Machilin A Inhibits Tumor Growth and Macrophage M2 Polarization Through the Reduction of Lactic Acid

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
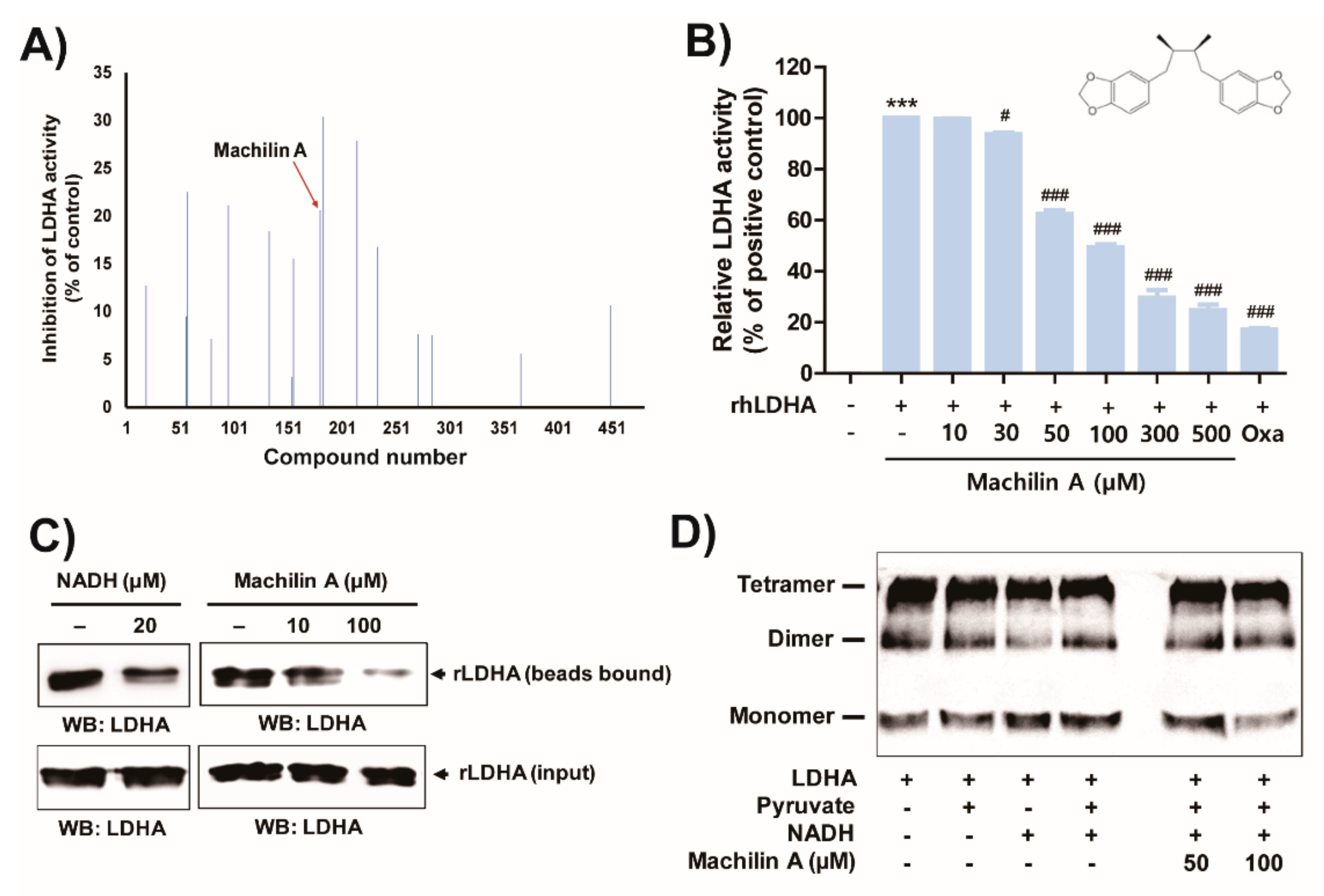
2.1. MA Inhibits In Vitro LDHA Activity by Binding of the Cofactor NADH to LDHA
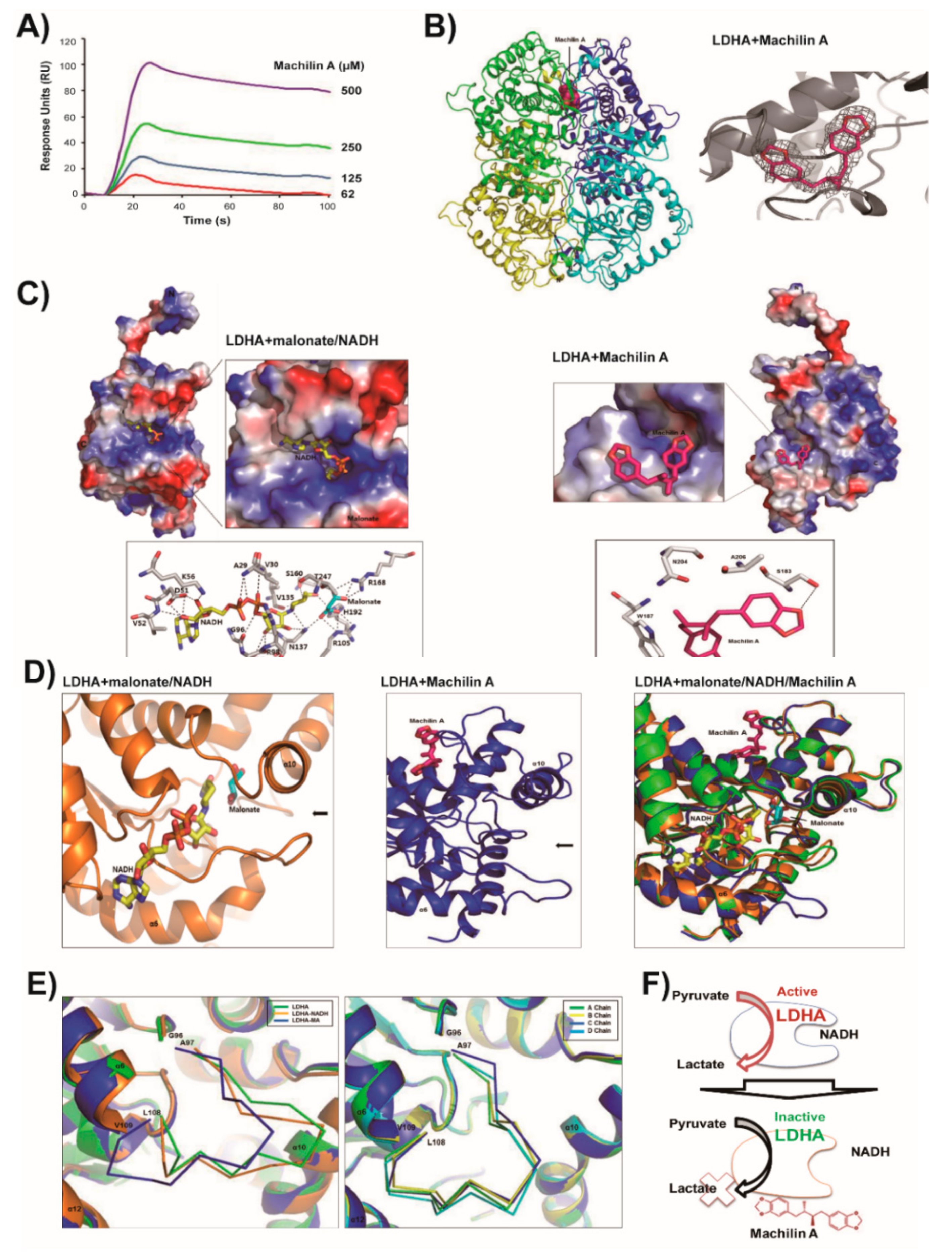
2.2. LDHA and Its NADH Binding Structures are Similar and In Closed Active-Site Conformation
2.3. MA Binds to LDHA In an Open Active-Site Conformation
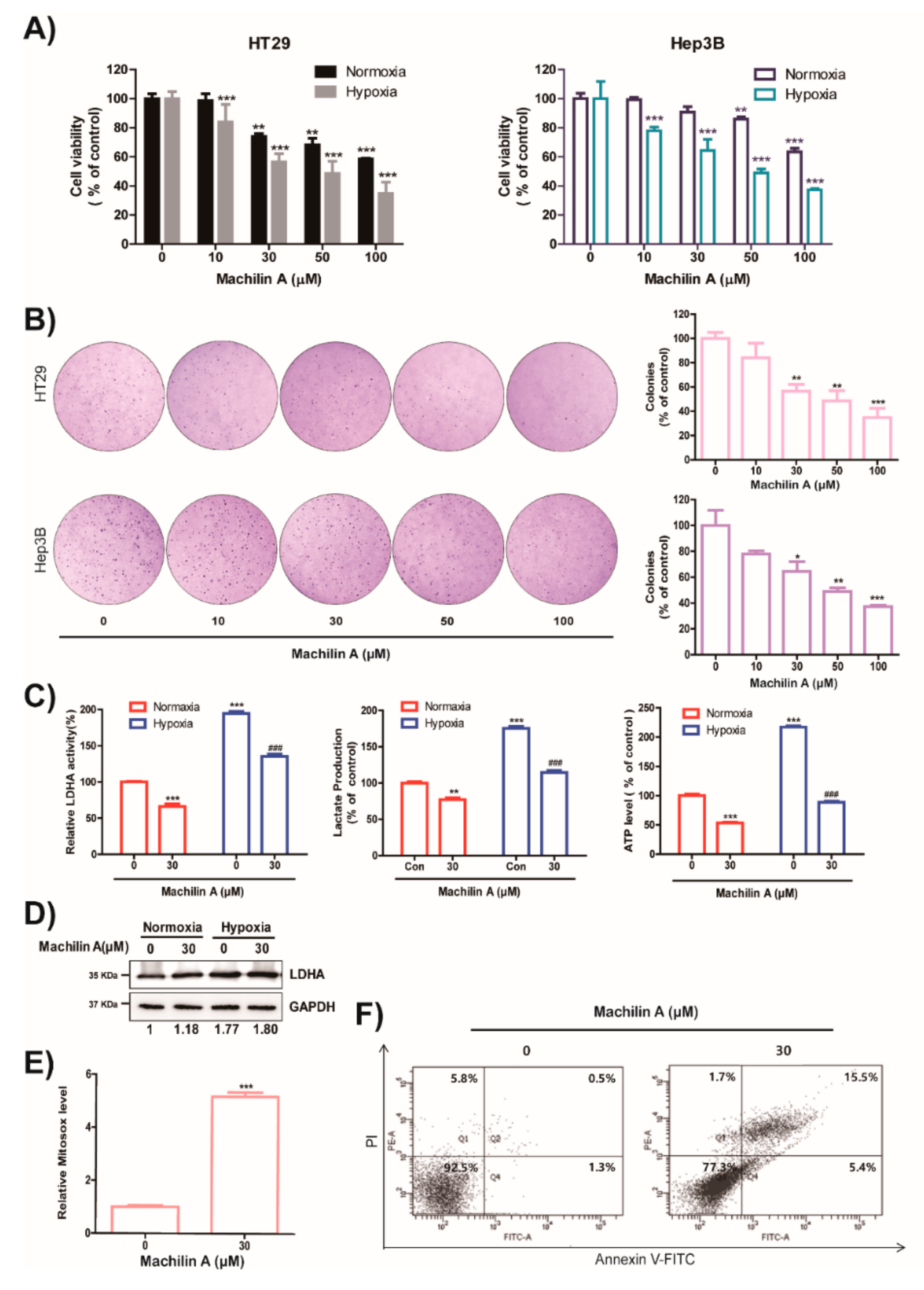
2.4. MA Reduces Growth of Cancer Cells, Colony Formation, LDH Activity, Lactate Production and ATP level in Hypoxic Condition
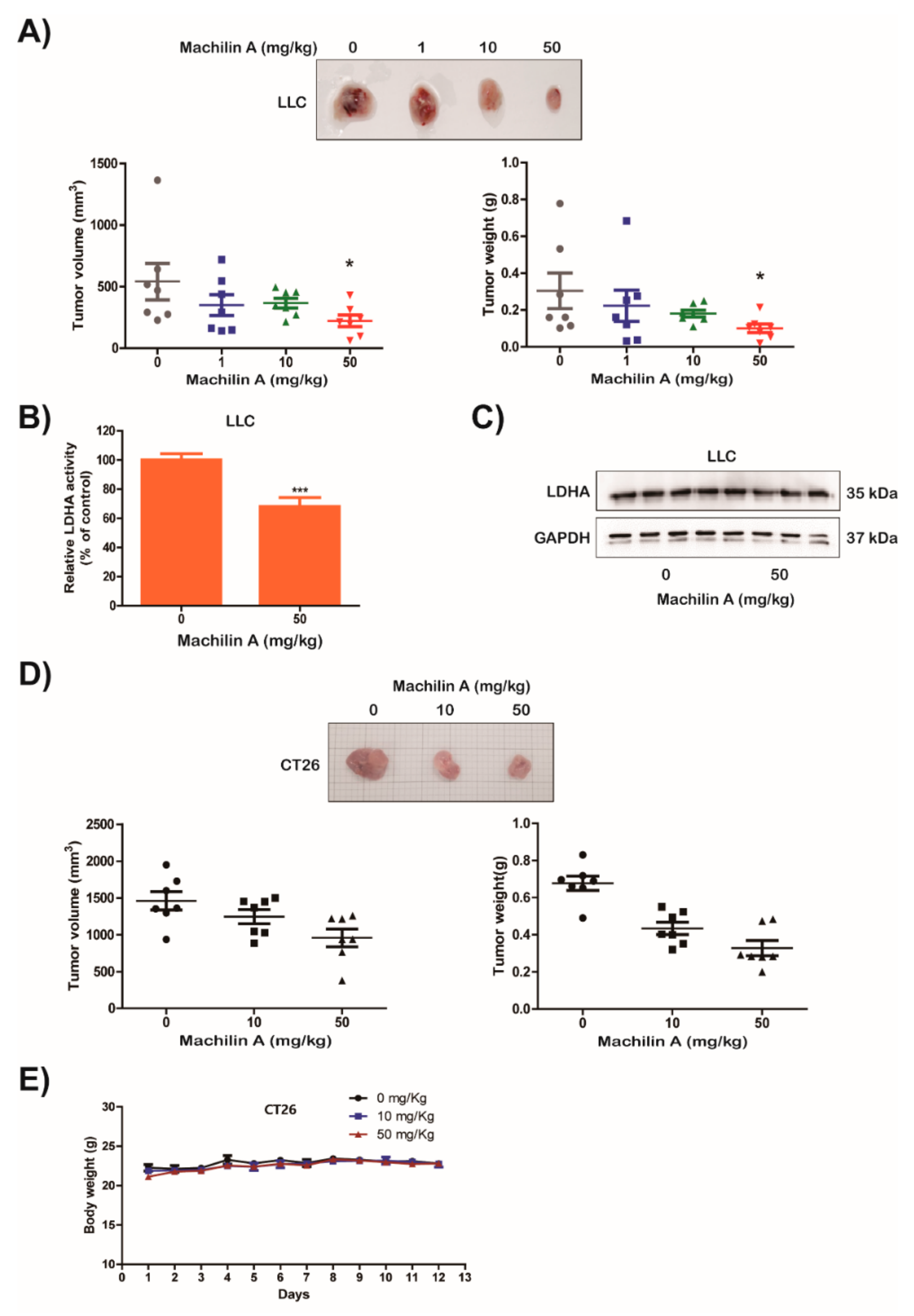
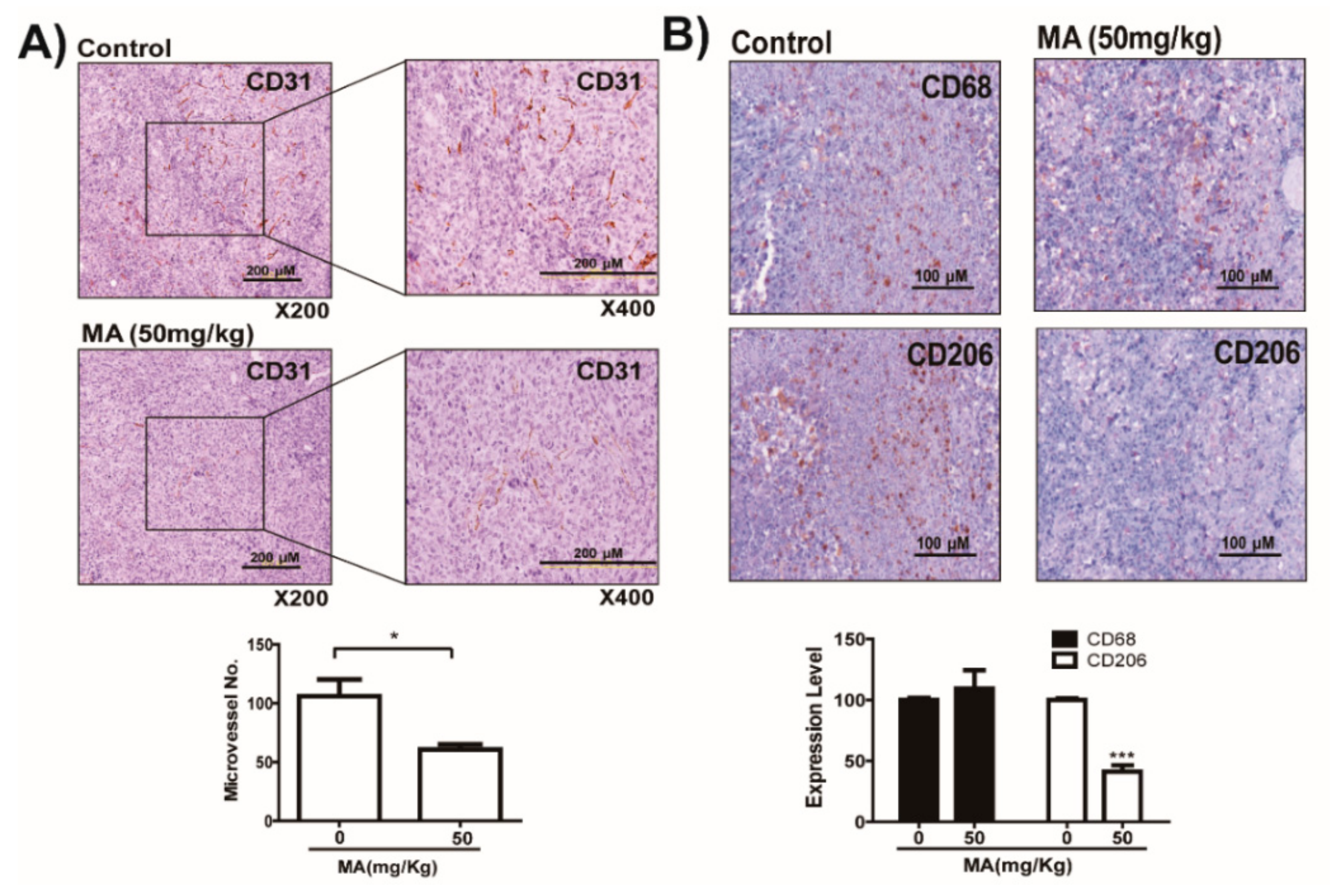
2.5. MA Suppresses LDHA Activity and In Vivo Growth of Lung Cancer LLC and CT26 Cells
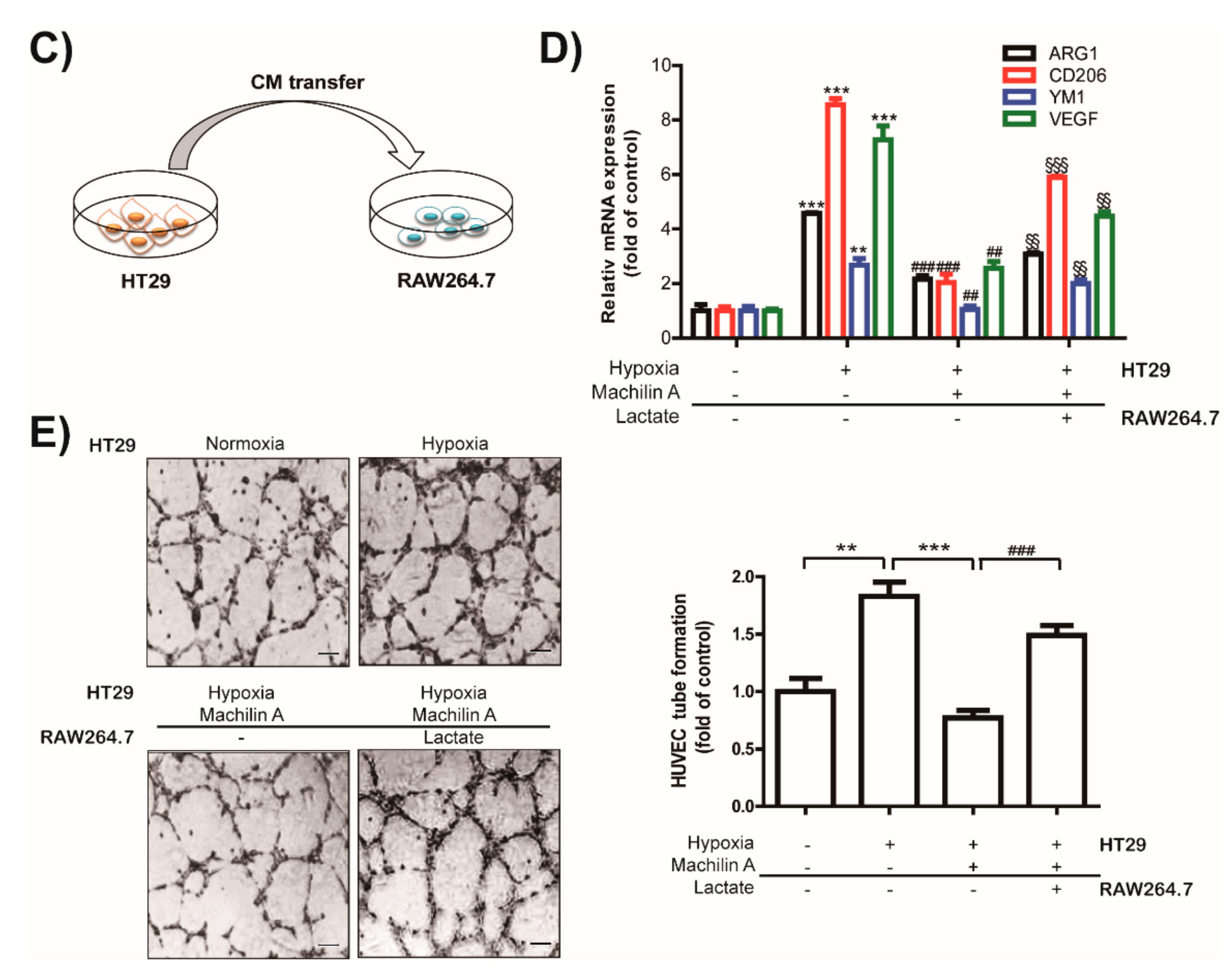
2.6. MA Inhibits Capillary Like-Tube Formation of Endothelial Cells Induced by Tumor-Derived Lactate Stimulated Macrophages
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Assay Compounds
4.3. LDH Activity Assay
4.4. NADH Binding Assay
4.5. Crosslinking Assay for Tetramer Formation of LDHA
4.6. Surface Plasmon Resonance (SPR) Biosensor Analysis
4.7. Expression and Purification of LDHA for Structural Study
4.8. Crystallization of LDHA, LDHA-NADH, and LDHA-MA Complexes
4.9. Data Collection, Structure Determination, and Refinement
4.10. Cell Culture, Hypoxia Condition and Determination of Cell Viability
4.11. Colony Formation Assay
4.12. Lactate Production Assay
4.13. Measurement of Intracellular ATP
4.14. Western Blot Analyses
4.15. Measurement of Mitochondrial Reactive Oxygen Species (ROS)
4.16. Detection of Apoptotic Cells
4.17. Tumor Allograft Model
4.18. Immunohistochemistry
4.19. Reverse-Transcription (RT)-PCR and Quantitative Real-Time RT-PCR (qRT-PCR)
4.20. Tube Formation Assay
4.21. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Bougioukas, G.; Didilis, V.; Gatter, K.C.; Harris, A.L. Lactate Dehydrogenase-5 (Ldh-5) Overexpression in Non-Small-Cell Lung Cancer Tissues Is Linked to Tumour Hypoxia, Angiogenic Factor Production and Poor Prognosis. Br. J. Cancer 2003, 89, 877–885. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Jagt, D.L.V.; Semenza, G.L.; Dang, C.V. Inhibition of Lactate Dehydrogenase a Induces Oxidative Stress and Inhibits Tumor Progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Hitosugi, T.; Chung, T.W.; Xie, J.; Ge, Q.; Gu, T.L.; Polakiewicz, R.D.; Chen, G.Z.; Boggon, T.J.; Lonial, S.; et al. Tyrosine Phosphorylation of Lactate Dehydrogenase a Is Important for Nadh/Nad(+) Redox Homeostasis in Cancer Cells. Mol. Cell Biol. 2011, 31, 4938–4950. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.; Thompson, C.B. Cancer’s Sweet Tooth. Cancer Cell 2006, 9, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Hanai, J.; Ren, J.G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting Lactate Dehydrogenase—A Inhibits Tumorigenesis and Tumor Progression in Mouse Models of Lung Cancer and Impacts Tumor-Initiating Cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hif-1 Mediates Metabolic Responses to Intratumoral Hypoxia and Oncogenic Mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting Lactate Metabolism for Cancer Therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Rellinger, E.J.; Craig, B.T.; Alvarez, A.L.; Dusek, H.L.; Kim, K.W.; Qiao, J.; Chung, D.H. Fx11 Inhibits Aerobic Glycolysis and Growth of Neuroblastoma Cells. Surgery 2017, 161, 747–752. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, X.; Yu, Y.; Li, J.; Hu, Q.; Xue, C.; Chen, J.; Shen, S.; Luo, Y.; Ren, F.; et al. Ldha Is a Direct Target of Mir-30d-5p and Contributes to Aggressive Progression of Gallbladder Carcinoma. Mol. Carcinog. 2018, 57, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of Ldh-a Expression Uncovers a Link between Glycolysis, Mitochondrial Physiology, Tumor Maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Durisova, V.; Vrbanova, A.; Ziegelhoffer, A.; Breier, A. Interaction of Cibacron Blue 3ga and Remazol Brilliant Blue R with the Nucleotide Binding Site of Lactate Dehydrogenase and (Na+ + K+)-Atpase. Gen. Physiol. Biophys. 1990, 9, 519–528. [Google Scholar] [PubMed]
- Dempster, S.; Harper, S.; Moses, J.E.; Dreveny, I. Structural Characterization of the Apo Form and Nadh Binary Complex of Human Lactate Dehydrogenase. Acta Crystallgr. D Biol. Crystallgr. 2014, 70, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Kolappan, S.; Shen, D.L.; Mosi, R.; Sun, J.; McEachern, E.J.; Vocadlo, D.J.; Craig, L. Structures of Lactate Dehydrogenase a (Ldha) in Apo, Ternary and Inhibitor-Bound Forms. Acta Crystallgr. D Biol. Crystallgr. 2015, 71, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Feldman-Salit, A.; Hering, S.; Messiha, H.L.; Veith, N.; Cojocaru, V.; Sieg, A.; Westerhoff, H.V.; Kreikemeyer, B.; Wade, R.C.; Fiedler, T. Regulation of the Activity of Lactate Dehydrogenases from Four Lactic Acid Bacteria. J. Biol. Chem. 2013, 288, 21295–21306. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Qiu, H.; Jackson, A.L.; Kilgore, J.E.; Zhong, Y.; Chan, L.L.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Jq1 Suppresses Tumor Growth through Downregulating Ldha in Ovarian Cancer. Oncotarget 2015, 6, 6915–6930. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, J.; Yu, Y.U.; Kim, Y.C. Inhibition of Phospholipase Cgamma1 and Cancer Cell Proliferation by Lignans and Flavans from Machilus Thunbergii. Arch. Pharm. Res. 2004, 27, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Park, B.Y.; Min, B.S.; Kwon, O.K.; Oh, S.R.; Ahn, K.S.; Kim, T.J.; Kim, D.Y.; Bae, K.; Lee, H.K. Increase of Caspase-3 Activity by Lignans from Machilus Thunbergii in Hl-60 Cells. Biol. Pharm. Bull. 2004, 27, 1305–1307. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ju, H.K.; Chang, H.W.; Jahng, Y.; Lee, S.H.; Son, J.K. Melanin Biosynthesis Inhibitors from the Bark of Machilus Thunbergii. Biol. Pharm. Bull. 2003, 26, 1039–1041. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, Y.U.; Kang, S.Y.; Park, H.Y.; Sung, S.H.; Lee, E.J.; Kim, S.Y.; Kim, Y.C. Antioxidant Lignans from Machilus Thunbergii Protect Ccl4-Injured Primary Cultures of Rat Hepatocytes. J. Pharm. Pharm. 2000, 52, 1163–1169. [Google Scholar] [CrossRef]
- Lee, S.U.; Shim, K.S.; Ryu, S.Y.; Min, Y.K.; Kim, S.H. Machilin a Isolated from Myristica Fragrans Stimulates Osteoblast Differentiation. Planta Med. 2009, 75, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; You, J.; Choi, H.G.; Kim, J.A.; Jee, J.G.; Lee, S. Selective Inhibitory Effects of Machilin a Isolated from Machilus Thunbergii on Human Cytochrome P450 1a and 2b6. Phytomedicine 2015, 22, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Perera, V.; Gross, A.S.; McLachlan, A.J. Measurement of Cyp1a2 Activity: A Focus on Caffeine as a Probe. Curr. Drug Metab. 2012, 13, 667–678. [Google Scholar] [CrossRef] [PubMed]
- McClendon, S.; Zhadin, N.; Callender, R. The Approach to the Michaelis Complex in Lactate Dehydrogenase: The Substrate Binding Pathway. Biophys. J. 2005, 89, 2024–2032. [Google Scholar] [CrossRef][Green Version]
- Johnson, L.N.; Barford, D. The Effects of Phosphorylation on the Structure and Function of Proteins. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 199–232. [Google Scholar] [CrossRef]
- Shi, Y.; Pinto, B.M. Human Lactate Dehydrogenase a Inhibitors: A Molecular Dynamics Investigation. PLoS ONE 2014, 9, e86365. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A Lactate-Induced Response to Hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate Influx through the Endothelial Cell Monocarboxylate Transporter Mct1 Supports an Nf-Kappab/Il-8 Pathway That Drives Tumor Angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. Ldha-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and Nk Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential Macrophage Programming in the Tumor Microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef]
- Kim, E.Y.; Chung, T.W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.T. A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death. Sci. Rep. 2019, 9, 3969. [Google Scholar] [CrossRef]
- Rani, R.; Kumar, V. Recent Update on Human Lactate Dehydrogenase Enzyme 5 (Hldh5) Inhibitors: A Promising Approach for Cancer Chemotherapy. J. Med. Chem. 2016, 59, 487–496. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; del Nagro, C.; et al. Metabolic Plasticity Underpins Innate and Acquired Resistance to Ldha Inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg Effect Supports Aggressiveness and Drug Resistance of Cancer Cells? Drug Resist. Updates 2018, 38, 1–11. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Omar, M.F.M.; Soong, R. The Warburg Effect and Drug Resistance. Br. J. Pharm. 2016, 173, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Choi, H.J.; Chung, T.W.; Jang, S.B.; Kim, K.; Ha, K.T. Expression and Efficient One-Step Chromatographic Purification of a Soluble Antagonist for Human Leukemia Inhibitory Factor Receptor in Escherichia Coli. J. Microbiol. Biotechnol. 2015, 25, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Navaza, J.; Saludjian, P. [33] Amore: An Automated Molecular Replacement Program Package. Methods Enzym. 1997, 276, 581–594. [Google Scholar]
- Kissinger, C.R.; Gehlhaar, D.K.; Fogel, D.B. Rapid Automated Molecular Replacement by Evolutionary Search. Acta Crystallgr. D Biol. Crystallgr. 1999, 55, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.A.; Zou, J.Y.; Cowan, S.W.; Kjeldgaard, M. Improved Methods for Building Protein Models in Electron Density Maps and the Location of Errors in These Models. Acta Crystallgr. A 1991, 47, 110–119. [Google Scholar] [CrossRef]
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural Basis for Altered Activity of M- and H-Isozyme Forms of Human Lactate Dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar] [CrossRef]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & Nmr System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallgr. D Biol. Crystallgr. 1998, 54, 905–921. [Google Scholar]
- Kraulis, P.J. Molscript—A Program to Produce Both Detailed and Schematic Plots of Protein Structures. J. Appl. Crystallgr. 1991, 24, 946–950. [Google Scholar] [CrossRef]
- Nichols, B.P.; Yanofsky, C. Nucleotide Sequences of Trpa of Salmonella Typhimurium and Escherichia Coli: An Evolutionary Comparison. Proc. Natl. Acad. Sci. USA 1979, 76, 5244–5248. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.A.; Murphy, M.E. Raster3d Version 2.0. A Program for Photorealistic Molecular Graphics. Acta Crystallgr. D Biol. Crystallgr. 1994, 50, 869–873. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The Pymol Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Chung, T.W.; Kim, S.J.; Choi, H.J.; Kim, K.J.; Kim, M.J.; Kim, S.H.; Lee, H.J.; Ko, J.H.; Lee, Y.C.; Suzuki, A.; et al. Ganglioside Gm3 Inhibits Vegf/Vegfr-2-Mediated Angiogenesis: Direct Interaction of Gm3 with Vegfr-2. Glycobiology 2009, 19, 229–239. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Cell Viability (μM) | LDHA Activity (μM) |
|---|---|---|
| WiDr | 17.19 ± 5.75 | 61.87 ± 8.02 |
| DLD-1 | 423.41 ± 17.55 | 129.46 ± 24.82 |
| RKO | 192.16 ± 15.19 | 73.67 ± 20.69 |
| HT29 | 107.01 ± 4.66 | 48.42 ± 4.33 |
| MCF-7 | 216.75 ± 12.88 | 61.18 ± 3.02 |
| HCC-95 | 228.38 ± 13.93 | 112.34 ± 10.11 |
| NCI-H1793 | 96.68 ± 5.32 | 63.99 ± 5.61 |
| Huh7 | 198.44 ± 26.70 | 85.07 ± 20.90 |
| HepG2 | 155.51 ± 23.24 | 100.78 ± 40.47 |
| Hep3B | 139.50 ± 8.55 | 48.30 ± 4.13 |
| Cell Line | MA 0 µM | MA 10 µM | MA 50 µM |
|---|---|---|---|
| Lactate (mM) | |||
| WiDr | 3.37 ± 0.06 | 3.22 ± 0.05 | 2.70 ± 0.06 |
| DLD-1 | 4.74 ± 0.05 | 3.68 ± 0.09 | 2.94 ± 0.17 |
| R.K.O | 2.94 ± 0.07 | 2.76 ± 0.06 | 2.48 ± 0.06 |
| HT29 | 3.68 ± 0.18 | 3.42 ± 0.37 | 2.73 ± 0.04 |
| MCF7 | 1.13 ± 0.14 | 1.07 ± 0.15 | 0.77 ± 0.19 |
| HCC-95 | 2.68 ± 0.09 | 2.25 ± 0.16 | 1.76 ± 0.10 |
| NCI-H1793 | 2.72 ± 0.48 | 2.36 ± 0.26 | 2.04 ± 0.38 |
| Huh7 | 1.58 ± 0.30 | 1.14 ± 0.32 | 0.67 ± 0.15 |
| HepG2 | 0.98 ± 0.20 | 0.51 ± 0.06 | 0.47 ± 0.07 |
| Hep3B | 1.20 ± 0.02 | 1.12 ± 0.04 | 0.73 ± 0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, T.-W.; Kim, E.-Y.; Han, C.W.; Park, S.Y.; Jeong, M.S.; Yoon, D.; Choi, H.-J.; Jin, L.; Park, M.-J.; Kwon, Y.J.; et al. Machilin A Inhibits Tumor Growth and Macrophage M2 Polarization Through the Reduction of Lactic Acid. Cancers 2019, 11, 963. https://doi.org/10.3390/cancers11070963
Chung T-W, Kim E-Y, Han CW, Park SY, Jeong MS, Yoon D, Choi H-J, Jin L, Park M-J, Kwon YJ, et al. Machilin A Inhibits Tumor Growth and Macrophage M2 Polarization Through the Reduction of Lactic Acid. Cancers. 2019; 11(7):963. https://doi.org/10.3390/cancers11070963
Chicago/Turabian StyleChung, Tae-Wook, Eun-Yeong Kim, Chang Woo Han, So Young Park, Mi Suk Jeong, Dahye Yoon, Hee-Jung Choi, Ling Jin, Mi-Ju Park, Yun Ju Kwon, and et al. 2019. "Machilin A Inhibits Tumor Growth and Macrophage M2 Polarization Through the Reduction of Lactic Acid" Cancers 11, no. 7: 963. https://doi.org/10.3390/cancers11070963
APA StyleChung, T.-W., Kim, E.-Y., Han, C. W., Park, S. Y., Jeong, M. S., Yoon, D., Choi, H.-J., Jin, L., Park, M.-J., Kwon, Y. J., Lee, H., Kim, K.-J., Park, K. H., Kim, S., Jang, S. B., & Ha, K.-T. (2019). Machilin A Inhibits Tumor Growth and Macrophage M2 Polarization Through the Reduction of Lactic Acid. Cancers, 11(7), 963. https://doi.org/10.3390/cancers11070963