A Novel Mutation of the NARROW LEAF 1 Gene Adversely Affects Plant Architecture in Rice (Oryza sativa L.)




Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characterization of the W149 Mutant
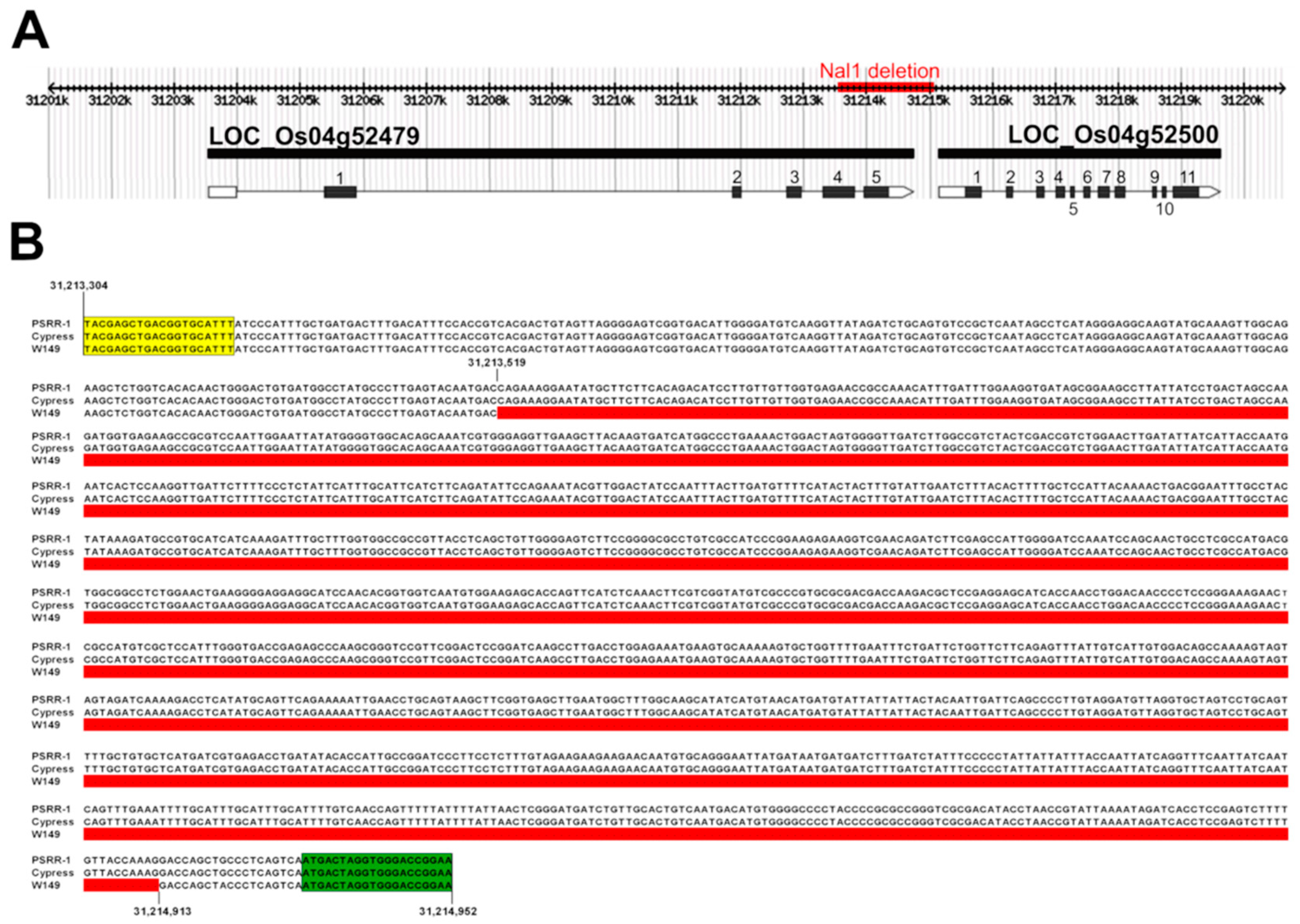
2.2. Bulked Segregant Analysis and Whole-Genome Sequencing Reveals Involvement of Nal1 Locus
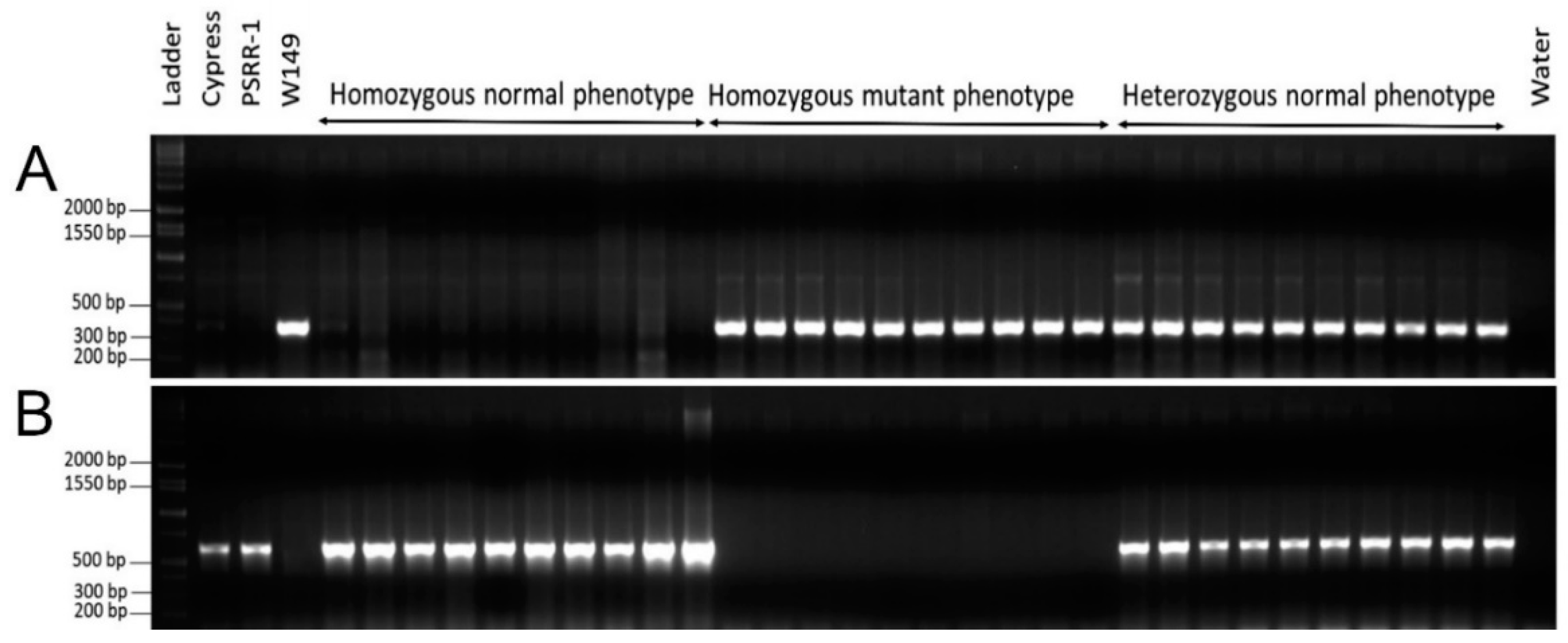
2.3. Genotyping and Phenotyping in Crosses Involving W149
2.4. Hormonal Response
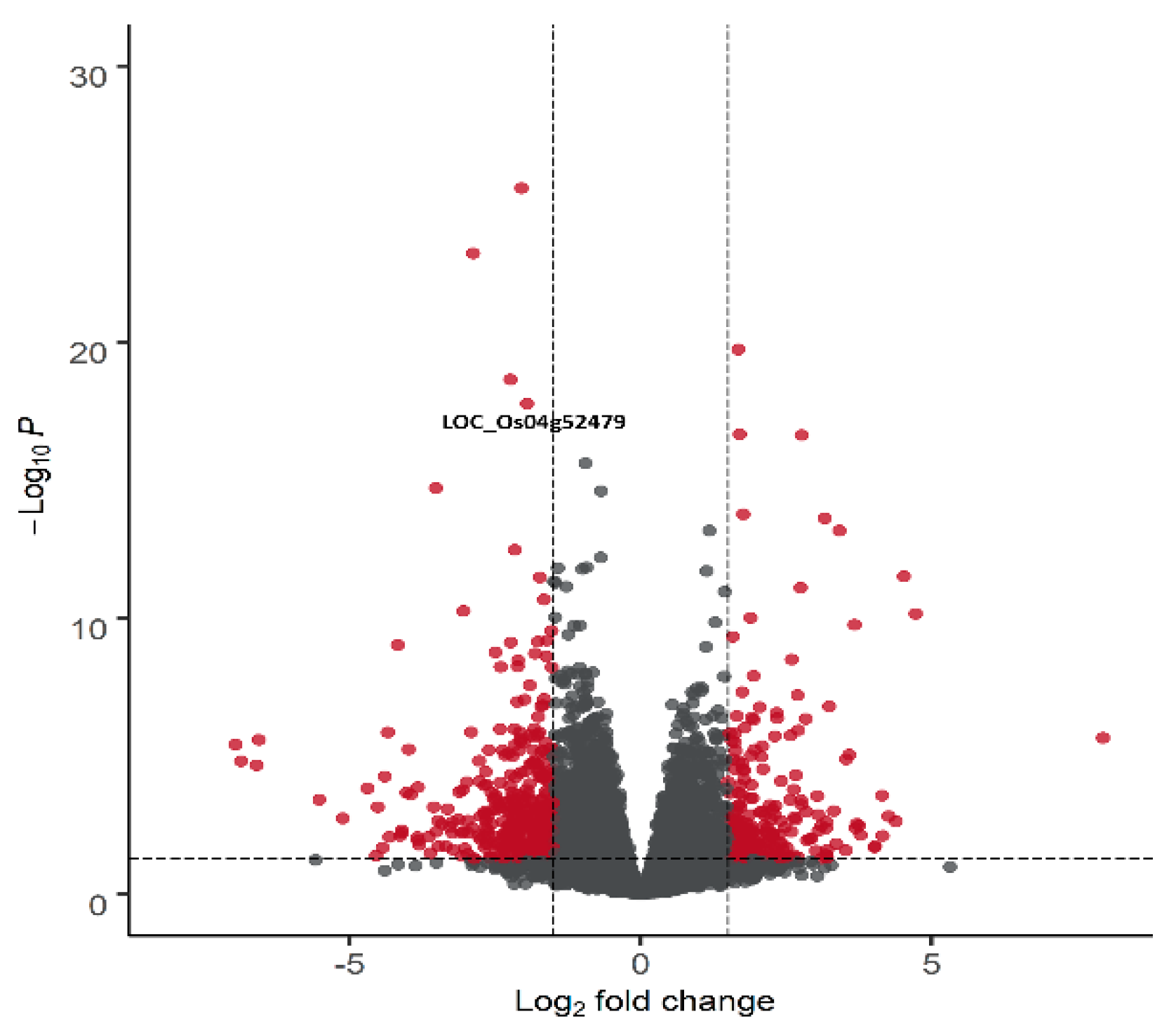
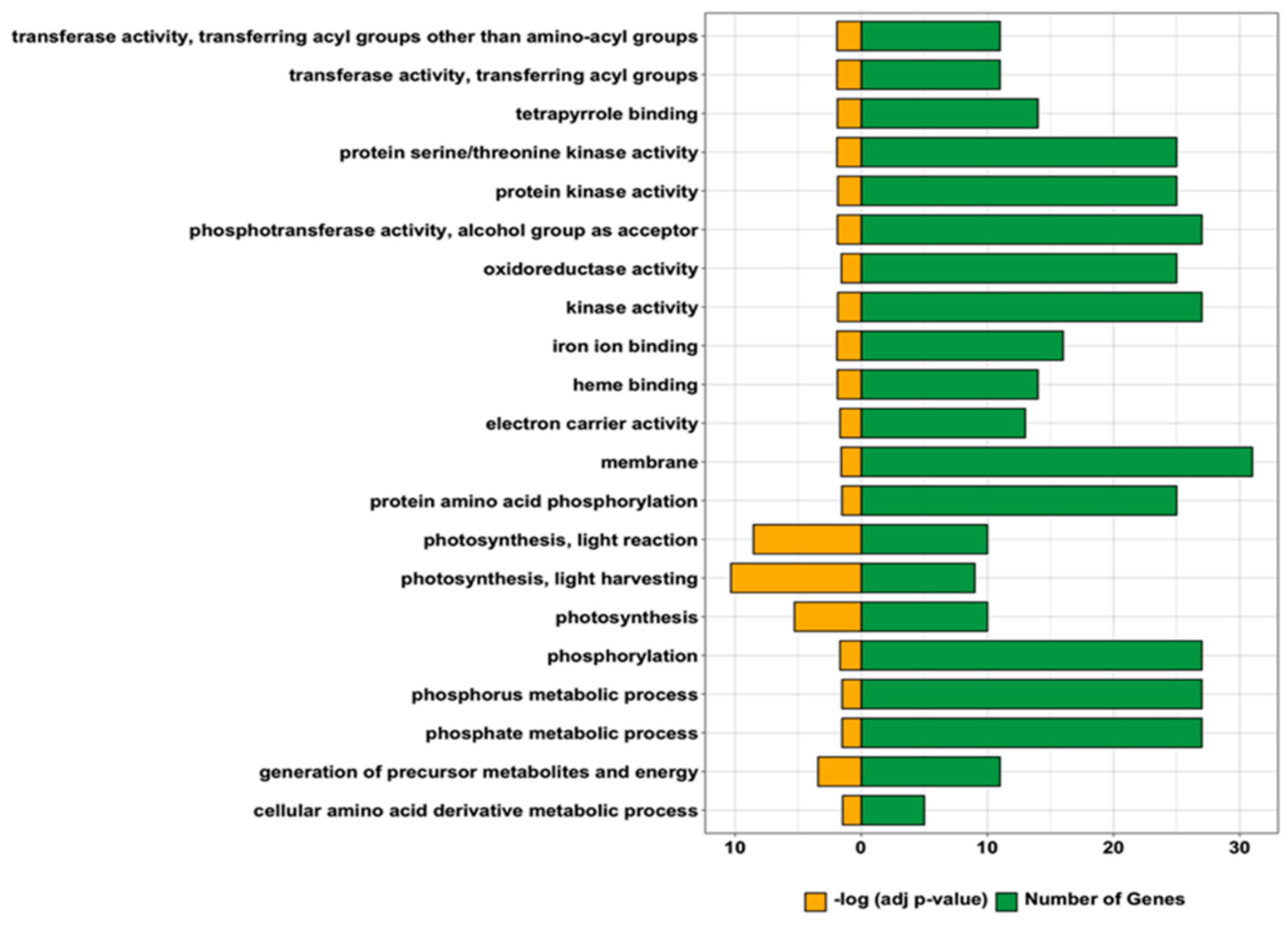
2.5. Comparative Transcriptomics
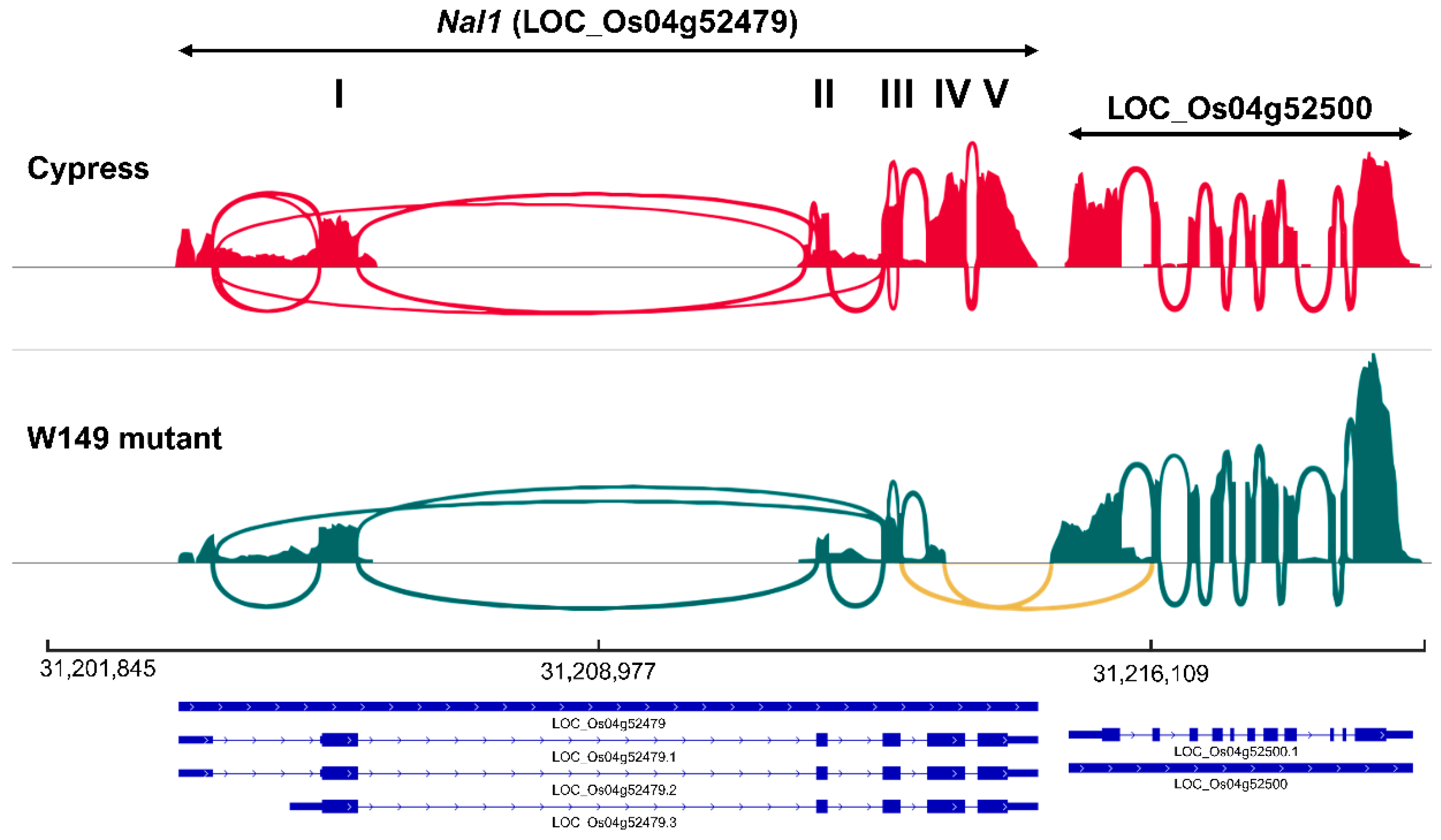
2.6. Identification and Validation of Chimeric nal1 Transcripts
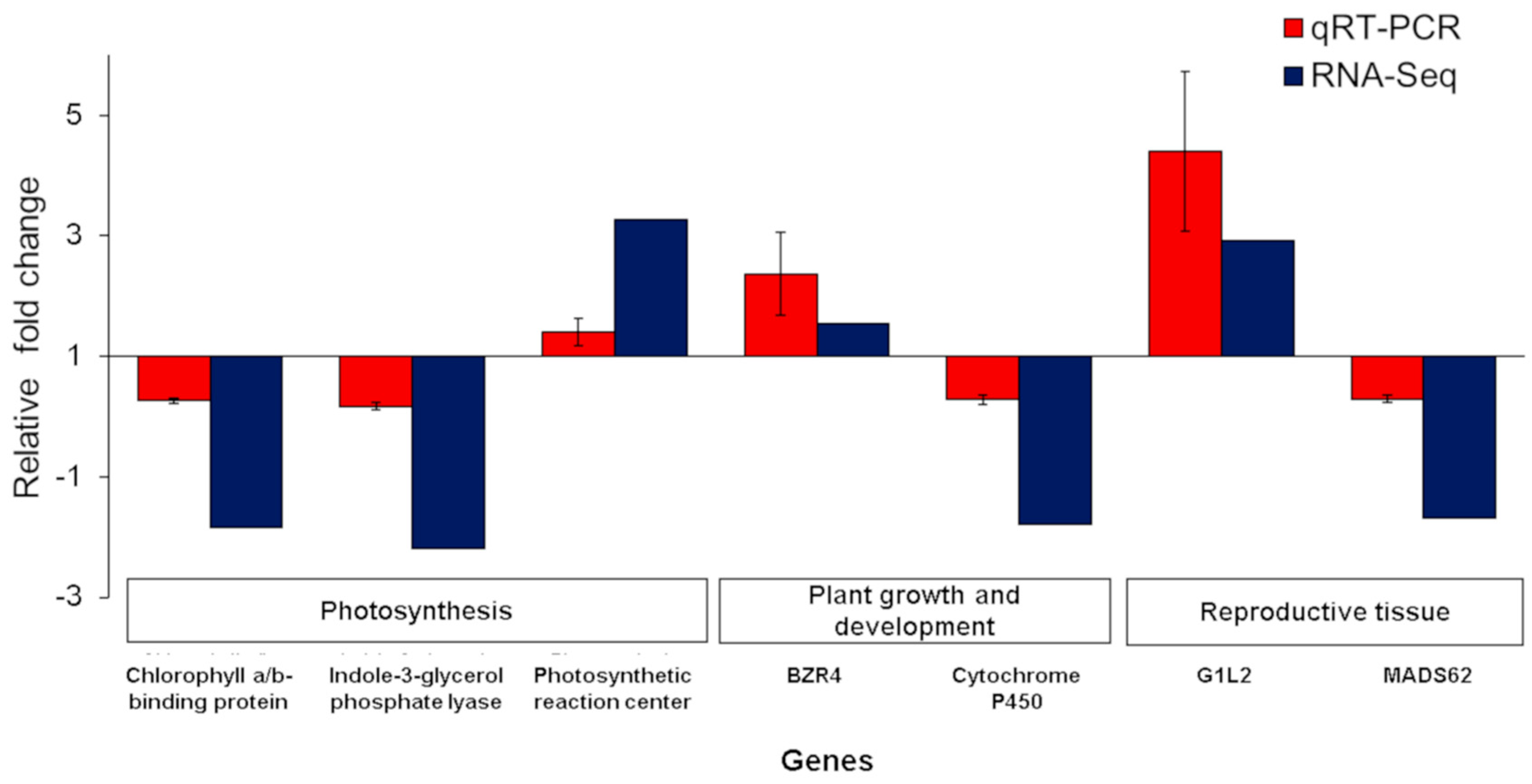
2.7. qRT-PCR of Differentially Expressed Genes
3. Discussion
3.1. Origin and Nature of the Mutation
3.2. Alteration in Plant Architecture Due to Pleiotropic Action of Nal1
3.3. Physiological and Molecular Basis of Mutant Phenotype
3.4. Role of Gibberellins (GA) and Brassinosteroid (BR)-Related Genes
4. Materials and Methods
4.1. Plant Materials and Cultivation
4.2. Origin of the W149 Mutant
4.3. Phenotypic Characterization of W149 Mutant
4.4. Hormonal Response
4.5. Mapping Populations for Cosegregation Test and Bulk Segregant Analysis (BSA)
4.6. Whole-Genome Sequencing (WGS) and Analysis of W149, Cypress, and PSRR-1
4.7. Confirmation of the Nal1 Deletion
4.8. Phenotyping and Genotyping of Nal1
- NalDel-W149-1F (5′-TGCCGCCCCATTATGGATTT-3′),
- NalDel-W149-1R (5′-GTACGCCAAACCGGATGAAG-3′),
- NalDel-CPRS-2F (5′-GGTGATAGCGGAAGCCTTATT-3′),
- and NalDel-CPRS-2R (5′-CGACATACCGACGAAGTTTGA-3′).
4.9. RNA-Seq Analysis
4.10. Validation of Gene Expression
4.11. Validation of Chimeric Nal1 Transcript
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GA | gibberellin |
| BR | brassinosteroid |
| QTL | quantitative trait loci |
| GPS | green for photosynthesis |
| FZP | frizzy panicle |
| UTR | untranslated region |
| LV | large vein |
| SV | small vein |
| IVD | interveinal distance |
| BSC | bundle sheath cells |
| VB | vascular bundle |
| MC | mesophyll cell |
| BSA | Bulked segregant analysis |
| SSR | simple sequence repeat |
| SNP | single nucleotide polymorphism |
| InDel | insertions-deletions |
| IBA | indole-3-butyric acid |
| NAA | 1-Naphthaleneacetic acid |
| 2,4-D | 2,4-dichlorophenoxyacetic acid |
| DEG | differentially expressed genes |
| GO | gene ontology |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| ns | nonsynonymous |
| ARF | auxin response factor |
| BP | biological process |
| MF | molecular function |
| CC | cellular component |
| SRA | Sequence Read Archive |
References
- Yang, X.C.; Hwa, C.M. Genetic modification of plant architecture and variety improvement in rice. Heredity 2008, 101, 396–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurata, N.; Miyoshi, K.; Nonomura, K.; Yamazaki, Y.; Ito, Y. Rice mutants and genes related to organ development, morphogenesis and physiological traits. Plant Cell Physiol. 2005, 46, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Guiderdoni, E.; An, G.; Hsing, Y.-I.C.; Han, C.-D.; Lee, M.C.; Yu, S.-M.; Upadhyaya, N.M.; Ramachandran, S.; Zhang, Q.; et al. Mutant resources in rice for functional genomics of the grasses. Plant Physiol. 2009, 149, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, J. Molecular basis of plant architecture. Annu. Rev. Plant Biol. 2008, 59, 253–279. [Google Scholar] [CrossRef]
- Spielmeyer, W.; Ellis, M.H.; Chandler, P.M. Semidwarf (sd-1), green revolution rice, contains a defective gibberellin 20-oxidase gene. Proc. Natl. Acad. Sci. USA 2002, 99, 9043–9048. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Hu, J.; Zhu, K.; Fang, Y.; Gao, Z.; He, Y.; Zhang, G.; Guo, L.; Zeng, D.; Dong, G.; et al. Identification and characterization of shortened uppermost internode 1, a gene negatively regulating uppermost internode elongation in rice. Plant Mol. Biol. 2011, 77, 475–487. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, L.; Zeng, D.; Gao, Z.; Guo, L.; Fang, Y.; Zhang, G.; Dong, G.; Yan, M.; Liu, J.; et al. Identification and characterization of narrow and rolled leaf 1, a novel gene regulating leaf morphology and plant architecture in rice. Plant Mol. Biol. 2010, 73, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Fujino, K.; Matsuda, Y.; Ozawa, K.; Nishimura, T.; Koshiba, T.; Fraaije, M.W.; Sekiguchi, H. Narrow leaf 7 controls leaf shape mediated by auxin in rice. Mol. Genet. Genom. 2008, 279, 499–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.H.; Yoo, S.C.; Zhang, H.; Pandeya, D.; Koh, H.J.; Hwang, J.Y.; Kim, G.T.; Paek, N.C. The rice narrow leaf2 and narrow leaf3 loci encode WUSCHEL-related homeobox 3A (OsWOX3A) and function in leaf, spikelet, tiller and lateral root development. New Phytol. 2013, 198, 1071–1084. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, Y.; Xue, D.; Wang, J.; Yan, M.; Liu, G.; Dong, G.; Zeng, D.; Lu, Z.; Zhu, X.; et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 2010, 42, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ikeda, M.; Matsubara, A.; Song, X.J.; Ito, M.; Asano, K.; Matsuoka, M.; Kitano, H.; Ashikari, M. OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 2010, 42, 545–549. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.-W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.R.; Mezey, J.G.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Qian, Q.; Bu, Q.; Li, S.; Chen, Q.; Sun, J.; Liang, W.; Zhou, Y.; Chu, C.; Li, X.; et al. Mutation of the rice narrow leaf1 gene, which encodes a novel protein, affects vein patterning and polar auxin transport. Plant Physiol. 2008, 147, 1947–1959. [Google Scholar] [CrossRef] [Green Version]
- Fujita, D.; Trijatmiko, K.R.; Tagle, A.G.; Sapasap, M.V.; Koide, Y.; Sasaki, K.; Tsakirpaloglou, N.; Gannaban, R.B.; Nishimura, T.; Yanagihara, S.; et al. NAL1 allele from a rice landrace greatly increases yield in modern indica cultivars. Proc. Natl. Acad. Sci. USA 2013, 110, 20431–20436. [Google Scholar] [CrossRef] [Green Version]
- Takai, T.; Adachi, S.; Taguchi-Shiobara, F.; Sanoh-Arai, Y.; Iwasawa, N.; Yoshinaga, S.; Hirose, S.; Taniguchi, Y.; Yamanouchi, U.; Wu, J.; et al. A natural variant of NAL1, selected in high-yield rice breeding programs, pleiotropically increases photosynthesis rate. Sci. Rep. 2013, 3, 2149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-H.; Li, S.-Y.; Wang, L.; Ye, W.-J.; Zeng, D.-L.; Rao, Y.-C.; Peng, Y.-L.; Hu, J.; Yang, Y.-L.; Xu, J.; et al. LSCHL4 from japonica cultivar, which is allelic to NAL1, increases yield of indica super rice 93–11. Mol. Plant 2014, 7, 1350–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Luo, J.; Shao, G.; Wei, X.; Tang, S.; Sheng, Z.; Song, J.; Hu, P. Fine mapping of a major QTL for flag leaf width in rice, qFLW4, which might be caused by alternative splicing of NAL1. Plant Cell Rep. 2012, 31, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.L.; Wang, Y.; Zhang, F.; Wu, Y.; Zheng, T.Q.; Wang, Y.H.; Zhao, X.Q.; Cui, Y.R.; Chen, K.; Zhang, Q.; et al. SS1 (NAL1)- and SS2-mediated genetic networks underlying source-sink and yield traits in rice (Oryza sativa L.). PLoS ONE 2015, 10, e0132060. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Fang, J.; Lou, L.; Zhao, J.; Yuan, S.; Yin, L.; Sun, W.; Peng, L.; Guo, B.; Li, X. Characterization of a null allelic mutant of the rice nal1 gene reveals its role in regulating cell division. PLoS ONE 2015, 10, e0118169. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.H.; Yoo, S.H.; Zhang, H.; Lim, J.H.; Paek, N.C. Rice NARROW LEAF1 regulates leaf and adventitious root development. Plant Mol. Biol. Rep. 2014, 32, 270–281. [Google Scholar] [CrossRef]
- Taguchi-Shiobara, F.; Ota, T.; Ebana, K.; Ookawa, T.; Yamasaki, M.; Tanabata, T.; Yamanouchi, U.; Wu, J.; Ono, N.; Nonoue, Y.; et al. Natural variation in the flag leaf morphology of rice due to a mutation of the NARROW LEAF 1 gene in Oryza sativa L. Genetics 2015, 201, 795–808. [Google Scholar] [CrossRef] [Green Version]
- Kubo, F.C.; Yasui, Y.; Kumamaru, T.; Sato, Y.; Hirano, H.Y. Genetic analysis of rice mutants responsible for narrow leaf phenotype and reduced vein number. Genes Genet. Syst. 2016, 91, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Zhao, Y.; Liu, F.; Chen, Q.; Qi, J. Narrow leaf 1 (NAL1) regulates leaf shape by affecting cell expansion in rice (Oryza sativa L.). Biochem. Biophys. Res. Commun. 2019, 516, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhao, S.; Fu, Y.; Sun, H.; Ma, X.; Tan, L.; Liu, F.; Sun, X.; Sun, H.; Gu, P.; et al. Variation in the regulatory region of FZP causes increases in secondary inflorescence branching and grain yield in rice domestication. Plant J. 2018, 96, 716–733. [Google Scholar] [CrossRef] [Green Version]
- Baack, E.J.; Rieseberg, L.H. A genomic view of introgression and hybrid speciation. Curr. Opin. Genet. Develop. 2007, 17, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Fu, D.; Gong, H.; Qian, W.; Xia, W.; Pires, J.C.; Li, R.; Long, Y.; Mason, A.S.; Yang, T.-J.; et al. De novo genetic variation associated with retrotransposon activation, genomic rearrangements, and trait variation in a recombinant inbred line population of Brassica napus derived from interspecific hybridization with Brassica rapa. Plant J. 2011, 68, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Bashir, T.; Mishra, R.C.; Hasan, M.M.; Mohanta, T.K.; Bae, H. Effect of hybridization on somatic mutations and genomic rearrangements in plants. Intl. J. Mol. Sci. 2018, 19, 3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, C.; Shankar, R.; Jain, M.; Subudhi, P.K. Genome-wide discovery of DNA polymorphisms by whole genome sequencing differentiates weedy and cultivated rice. Sci. Rep. 2018, 8, 14218. [Google Scholar] [CrossRef]
- Subudhi, P.K.; De Leon, T.; Singh, P.K.; Parco, A.; Cohn, M.A.; Sasaki, T. A chromosome segment substitution library of weedy rice for genetic dissection of complex agronomic and domestication traits. PLoS ONE 2015, 10, e0130650. [Google Scholar] [CrossRef] [Green Version]
- Thomson, T.M.; Lozano, J.J.; Loukili, N.; Carrió, R.; Serras, F.; Cormand, B.; Valeri, M.; DíazV, M.; Abril, J.; Burset, M.; et al. Fusion of the human gene for the polyubiquitination coeffector UEV1 with Kua, a newly identified gene. Genome Res. 2000, 10, 1743–1756. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Tang, Y.; Liu, J.; Tan, L.; Jiang, J.; Wang, M.; Zhu, Z.; Sun, X.; Sun, C. Emergence of a novel chimeric gene underlying grain number in rice. Genetics 2017, 205, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, H.; Fan, C.; Li, J.; Shi, J.; Cai, Z.; Zhang, G.; Liu, D.; Zhang, J.; Vang, S.; et al. High rate of chimeric gene origination by retroposition in plant genomes. Plant Cell 2006, 18, 1791–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, M.; Betrán, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef]
- Itoh, J.; Hibara, K.; Sato, Y.; Nagato, Y. Developmental role and auxin responsiveness of Class III homeodomain leucine zipper gene family members in rice. Plant Physiol. 2008, 147, 1960–1975. [Google Scholar] [CrossRef] [Green Version]
- Itoh, J.; Nonomura, K.; Ikeda, K.; Yamaki, S.; Inukai, Y.; Yamagishi, H.; Kitano, H.; Nagato, Y. Rice plant development: From zygote to spikelet. Plant Cell Physiol. 2005, 46, 23–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathan, J.; Bhattacharya, J.; Ranjan, A. Enhancing crop yield by optimizing plant developmental features. Development 2016, 143, 3283–3294. [Google Scholar] [CrossRef] [Green Version]
- Frey, M.; Chomet, P.; Glawischnig, E.; Stettner, C.; Grün, S.; Winklmair, A.; Eisenreich, W.; Bacher, A.; Meeley, R.B.; Briggs, S.P.; et al. Analysis of a chemical plant defense mechanism in grasses. Science 1997, 277, 696–699. [Google Scholar] [CrossRef]
- Lu, G.; Coneva, V.; Casaretto, J.A.; Ying, S.; Mahmood, K.; Liu, F.; Nambara, E.; Bi, Y.M.; Rothstein, S.J. OsPIN5b modulates rice (Oryza sativa) plant architecture and yield by changing auxin homeostasis, transport and distribution. Plant J. 2015, 83, 913–925. [Google Scholar] [CrossRef]
- Saini, S.; Sharma, I.; Pati, P.K. Versatile roles of brassinosteroid in plants in the context of its homoeostasis, signaling and crosstalks. Front. Plant Sci. 2015, 6, 950. [Google Scholar] [CrossRef] [Green Version]
- Schwechheimer, C. Gibberellin signaling in plants—The extended version. Front. Plant Sci. 2011, 2, 107. [Google Scholar] [CrossRef] [Green Version]
- Tong, H.; Xiao, Y.; Liu, D.; Gao, S.; Liu, L.; Yin, Y.; Jin, Y.; Qian, Q.; Chua, C. Brassinosteroid regulates cell elongation by modulating gibberellin metabolism in rice. Plant Cell 2014, 26, 376–4393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, N.R. Taking hormone crosstalk to a new level: Brassinosteroids regulate gibberellin biosynthesis. Plant Cell 2015, 27, 2081. [Google Scholar] [CrossRef] [Green Version]
- Ueguchi-Tanaka, M.; Nakajima, M.; Motoyuki, A.; Matsuoka, M. Gibberellin receptor and its role in gibberellin signaling in plants. Annu. Rev. Plant Biol. 2007, 58, 183–198. [Google Scholar] [CrossRef] [Green Version]
- Nahirnak, V.; Almasia, N.I.; Hopp, H.E.; Vazquez-Rovere, C. Snakin/GASA proteins: Involvement in hormone crosstalk and redox homeostasis. Plant Signal. Behav. 2012, 7, 1004–1008. [Google Scholar] [CrossRef] [Green Version]
- Khew, C.Y.; Teo, C.J.; Chan, W.S.; Wong, H.L.; Namasivayam, P.; Ho, C.L. Brassinosteroid insensitive 1-associated kinase 1 (OsI-BAK1) is associated with grain filling and leaf development in rice. J. Plant Physiol. 2015, 182, 23–32. [Google Scholar] [CrossRef] [PubMed]
- He, J.X.; Gendron, J.M.; Sun, Y.; Gampala, S.S.L.; Gendron, N.; Sun, C.Q.; Wang, Z.Y. BZR1 is a transcriptional repressor with dual roles in brassinosteroid homeostasis and growth responses. Science 2005, 307, 1634–1638. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Y.; Bai, M.; Oh, E.; Zhu, J.Y. Brassinosteroid signaling network and regulation of photomorphogenesis. Annu. Rev. Genet. 2012, 46, 701–724. [Google Scholar] [CrossRef] [PubMed]
- Caño-Delgado, A.; Yin, Y.; Yu, C.; Vafeados, D.; Mora-García, S.; Cheng, J.C.; Nam, K.H.; Li, J.; Chory, J. BRL1 and BRL3 are novel brassinosteroid receptors that function in vascular differentiation in Arabidopsis. Development 2004, 131, 5341–5351. [Google Scholar] [CrossRef] [Green Version]
- Linscombe, S.D.; Jodari, F.; Mckenzie, K.S.; Bollich, P.K.; White, L.M.; Groth, D.E.; Dunand, R.T. Registration of Cypress rice. Crop. Sci. 1993, 33, 355. [Google Scholar] [CrossRef]
- Linscombe, S.D.; Jodari, F.; Mckenzie, K.S.; Bollich, P.K.; White, L.M.; Groth, D.E.; Dunand, R.T. Registration of Bengal rice. Crop. Sci. 1993, 33, 645–646. [Google Scholar] [CrossRef]
- Subudhi, P.K.; Parco, A.; Singh, P.K.; De Leon, T.; Karan, R.; Biradar, H.; Cohn, M.A.; Brar, D.S.; Sasaki, T. Genetic architecture of seed dormancy in US weedy rice in different genetic backgrounds. Crop. Sci. 2012, 52, 2564–2575. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bullet. 1987, 19, 11–15. [Google Scholar]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, Y.; De La Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Eisfeldt, J.; Vezzi, F.; Olason, P.; Nilsson, D.; Lindstrand, A. TIDDIT, an efficient and comprehensive structural variant caller for massive parallel sequencing data. F1000 Res. 2017, 6, 664. [Google Scholar] [CrossRef]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.; Lu, X.; Ruden, D. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucl. Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Frazee, A.C.; Collado-Torres, L.; Jaffe, A.E.; Leek, J.T. Ballgown: Flexible, Isoform-Level Differential Expression Analysis. R Package Version 2.18.0. 2019. Available online: https://rdrr.io/bioc/ballgown/ (accessed on 29 October 2020).
- Willems, E.; Leyns, L.; Vandesompele, J. Standardization of real-time PCR gene expression data from independent biological replicates. Anal. Biochem. 2008, 379, 127–129. [Google Scholar] [CrossRef] [PubMed]
- McSteen, P. Hormonal regulation of branching in grasses. Plant Physiol. 2009, 149, 46–55. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | Cypress | W149 |
|---|---|---|
| Plant height (cm) | 81.1 ± 1.6 (10) | 50.7 ± 1.9 * (10) |
| Panicle length (cm) | 18.6 ± 0.3 (10) | 11.3 ± 0.3 * (10) |
| Leaf length (cm) | 43.0 ± 2.0 (10) | 22.9 ± 0.7 * (10) |
| Leaf width (cm) | 1.2 ± 0.0 (10) | 0.5 ± 0.0 * (10) |
| Leaf greenness (SPAD value) | 41.9 ± 0.8 (10) | 50.9 ± 0.6 * (10) |
| Number of large veins (LV) | 9.6 ± 0.3 (5) | 9.2 ± 0.4 ns (5) |
| Number of small veins (SV) | 52.4 ± 0.7 (5) | 29.6 ± 0.5 * (5) |
| Number of SV between LV | 6.14 ± 0.3 (7) | 3.6 ± 0.2 * (7) |
| Leaf midrib | Pronounced | Not pronounced |
| Leaf thickness (µm) | 90.2 ± 3.3 (9) | 139.1 ± 0.6 * (9) |
| Interveinal distance (µm) | 198.1 ± 4.4 (9) | 152.3 ± 4.1 * (9) |
| Mesophyll cell (MC) area (µm2) | 10683.1 ± 254.5 (9) | 11941.7 ± 717.6 ns (9) |
| Lateral MC number | 10.1 ± 0.3 (9) | 5.0 ± 0.1 * (9) |
| MC layer number | 3.3 ± 0.2 (9) | 4.7 ± 0.2 * (9) |
| Vascular area (µm2) | 589.0 ± 29.4 (18) | 963.0 ± 75.3 * (18) |
| Bundle sheath cell (BSC) area (µm2) | 1557.6 ± 22.7 (18) | 2759.4 ± 131.8 * (18) |
| Vascular bundle + BSC area (µm2) | 2146.6 ± 31.71 (18) | 3722.4 ± 190.6 * (18) |
| Bundle sheath cell extension number | 1.9 ± 0.2 (18) | 5.2 ± 0.3 *(18) |
| Panicle neck (mm2) | 0.7 ± 0.0 (3) | 0.2 ± 0.0 * (3) |
| Number of grains per panicle | 270.2 ± 24.3 (9) | 89.6 ± 10.0 * (9) |
| 100-grain weight (g) | 2.2 ± 0.04 (9) | 1.8 ± 0.05 * (9) |
| Total grain weight per panicle (g) | 5.9 ± 0.56 (9) | 1.6 ± 0.21 * (9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subudhi, P.K.; Garcia, R.S.; Coronejo, S.; De Leon, T.B. A Novel Mutation of the NARROW LEAF 1 Gene Adversely Affects Plant Architecture in Rice (Oryza sativa L.). Int. J. Mol. Sci. 2020, 21, 8106. https://doi.org/10.3390/ijms21218106
Subudhi PK, Garcia RS, Coronejo S, De Leon TB. A Novel Mutation of the NARROW LEAF 1 Gene Adversely Affects Plant Architecture in Rice (Oryza sativa L.). International Journal of Molecular Sciences. 2020; 21(21):8106. https://doi.org/10.3390/ijms21218106
Chicago/Turabian StyleSubudhi, Prasanta K., Richard S. Garcia, Sapphire Coronejo, and Teresa B. De Leon. 2020. "A Novel Mutation of the NARROW LEAF 1 Gene Adversely Affects Plant Architecture in Rice (Oryza sativa L.)" International Journal of Molecular Sciences 21, no. 21: 8106. https://doi.org/10.3390/ijms21218106