4046. [Google Scholar]
- Medrano, E.E.; Im, S.; Yang, F.; Abdel-Malek, Z.A. Ultraviolet B light induces G1 arrest in human melanocytes by prolonged inhibition of retinoblastoma protein phosphorylation associated with long-term expression of the p21Waf-1/SDI-1/Cip-1 protein. Cancer Res. 1995, 55, 4047–4052. [Google Scholar]
- Choi, S.-Y.; Bin, B.-H.; Kim, W.; Lee, E.; Lee, T.R.; Cho, E.-G. Exposure of human melanocytes to UVB twice and subsequent incubation leads to cellular senescence and senescence-associated pigmentation through the prolonged p53 expression. J. Dermatol. Sci. 2018, 90, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Murase, D.; Hachiya, A.; Amano, Y.; Ohuchi, A.; Kitahara, T.; Takema, Y. The essential role of p53 in hyperpigmentation of the skin via regulation of paracrine melanogenic cytokine receptor signaling. J. Biol. Chem. 2009, 284, 4343–4353. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Anumanthan, G.; Scheaffer, S.; Cornelius, L.A. HMGB1/RAGE mediates UVB-induced secretory inflammatory response and resistance to apoptosis in human melanocytes. J. Investig. Dermatol. 2019, 139, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Luan, Z.-G.; Zhang, H.; Yang, P.-T.; Ma, X.-C.; Zhang, C.; Guo, R.-X. HMGB1 activates nuclear factor-κB signaling by RAGE and increases the production of TNF-α in human umbilical vein endothelial cells. Immunobiology 2010, 215, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Mou, K.; Liu, W.; Miao, Y.; Cao, F.; Li, P. HMGB 1 deficiency reduces H2O2-induced oxidative damage in human melanocytes via the Nrf2 pathway. J. Cell Mol. Med. 2018, 22, 6148–6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Jarrold, B.; Zhao, W.; Deng, G.; Moulton, L.; Laughlin, T.; Hakozaki, T. The combination of sucrose dilaurate and sucrose laurate suppresses HMGB1: An enhancer of melanocyte dendricity and melanosome transfer to keratinocytes. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 3–11. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2012, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, G.; Jurk, D.; Marques, F.M.; Correia-Melo, C.; Hardy, T.L.D.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, B.K.; Bishop, C.L. Current Understanding of the Role of Senescent Melanocytes in Skin Ageing. Biomedicines 2022, 10, 3111. https://doi.org/10.3390/biomedicines10123111
Hughes BK, Bishop CL. Current Understanding of the Role of Senescent Melanocytes in Skin Ageing. Biomedicines. 2022; 10(12):3111. https://doi.org/10.3390/biomedicines10123111
Chicago/Turabian StyleHughes, Bethany K., and Cleo L. Bishop. 2022. "Current Understanding of the Role of Senescent Melanocytes in Skin Ageing" Biomedicines 10, no. 12: 3111. https://doi.org/10.3390/biomedicines10123111
APA StyleHughes, B. K., & Bishop, C. L. (2022). Current Understanding of the Role of Senescent Melanocytes in Skin Ageing. Biomedicines, 10(12), 3111. https://doi.org/10.3390/biomedicines10123111