
GDF15 and Cardiac Cells: Current Concepts and New Insights


{kind=link}
{kind=link}
{kind=link}
Abstract
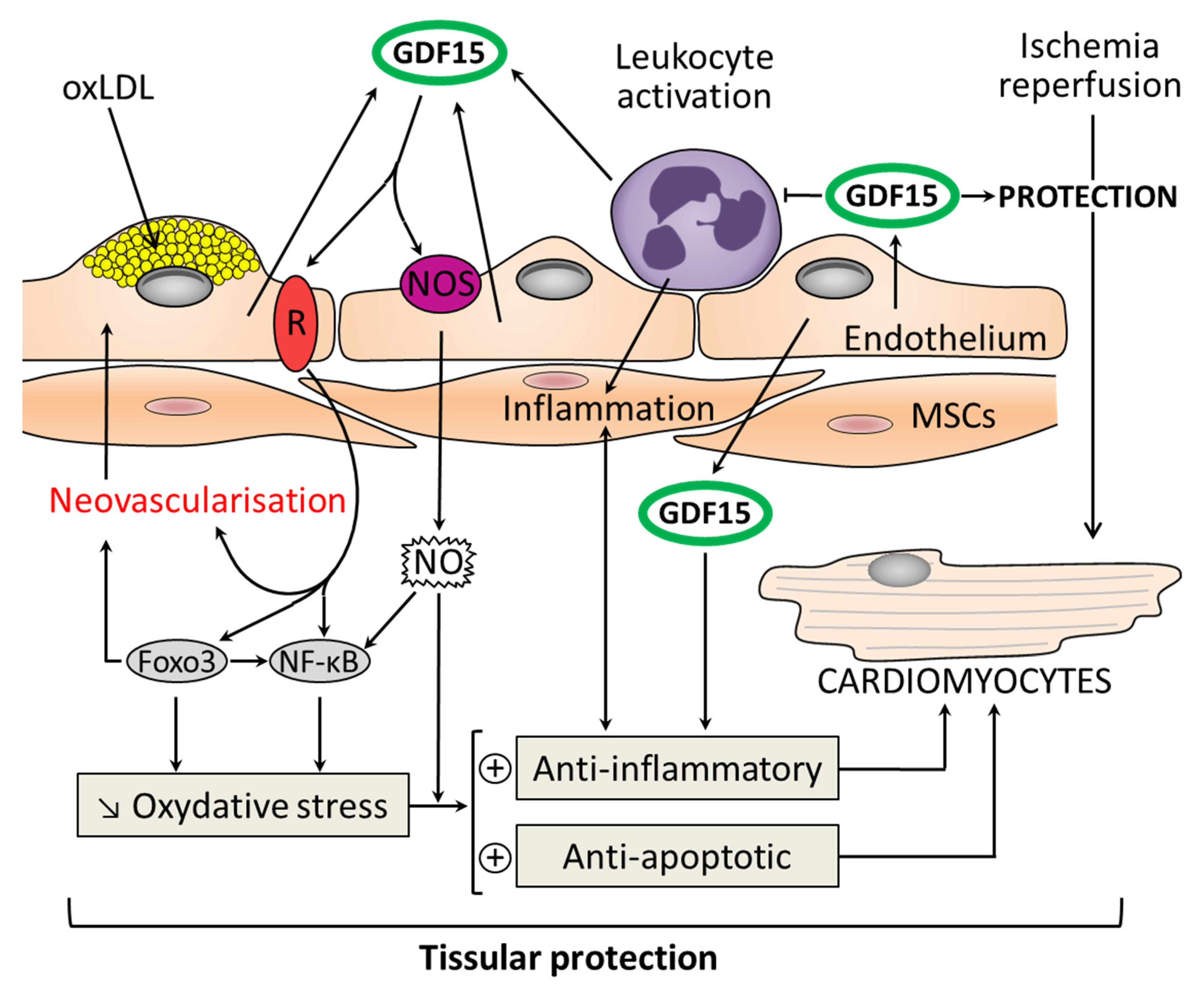
:1. Introduction
2. Cardiac Cellular Senescence and Tissue Regeneration
3. Cardiac Remodeling and Progressive Heart Dysfunction
4. Basic Biology of GDF15
4.1. Synthesis, Structure, Secretion, and Distribution of GDF15
4.2. Distribution of GDF15
4.3. GDF15 and Cellular Senescence
5. GDF15 Signaling—Molecular Mechanisms Underlying GDF15 Activity
5.1. GDF15: Interaction with Smad Signaling and miRNA
5.2. Molecular Mechanisms of GDF15 and Oxygen Metabolism
5.3. GDF15: A Regulator of Mitochondrial Functions
5.4. GFRAL: An Orphan Member of the GDNF Receptor Family Identified as the Receptor for GDF15
6. GDF15 as a Metabolic Regulator in Relation to Inflammation
7. Role of GDF15 in Adapting the Body to Metabolic Conditions: Effect on the Homeostasis of the Heart
8. Perspectives for Clinical Applications of GDF15 in Cardiovascular Domain
9. GDF15 as an Emerging Biomarker Reflecting Mitochondrial Function
10. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galdos, F.X.; Guo, Y.; Paige, S.L.; VanDusen, N.J.; Wu, S.M.; Pu, W.T. Cardiac Regeneration: Lessons From Development. Circ. Res. 2017, 120, 941–959. [Google Scholar] [CrossRef] [Green Version]
- Harary, I.; Farley, B. In vitro studies on single beating rat heart cells. II. Intercellular communication. Exp. Cell Res. 1963, 29, 466–474. [Google Scholar] [CrossRef]
- Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Squiers, G.T.; McLellan, M.A.; Ilinykh, A.; Branca, J.; Rosenthal, N.A.; Pinto, A.R. Cardiac Cellularity is Dependent upon Biological Sex and is Regulated by Gonadal Hormones. Cardiovasc. Res. 2020, cvaa265. [Google Scholar] [CrossRef] [PubMed]
- Saucerman, J.J.; Tan, P.M.; Buchholz, K.S.; McCulloch, A.D.; Omens, J.H. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 2019, 16, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Rupert, C.E.; Coulombe, K.L. The roles of neuregulin-1 in cardiac development, homeostasis, and disease. Biomark. Insights 2015, 10 (Suppl. 1), BMI-S20061. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef] [PubMed]
- Tirziu, D.; Giordano, F.J.; Simons, M. Cell communications in the heart. Circulation 2010, 122, 928–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, F.; Ross, R.S.; Chen, J. Cell-cell connection to cardiac disease. Trends Cardiovasc. Med. 2009, 19, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Pope, A.J.; Sands, G.B.; Smaill, B.H.; LeGrice, I.J. Three-dimensional transmural organization of perimysial collagen in the heart. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1243–H1252. [Google Scholar] [CrossRef]
- Conigliaro, A.; Fontana, S.; Raimondo, S.; Alessandro, R. Exosomes: Nanocarriers of Biological Messages. Adv. Exp. Med. Biol. 2017, 998, 23–43. [Google Scholar] [PubMed]
- Li, N.; Rochette, L.; Wu, Y.; Rosenblatt-Velin, N. New Insights into the Role of Exosomes in the Heart After Myocardial Infarction. J. Cardiovasc. Transl. Res. 2019, 12, 18–27. [Google Scholar] [CrossRef]
- Rakusan, K.; Cicutti, N.; Flanagan, M.F. Changes in the microvascular network during cardiac growth, development, and aging. Cell. Mol. Biol. Res. 1994, 40, 117–122. [Google Scholar]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef]
- McClatchy, D.B.; Ma, Y.; Liem, D.A.; Ng, D.C.M.; Ping, P.; Yates, J.R., 3rd. Quantitative temporal analysis of protein dynamics in cardiac remodeling. J. Mol. Cell. Cardiol. 2018, 121, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Leri, A.; Kajstura, J.; Anversa, P. Mechanisms of myocardial regeneration. Trends Cardiovasc. Med. 2011, 21, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Karpac, J.; Jasper, H. Promoting longevity by maintaining metabolic and proliferative homeostasis. J. Exp. Biol. 2014, 217, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Oudot, A.; Martin, C.; Busseuil, D.; Vergely, C.; Demaison, L.; Rochette, L. NADPH oxidases are in part responsible for increased cardiovascular superoxide production during aging. Free Radic. Biol. Med. 2006, 40, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- Saliques, S.; Teyssier, J.R.; Vergely, C.; Lorgis, L.; Lorin, J.; Farnier, M.; Donzel, A.; Sicard, P.; Berchoud, J.; Lagrost, A.C.; et al. Circulating leukocyte telomere length and oxidative stress: A new target for statin therapy. Atherosclerosis 2011, 219, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Kirkwood, T.B.L.; Bohr, V.A. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 2019, 54, 100940. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef]
- Segers, V.F.; Lee, R.T. Protein therapeutics for cardiac regeneration after myocardial infarction. J. Cardiovasc. Transl. Res. 2010, 3, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Koninckx, R.; Daniëls, A.; Windmolders, S.; Mees, U.; Macianskiene, R.; Mubagwa, K.; Steels, P.; Jamaer, L.; Dubois, J.; Robic, B.; et al. The cardiac atrial appendage stem cell: A new and promising candidate for myocardial repair. Cardiovasc. Res. 2013, 97, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Mazini, L.; Rochette, L.; Amine, M.; Malka, G. Regenerative Capacity of Adipose Derived Stem Cells (ADSCs), Comparison with Mesenchymal Stem Cells (MSCs). Int. J. Mol. Sci. 2019, 20, 2523. [Google Scholar] [CrossRef] [Green Version]
- Gnecchi, M.; Danieli, P.; Malpasso, G.; Ciuffreda, M.C. Paracrine Mechanisms of Mesenchymal Stem Cells in Tissue Repair. Methods Mol. Biol. 2016, 1416, 123–146. [Google Scholar] [PubMed]
- Lupu, I.E.; De Val, S.; Smart, N. Coronary vessel formation in development and disease: Mechanisms and insights for therapy. Nat. Rev. Cardiol. 2020, 17, 790–806. [Google Scholar] [CrossRef] [PubMed]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.J.; Kelly, D.P. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin. Exp. Pharmacol. Physiol. 2002, 29, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.C.; Sicard, R.E.; Schuler, H.G. Palmitate oxidation by isolated working fetal and newborn pig hearts. Am. J. Physiol. 1989, 256, E315–E321. [Google Scholar] [CrossRef]
- Cardoso, A.C.; Lam, N.T.; Savla, J.J.; Nakada, Y.; Pereira, A.H.M.; Elnwasany, A.; Menendez-Montes, I.; Ensley, E.L.; Petric, U.B.; Sharma, G.; et al. Mitochondrial Substrate Utilization Regulates Cardiomyocyte Cell Cycle Progression. Nat. Metab. 2020, 2, 167–178. [Google Scholar] [CrossRef]
- Barreto, S.; Hamel, L.; Schiatti, T.; Yang, Y.; George, V. Cardiac Progenitor Cells from Stem Cells: Learning from Genetics and Biomaterials. Cells 2019, 8, 1536. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, L.; Guenancia, C.; Gudjoncik, A.; Hachet, O.; Zeller, M.; Cottin, Y.; Vergely, C. Anthracyclines/trastuzumab: New aspects of cardiotoxicity and molecular mechanisms. Trends Pharmacol. Sci. 2015, 36, 326–348. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Pittet, M.J.; Swirski, F.K. Monocytes: Protagonists of infarct inflammation and repair after myocardial infarction. Circulation 2010, 121, 2437–2445. [Google Scholar] [CrossRef]
- Tiwari, A.; Mukherjee, B.; Dixit, M. MicroRNA Key to Angiogenesis Regulation: MiRNA Biology and Therapy. Curr. Cancer Drug Targets 2018, 18, 266–277. [Google Scholar] [CrossRef]
- Wang, X.; Song, C.; Zhou, X.; Han, X.; Li, J.; Wang, Z.; Shang, H.; Liu, Y.; Cao, H. Mitochondria Associated MicroRNA Expression Profiling of Heart Failure. BioMed Res. Int. 2017, 2017, 4042509. [Google Scholar] [CrossRef] [PubMed]
- Bostjancic, E.; Zidar, N.; Stajer, D.; Glavac, D. MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology 2010, 115, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Eskildsen, T.V.; Schneider, M.; Sandberg, M.B.; Skov, V.; Bronnum, H.; Thomassen, M.; Kruse, T.A.; Andersen, D.C.; Sheikh, S.P. The microRNA-132/212 family fine-tunes multiple targets in Angiotensin II signalling in cardiac fibroblasts. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 1288–1297. [Google Scholar] [CrossRef]
- Gareri, C.; De Rosa, S.; Indolfi, C. MicroRNAs for Restenosis and Thrombosis After Vascular Injury. Circ. Res. 2016, 118, 1170–1184. [Google Scholar] [CrossRef] [Green Version]
- Herpin, A.; Lelong, C.; Favrel, P. Transforming growth factor-beta-related proteins: An ancestral and widespread superfamily of cytokines in metazoans. Dev. Comp. Immunol. 2004, 28, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Tobin, J.F.; Celeste, A.J. Bone morphogenetic proteins and growth differentiation factors as drug targets in cardiovascular and metabolic disease. Drug Discov. Today 2006, 11, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Unsicker, K.; Spittau, B.; Krieglstein, K. The multiple facets of the TGF-beta family cytokine growth/differentiation factor-15/macrophage inhibitory cytokine-1. Cytokine Growth Factor Rev. 2013, 24, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Growth and differentiation factor 11 (GDF11): Functions in the regulation of erythropoiesis and cardiac regeneration. Pharmacol. Ther. 2015, 156, 26–33. [Google Scholar] [CrossRef]
- Lawton, L.N.; Bonaldo, M.F.; Jelenc, P.C.; Qiu, L.; Baumes, S.A.; Marcelino, R.A.; de Jesus, G.M.; Wellington, S.; Knowles, J.A.; Warburton, D.; et al. Identification of a novel member of the TGF-beta superfamily highly expressed in human placenta. Gene 1997, 203, 17–26. [Google Scholar] [CrossRef]
- Ho, J.E.; Mahajan, A.; Chen, M.H.; Larson, M.G.; McCabe, E.L.; Ghorbani, A.; Cheng, S.; Johnson, A.D.; Lindgren, C.M.; Kempf, T.; et al. Clinical and genetic correlates of growth differentiation factor 15 in the community. Clin. Chem. 2012, 58, 1582–1591. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Thalamuthu, A.; Ho, J.E.; Mahajan, A.; Ek, W.E.; Brown, D.A.; Breit, S.N.; Wang, T.J.; Gyllensten, U.; Chen, M.H.; et al. A Meta-Analysis of Genome-Wide Association Studies of Growth Differentiation Factor-15 Concentration in Blood. Front. Genet. 2018, 9, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.A.; Breit, S.N.; Buring, J.; Fairlie, W.D.; Bauskin, A.R.; Liu, T.; Ridker, P.M. Concentration in plasma of macrophage inhibitory cytokine-1 and risk of cardiovascular events in women: A nested case-control study. Lancet 2002, 359, 2159–2163. [Google Scholar] [CrossRef]
- Kempf, T.; Horn-Wichmann, R.; Brabant, G.; Peter, T.; Allhoff, T.; Klein, G.; Drexler, H.; Johnston, N.; Wallentin, L.; Wollert, K.C. Circulating concentrations of growth-differentiation factor 15 in apparently healthy elderly individuals and patients with chronic heart failure as assessed by a new immunoradiometric sandwich assay. Clin. Chem. 2007, 53, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Wen, W.; Sachdev, P.S. Macrophage inhibitory cytokine-1/growth differentiation factor 15 as a marker of cognitive ageing and dementia. Curr. Opin. Psychiatry 2016, 29, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Rothenbacher, D.; Dallmeier, D.; Christow, H.; Koenig, W.; Denkinger, M.; Klenk, J. Association of growth differentiation factor 15 with other key biomarkers, functional parameters and mortality in community-dwelling older adults. Age Ageing 2019, 48, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chrysovergis, K.; Kosak, J.; Kissling, G.; Streicker, M.; Moser, G.; Li, R.; Eling, T.E. hNAG-1 increases lifespan by regulating energy metabolism and insulin/IGF-1/mTOR signaling. Aging 2014, 6, 690–704. [Google Scholar] [CrossRef] [Green Version]
- Kleinert, M.; Clemmensen, C.; Sjoberg, K.A.; Carl, C.S.; Jeppesen, J.F.; Wojtaszewski, J.F.P.; Kiens, B.; Richter, E.A. Exercise increases circulating GDF15 in humans. Mol. Metab. 2018, 9, 187–191. [Google Scholar] [CrossRef]
- Nair, N.; Gongora, E. Correlations of GDF-15 with sST2, MMPs, and worsening functional capacity in idiopathic dilated cardiomyopathy: Can we gain new insights into the pathophysiology? J. Circ. Biomark. 2018, 7, 1849454417751735. [Google Scholar] [CrossRef] [Green Version]
- Bootcov, M.R.; Bauskin, A.R.; Valenzuela, S.M.; Moore, A.G.; Bansal, M.; He, X.Y.; Zhang, H.P.; Donnellan, M.; Mahler, S.; Pryor, K.; et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 11514–11519. [Google Scholar] [CrossRef] [Green Version]
- Wollert, K.C.; Kempf, T.; Wallentin, L. Growth Differentiation Factor 15 as a Biomarker in Cardiovascular Disease. Clin. Chem. 2017, 63, 140–151. [Google Scholar] [CrossRef]
- Planavila, A.; Fernandez-Sola, J.; Villarroya, F. Cardiokines as Modulators of Stress-Induced Cardiac Disorders. Adv. Protein Chem. Struct. Biol. 2017, 108, 227–256. [Google Scholar] [PubMed]
- Mazagova, M.; Buikema, H.; Landheer, S.W.; Vavrinec, P.; Buiten, A.; Henning, R.H.; Deelman, L.E. Growth differentiation factor 15 impairs aortic contractile and relaxing function through altered caveolar signaling of the endothelium. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H709–H718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, S.I.; Winkens, B.; Goldschmeding, R.; van Geffen, A.J.; Nous, F.M.; van Kuik, J.; van der Weide, P.; Klopping, C.; Kirkels, J.H.; Lahpor, J.R.; et al. Circulating growth differentiation factor-15 correlates with myocardial fibrosis in patients with non-ischaemic dilated cardiomyopathy and decreases rapidly after left ventricular assist device support. Eur. J. Heart Fail. 2012, 14, 1249–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalloz, F.; Maingon, P.; Cottin, Y.; Briot, F.; Horiot, J.C.; Rochette, L. Effects of combined irradiation and doxorubicin treatment on cardiac function and antioxidant defenses in the rat. Free Radic. Biol. Med. 1999, 26, 785–800. [Google Scholar] [CrossRef]
- Becker, B.V.; Majewski, M.; Abend, M.; Palnek, A.; Nestler, K.; Port, M.; Ullmann, R. Gene expression changes in human iPSC-derived cardiomyocytes after X-ray irradiation. Int. J. Radiat. Biol. 2018, 94, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Daniels, L.B.; Clopton, P.; Laughlin, G.A.; Maisel, A.S.; Barrett-Connor, E. Growth-differentiation factor-15 is a robust, independent predictor of 11-year mortality risk in community-dwelling older adults: The Rancho Bernardo Study. Circulation 2011, 123, 2101–2110. [Google Scholar] [CrossRef] [Green Version]
- Fujita, Y.; Taniguchi, Y.; Shinkai, S.; Tanaka, M.; Ito, M. Secreted growth differentiation factor 15 as a potential biomarker for mitochondrial dysfunctions in aging and age-related disorders. Geriatr. Gerontol. Int. 2016, 16 (Suppl. 1), 17–29. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, C.H.; Jeong, J.H.; Park, M.; Kim, K.S. GDF15 contributes to radiation-induced senescence through the ROS-mediated p16 pathway in human endothelial cells. Oncotarget 2016, 7, 9634–9644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Y.L.; Hilal, S.; Chong, J.P.; Ng, Y.X.; Liew, O.W.; Xu, X.; Ikram, M.K.; Venketasubramanian, N.; Richards, A.M.; Lai, M.K.; et al. Growth differentiation factor-15 and white matter hyperintensities in cognitive impairment and dementia. Medicine 2016, 95, e4566. [Google Scholar] [CrossRef]
- Villeda, S.A.; Plambeck, K.E.; Middeldorp, J.; Castellano, J.M.; Mosher, K.I.; Luo, J.; Smith, L.K.; Bieri, G.; Lin, K.; Berdnik, D.; et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 2014, 20, 659–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Wang, Y.; Guan, K.; Sun, Y. PTGF-beta, a type beta transforming growth factor (TGF-beta) superfamily member, is a p53 target gene that inhibits tumor cell growth via TGF-beta signaling pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Lu, D.; Fu, Y.; Zhang, J.; Huang, X.; Cao, S.; Xu, D.; Bin, J.; Kitakaze, M.; Huang, Q.; et al. Olmesartan prevents cardiac rupture in mice with myocardial infarction by modulating growth differentiation factor 15 and p53. Br. J. Pharmacol. 2014, 171, 3741–3753. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Ma, Y.M.; Zheng, P.S.; Zhang, P. GDF15 promotes the proliferation of cervical cancer cells by phosphorylating AKT1 and Erk1/2 through the receptor ErbB2. J. Exp. Clin. Cancer Res. CR 2018, 37, 80. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Meloux, A.; Zeller, M.; Cottin, Y.; Vergely, C. Functional roles of GDF15 in modulating microenvironment to promote carcinogenesis. Biochim. Et Biophys. Acta Mol. Basis Dis. 2020, 1866, 165798. [Google Scholar] [CrossRef]
- Xu, J.; Kimball, T.R.; Lorenz, J.N.; Brown, D.A.; Bauskin, A.R.; Klevitsky, R.; Hewett, T.E.; Breit, S.N.; Molkentin, J.D. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ. Res. 2006, 98, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.; Chen, Y.G. TGF-beta Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, 022061. [Google Scholar] [CrossRef]
- Jones, M.F.; Li, X.L.; Subramanian, M.; Shabalina, S.A.; Hara, T.; Zhu, Y.; Huang, J.; Yang, Y.; Wakefield, L.M.; Prasanth, K.V.; et al. Growth differentiation factor-15 encodes a novel microRNA 3189 that functions as a potent regulator of cell death. Cell Death Differ. 2015, 22, 1641–1653. [Google Scholar] [CrossRef] [Green Version]
- Magnussen, C.; Blankenberg, S. Biomarkers for heart failure: Small molecules with high clinical relevance. J. Intern. Med. 2018, 283, 530–543. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Yin, D.; Liu, Z. GDF-15 promotes angiogenesis through modulating p53/HIF-1alpha signaling pathway in hypoxic human umbilical vein endothelial cells. Mol. Biol. Rep. 2012, 39, 4017–4022. [Google Scholar] [CrossRef]
- Dong, G.; Zheng, Q.D.; Ma, M.; Wu, S.F.; Zhang, R.; Yao, R.R.; Dong, Y.Y.; Ma, H.; Gao, D.M.; Ye, S.L.; et al. Angiogenesis enhanced by treatment damage to hepatocellular carcinoma through the release of GDF15. Cancer Med. 2018, 7, 820–830. [Google Scholar] [CrossRef] [Green Version]
- Neary, M.T.; Ng, K.E.; Ludtmann, M.H.; Hall, A.R.; Piotrowska, I.; Ong, S.B.; Hausenloy, D.J.; Mohun, T.J.; Abramov, A.Y.; Breckenridge, R.A. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J. Mol. Cell. Cardiol. 2014, 74, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabo, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [Green Version]
- Nan, J.; Zhu, W.; Rahman, M.S.; Liu, M.; Li, D.; Su, S.; Zhang, N.; Hu, X.; Yu, H.; Gupta, M.P.; et al. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim. Et Biophys. Acta Mol. Cell Res. 2017, 1864, 1260–1273. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Diaz, M.; Nicolas-Avila, J.A.; Cordero, M.D.; Hidalgo, A. Mitochondrial Adaptations in the Growing Heart. Trends Endocrinol. Metab. TEM 2020, 31, 308–319. [Google Scholar] [CrossRef]
- Tiwari, K.K.; Moorthy, B.; Lingappan, K. Role of GDF15 (growth and differentiation factor 15) in pulmonary oxygen toxicity. Toxicol. In Vitro An Int. J. Publ. Assoc. BIBRA 2015, 29, 1369–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, R.M.; Picard, M.; Derbeneva, O.; Leipzig, J.; McManus, M.J.; Gouspillou, G.; Barbat-Artigas, S.; Dos Santos, C.; Hepple, R.T.; Murdock, D.G.; et al. Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc. Natl. Acad. Sci. USA 2017, 114, 2705–2710. [Google Scholar] [CrossRef] [Green Version]
- Wall, C.E.; Whyte, J.; Suh, J.M.; Fan, W.; Collins, B.; Liddle, C.; Yu, R.T.; Atkins, A.R.; Naviaux, J.C.; Li, K.; et al. High-fat diet and FGF21 cooperatively promote aerobic thermogenesis in mtDNA mutator mice. Proc. Natl. Acad. Sci. USA 2015, 112, 8714–8719. [Google Scholar] [CrossRef] [Green Version]
- Yatsuga, S.; Fujita, Y.; Ishii, A.; Fukumoto, Y.; Arahata, H.; Kakuma, T.; Kojima, T.; Ito, M.; Tanaka, M.; Saiki, R.; et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 2015, 78, 814–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.K.; Kim, J.T.; Kim, H.W.; Kwon, M.; Kim, S.Y.; Shong, M.; Kim, K.S.; Yi, H.S. GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Sci. Rep. 2017, 7, 17238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, L.; Meloux, A.; Zeller, M.; Malka, G.; Cottin, Y.; Vergely, C. Mitochondrial SLC25 Carriers: Novel Targets for Cancer Therapy. Molecules 2020, 25, 2417. [Google Scholar] [CrossRef]
- Rochette, L.; Meloux, A.; Zeller, M.; Cottin, Y.; Vergely, C. Role of humanin, a mitochondrial-derived peptide, in cardiovascular disorders. Arch. Cardiovasc. Dis. 2020, 113, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Ostan, R.; Fabbri, C.; Santoro, A.; Guidarelli, G.; Vitale, G.; Mari, D.; Sevini, F.; Capri, M.; Sandri, M.; et al. Human Aging and Longevity Are Characterized by High Levels of Mitokines. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2019, 74, 600–607. [Google Scholar] [CrossRef]
- Tsygankova, P.G.; Itkis, Y.S.; Krylova, T.D.; Kurkina, M.V.; Bychkov, I.O.; Ilyushkina, A.A.; Zabnenkova, V.V.; Mikhaylova, S.V.; Pechatnikova, N.L.; Sheremet, N.L.; et al. Plasma FGF-21 and GDF-15 are elevated in different inherited metabolic diseases and are not diagnostic for mitochondrial disorders. J. Inherit. Metab. Dis. 2019, 42, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Del Dotto, V.; Romagnoli, M.; La Morgia, C.; Di Vito, L.; Capristo, M.; Valentino, M.L.; Carelli, V. Expanding and validating the biomarkers for mitochondrial diseases. J. Mol. Med. 2020, 98, 1467–1478. [Google Scholar] [CrossRef]
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. ccf-mtDNA as a Potential Link Between the Brain and Immune System in Neuro-Immunological Disorders. Front. Immunol. 2019, 10, 1064. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, J.M.; Auranen, M.; Darin, N.; Sofou, K.; Bindoff, L.; Hikmat, O.; Uusimaa, J.; Vieira, P.; Tulinius, M.; Lonnqvist, T.; et al. Diagnostic value of serum biomarkers FGF21 and GDF15 compared to muscle sample in mitochondrial disease. J. Inherit. Metab. Dis. 2020, 44, 469–480. [Google Scholar] [CrossRef]
- Ji, X.; Zhao, L.; Ji, K.; Zhao, Y.; Li, W.; Zhang, R.; Hou, Y.; Lu, J.; Yan, C. Growth Differentiation Factor 15 Is a Novel Diagnostic Biomarker of Mitochondrial Diseases. Mol. Neurobiol. 2017, 54, 8110–8116. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017, 23, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.Y.; Crawley, S.; Chen, M.; Ayupova, D.A.; Lindhout, D.A.; Higbee, J.; Kutach, A.; Joo, W.; Gao, Z.; Fu, D.; et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 2017, 550, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef]
- Yang, L.; Chang, C.C.; Sun, Z.; Madsen, D.; Zhu, H.; Padkjaer, S.B.; Wu, X.; Huang, T.; Hultman, K.; Paulsen, S.J.; et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 2017, 23, 1158–1166. [Google Scholar] [CrossRef]
- Sariola, H.; Saarma, M. Novel functions and signalling pathways for GDNF. J. Cell Sci. 2003, 116, 3855–3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fielder, G.C.; Yang, T.W.; Razdan, M.; Li, Y.; Lu, J.; Perry, J.K.; Lobie, P.E.; Liu, D.X. The GDNF Family: A Role in Cancer? Neoplasia 2018, 20, 99–117. [Google Scholar] [CrossRef]
- Li, Z.; Wang, B.; Wu, X.; Cheng, S.Y.; Paraoan, L.; Zhou, J. Identification, expression and functional characterization of the GRAL gene. J. Neurochem. 2005, 95, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Clemmensen, C.; Muller, T.D.; Woods, S.C.; Berthoud, H.R.; Seeley, R.J.; Tschop, M.H. Gut-Brain Cross-Talk in Metabolic Control. Cell 2017, 168, 758–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Redox Functions of Heme Oxygenase-1 and Biliverdin Reductase in Diabetes. Trends Endocrinol. Metab. TEM 2018, 29, 74–85. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Et Biophys. Acta 2014, 1840, 2709–2729. [Google Scholar] [CrossRef]
- Hesselink, M.K.; Schrauwen-Hinderling, V.; Schrauwen, P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 633–645. [Google Scholar] [CrossRef]
- Chung, H.K.; Ryu, D.; Kim, K.S.; Chang, J.Y.; Kim, Y.K.; Yi, H.S.; Kang, S.G.; Choi, M.J.; Lee, S.E.; Jung, S.B.; et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 2017, 216, 149–165. [Google Scholar] [CrossRef]
- Ding, Q.; Mracek, T.; Gonzalez-Muniesa, P.; Kos, K.; Wilding, J.; Trayhurn, P.; Bing, C. Identification of macrophage inhibitory cytokine-1 in adipose tissue and its secretion as an adipokine by human adipocytes. Endocrinology 2009, 150, 1688–1696. [Google Scholar] [CrossRef]
- Coll, A.P.; Chen, M.; Taskar, P.; Rimmington, D.; Patel, S.; Tadross, J.A.; Cimino, I.; Yang, M.; Welsh, P.; Virtue, S.; et al. GDF15 mediates the effects of metformin on body weight and energy balance. Nature 2020, 578, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, L.; Qin, W.; Zhang, G.; Yuan, J.; Wang, F. Adaptive induction of growth differentiation factor 15 attenuates endothelial cell apoptosis in response to high glucose stimulus. PLoS ONE 2013, 8, e65549. [Google Scholar] [CrossRef]
- Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C.; Rochette, L. Iron, oxidative stress, and redox signaling in the cardiovascular system. Mol. Nutr. Food Res. 2014, 58, 1721–1738. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C. The iron-regulatory hormone hepcidin: A possible therapeutic target? Pharmacol. Ther. 2015, 146, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fang, Z.; Zhu, Z.; Liu, J.; Chen, H. Reciprocal regulation between hepcidin and erythropoiesis and its therapeutic application in erythroid disorders. Exp. Hematol. 2017, 52, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Wang, H.; Liu, C.; Cao, Q.; Fu, R.; Wang, T.; Qi, W.; Shao, Z. Transforming growth factor 15 increased in severe aplastic anemia patients. Hematology 2017, 22, 548–553. [Google Scholar] [CrossRef] [Green Version]
- Luan, H.H.; Wang, A.; Hilliard, B.K.; Carvalho, F.; Rosen, C.E.; Ahasic, A.M.; Herzog, E.L.; Kang, I.; Pisani, M.A.; Yu, S.; et al. GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 2019, 178, 1231–1244. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, H.G.; Lee, J.S.; Kim, W.Y.; Lee, M.M.; Kim, Y.H.; Lee, J.O.; Kim, H.S.; Son, C.G. Intermittent restraint-induced sympathetic activation attenuates hepatic steatosis and inflammation in a high-fat diet-fed mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G811–G823. [Google Scholar] [CrossRef]
- Takahashi, Y. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int. J. Mol. Sci. 2017, 18, 1477. [Google Scholar] [CrossRef]
- Wang, T.; Liu, J.; McDonald, C.; Lupino, K.; Zhai, X.; Wilkins, B.J.; Hakonarson, H.; Pei, L. GDF15 is a heart-derived hormone that regulates body growth. EMBO Mol. Med. 2017, 9, 1150–1164. [Google Scholar] [CrossRef] [PubMed]
- Fluschnik, N.; Ojeda, F.; Zeller, T.; Jorgensen, T.; Kuulasmaa, K.; Becher, P.M.; Sinning, C.; Blankenberg, S.; Westermann, D. Predictive value of long-term changes of growth differentiation factor-15 over a 27-year-period for heart failure and death due to coronary heart disease. PLoS ONE 2018, 13, e0197497. [Google Scholar] [CrossRef] [Green Version]
- George, M.; Jena, A.; Srivatsan, V.; Muthukumar, R.; Dhandapani, V.E. GDF 15--A Novel Biomarker in the Offing for Heart Failure. Curr. Cardiol. Rev. 2016, 12, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Guo, Y.; Yu, H.; Zheng, L.; Mi, L.; Gao, W. Growth differentiation factor 15 in different stages of heart failure: Potential screening implications. Biomarkers 2010, 15, 671–676. [Google Scholar] [CrossRef]
- Dinh, W.; Futh, R.; Lankisch, M.; Hess, G.; Zdunek, D.; Scheffold, T.; Kramer, F.; Klein, R.M.; Barroso, M.C.; Nickl, W. Growth-differentiation factor-15: A novel biomarker in patients with diastolic dysfunction? Arq. Bras. Cardiol. 2011, 97, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cui, Y.; Huang, A.; Li, Q.; Jia, W.; Liu, K.; Qi, X. Additional Diagnostic Value of Growth Differentiation Factor-15 (GDF-15) to N-Terminal B-Type Natriuretic Peptide (NT-proBNP) in Patients with Different Stages of Heart Failure. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 4992–4999. [Google Scholar] [CrossRef] [PubMed]
- Mendez Fernandez, A.B.; Ferrero-Gregori, A.; Garcia-Osuna, A.; Mirabet-Perez, S.; Pirla-Buxo, M.J.; Cinca-Cuscullola, J.; Ordonez-Llanos, J.; Roig Minguell, E. Growth differentiation factor 15 as mortality predictor in heart failure patients with non-reduced ejection fraction. ESC Heart Fail. 2020, 7, 2223–2229. [Google Scholar] [CrossRef]
- Larissi, K.; Politou, M.; Margeli, A.; Poziopoulos, C.; Flevari, P.; Terpos, E.; Papassotiriou, I.; Voskaridou, E. The Growth Differentiation Factor-15 (GDF-15) levels are increased in patients with compound heterozygous sickle cell and beta-thalassemia (HbS/beta(thal)), correlate with markers of hemolysis, iron burden, coagulation, endothelial dysfunction and pulmonary hypertension. Blood Cells Mol. Dis. 2019, 77, 137–141. [Google Scholar]
- Ecarnot-Laubriet, A.; Rochette, L.; Vergely, C.; Sicard, P.; Teyssier, J.R. The activation pattern of the antioxidant enzymes in the right ventricle of rat in response to pressure overload is of heart failure type. Heart Dis. 2003, 5, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef]
- Mason, J.C. Cytoprotective pathways in the vascular endothelium. Do they represent a viable therapeutic target? Vasc. Pharmacol. 2016, 86, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.J.; Lee, J.H.; Kim, Y.M.; Oh, G.T.; Lee, H. Macrophage inhibitory cytokine-1 stimulates proliferation of human umbilical vein endothelial cells by up-regulating cyclins D1 and E through the PI3K/Akt-, ERK-, and JNK-dependent AP-1 and E2F activation signaling pathways. Cell. Signal. 2012, 24, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Kempf, T.; Zarbock, A.; Widera, C.; Butz, S.; Stadtmann, A.; Rossaint, J.; Bolomini-Vittori, M.; Korf-Klingebiel, M.; Napp, L.C.; Hansen, B.; et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat. Med. 2011, 17, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Artz, A.; Butz, S.; Vestweber, D. GDF-15 inhibits integrin activation and mouse neutrophil recruitment through the ALK-5/TGF-betaRII heterodimer. Blood 2016, 128, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Doise, J.M.; Aho, L.S.; Quenot, J.P.; Guilland, J.C.; Zeller, M.; Vergely, C.; Aube, H.; Blettery, B.; Rochette, L. Plasma antioxidant status in septic critically ill patients: A decrease over time. Fundam. Clin. Pharmacol. 2008, 22, 203–209. [Google Scholar] [CrossRef]
- Lecour, S.; Chevet, D.; Maupoil, V.; Moisant, M.; Bernard, C.; Zahnd, J.P.; Touchard, G.; Briot, F.; Rochette, L. Intrarenal detection of nitric oxide using electron spin resonance spectroscopy in hypertensive lipopolysaccharide-treated rats. J. Cardiovasc. Pharmacol. 2002, 40, 9–17. [Google Scholar] [CrossRef]
- Wischhusen, J.; Melero, I.; Fridman, W.H. Growth/Differentiation Factor-15 (GDF-15): From Biomarker to Novel Targetable Immune Checkpoint. Front. Immunol. 2020, 11, 951. [Google Scholar] [CrossRef]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef] [Green Version]
- Bonaterra, G.A.; Zugel, S.; Thogersen, J.; Walter, S.A.; Haberkorn, U.; Strelau, J.; Kinscherf, R. Growth differentiation factor-15 deficiency inhibits atherosclerosis progression by regulating interleukin-6-dependent inflammatory response to vascular injury. J. Am. Heart Assoc. 2012, 1, e002550. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Zhang, T.; Guo, J.; Peng, Y.F.; Wei, Y.S. The Association of Growth Differentiation Factor-15 Gene Polymorphisms with Growth Differentiation Factor-15 Serum Levels and Risk of Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2017, 26, 2111–2119. [Google Scholar] [CrossRef]
- Kempf, T.; Eden, M.; Strelau, J.; Naguib, M.; Willenbockel, C.; Tongers, J.; Heineke, J.; Kotlarz, D.; Xu, J.; Molkentin, J.D.; et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ. Res. 2006, 98, 351–360. [Google Scholar] [CrossRef]
- Kempf, T.; Wollert, K.C. Growth differentiation factor-15: A new biomarker in cardiovascular disease. Herz 2009, 34, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pan, K.; Liu, Q.; Zhou, X.; Jiang, T.; Li, Y. Growth differentiation factor 15 may protect the myocardium from noreflow by inhibiting the inflammatorylike response that predominantly involves neutrophil infiltration. Mol. Med. Rep. 2016, 13, 623–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Moszczynski, L.A.; Liu, Q.; Jiang, J.; Zhao, D.; Quan, D.; Mele, T.; McAlister, V.; Jevnikar, A.; Baek, S.J.; et al. Over-expression of growth differentiation factor 15 (GDF15) preventing cold ischemia reperfusion (I/R) injury in heart transplantation through Foxo3a signaling. Oncotarget 2017, 8, 36531–36544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Ago, T.; Sadoshima, J. GDF15, a cardioprotective TGF-beta superfamily protein. Circ. Res. 2006, 98, 294–297. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.; Wollert, K.C.; Larson, M.G.; Coglianese, E.; McCabe, E.L.; Cheng, S.; Ho, J.E.; Fradley, M.G.; Ghorbani, A.; Xanthakis, V.; et al. Prognostic utility of novel biomarkers of cardiovascular stress: The Framingham Heart Study. Circulation 2012, 126, 1596–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, D.; Hagstrom, E.; James, S.K.; Becker, R.C.; Cannon, C.P.; Himmelmann, A.; Katus, H.A.; Maurer, G.; Lopez-Sendon, J.L.; Steg, P.G.; et al. Growth Differentiation Factor 15 at 1 Month After an Acute Coronary Syndrome Is Associated With Increased Risk of Major Bleeding. J. Am. Heart Assoc. 2017, 6, e005580. [Google Scholar] [CrossRef]
- Hagstrom, E.; Held, C.; Stewart, R.A.; Aylward, P.E.; Budaj, A.; Cannon, C.P.; Koenig, W.; Krug-Gourley, S.; Mohler, E.R., 3rd; Steg, P.G.; et al. Growth Differentiation Factor 15 Predicts All-Cause Morbidity and Mortality in Stable Coronary Heart Disease. Clin. Chem. 2017, 63, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Kahli, A.; Guenancia, C.; Zeller, M.; Grosjean, S.; Stamboul, K.; Rochette, L.; Girard, C.; Vergely, C. Growth differentiation factor-15 (GDF-15) levels are associated with cardiac and renal injury in patients undergoing coronary artery bypass grafting with cardiopulmonary bypass. PLoS ONE 2014, 9, e105759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchot, O.; Guenancia, C.; Kahli, A.; Pujos, C.; Malapert, G.; Vergely, C.; Laurent, G. Low Circulating Levels of Growth Differentiation Factor-15 Before Coronary Artery Bypass Surgery May Predict Postoperative Atrial Fibrillation. J. Cardiothorac. Vasc. Anesth. 2015, 29, 1131–1139. [Google Scholar] [CrossRef]
- Guenancia, C.; Kahli, A.; Laurent, G.; Hachet, O.; Malapert, G.; Grosjean, S.; Girard, C.; Vergely, C.; Bouchot, O. Pre-operative growth differentiation factor 15 as a novel biomarker of acute kidney injury after cardiac bypass surgery. Int. J. Cardiol. 2015, 197, 66–71. [Google Scholar] [CrossRef]
- Wallentin, L.; Hijazi, Z.; Andersson, U.; Alexander, J.H.; De Caterina, R.; Hanna, M.; Horowitz, J.D.; Hylek, E.M.; Lopes, R.D.; Asberg, S.; et al. Growth differentiation factor 15, a marker of oxidative stress and inflammation, for risk assessment in patients with atrial fibrillation: Insights from the Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial. Circulation 2014, 130, 1847–1858. [Google Scholar]
- Bonaca, M.P.; Morrow, D.A.; Braunwald, E.; Cannon, C.P.; Jiang, S.; Breher, S.; Sabatine, M.S.; Kempf, T.; Wallentin, L.; Wollert, K.C. Growth differentiation factor-15 and risk of recurrent events in patients stabilized after acute coronary syndrome: Observations from PROVE IT-TIMI 22. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Widera, C.; Giannitsis, E.; Kempf, T.; Korf-Klingebiel, M.; Fiedler, B.; Sharma, S.; Katus, H.A.; Asaumi, Y.; Shimano, M.; Walsh, K.; et al. Identification of follistatin-like 1 by expression cloning as an activator of the growth differentiation factor 15 gene and a prognostic biomarker in acute coronary syndrome. Clin. Chem. 2012, 58, 1233–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meloux, A.; Bejot, Y.; Rochette, L.; Cottin, Y.; Vergely, C. Brain-Heart Interactions During Ischemic Processes: Clinical and Experimental Evidences. Stroke 2020, 51, 679–686. [Google Scholar] [CrossRef]
- Jensen, M.K.; Bertoia, M.L.; Cahill, L.E.; Agarwal, I.; Rimm, E.B.; Mukamal, K.J. Novel metabolic biomarkers of cardiovascular disease. Nat. Rev. Endocrinol. 2014, 10, 659–672. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Patel, S.K.; Velkoska, E.; Freeman, M.; Wai, B.; Lancefield, T.F.; Burrell, L.M. From gene to protein-experimental and clinical studies of ACE2 in blood pressure control and arterial hypertension. Front. Physiol. 2014, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; Lindback, J.; Eriksson, N.; Hijazi, Z.; Eikelboom, J.W.; Ezekowitz, M.D.; Granger, C.B.; Lopes, R.D.; Yusuf, S.; Oldgren, J.; et al. Angiotensin-converting enzyme 2 (ACE2) levels in relation to risk factors for COVID-19 in two large cohorts of patients with atrial fibrillation. Eur. Heart J. 2020, 41, 4037–4046. [Google Scholar] [CrossRef] [PubMed]
- Bohn, M.K.; Lippi, G.; Horvath, A.; Sethi, S.; Koch, D.; Ferrari, M.; Wang, C.B.; Mancini, N.; Steele, S.; Adeli, K. Molecular, serological, and biochemical diagnosis and monitoring of COVID-19: IFCC taskforce evaluation of the latest evidence. Clin. Chem. Lab. Med. 2020, 58, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Myhre, P.L.; Prebensen, C.; Strand, H.; Roysland, R.; Jonassen, C.M.; Rangberg, A.; Sorensen, V.; Sovik, S.; Rosjo, H.; Svensson, M.; et al. Growth Differentiation Factor-15 Provides Prognostic Information Superior to Established Cardiovascular and Inflammatory Biomarkers in Unselected Patients Hospitalized with COVID-19. Circulation 2020, 142, 2128–2137. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Nishiyama, M.; Tokumoto, S.; Ishida, Y.; Tomioka, K.; Aoki, K.; Seino, Y.; Toyoshima, D.; Takeda, H.; Kurosawa, H.; et al. Elevated cytokine, chemokine, and growth and differentiation factor-15 levels in hemorrhagic shock and encephalopathy syndrome: A retrospective observational study. Cytokine 2020, 137, 155324. [Google Scholar] [CrossRef]
- Sidorova, Y.A.; Bespalov, M.M.; Wong, A.W.; Kambur, O.; Jokinen, V.; Lilius, T.O.; Suleymanova, I.; Karelson, G.; Rauhala, P.V.; Karelson, M.; et al. A Novel Small Molecule GDNF Receptor RET Agonist, BT13, Promotes Neurite Growth from Sensory Neurons in Vitro and Attenuates Experimental Neuropathy in the Rat. Front. Pharmacol. 2017, 8, 365. [Google Scholar] [CrossRef]
- Hedstrom, K.L.; Murtie, J.C.; Albers, K.; Calcutt, N.A.; Corfas, G. Treating small fiber neuropathy by topical application of a small molecule modulator of ligand-induced GFRalpha/RET receptor signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 2325–2330. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Fan, Z.; Li, L.; Lu, J.; Zhai, Y.; Zhao, J. The chemokine system and its role in obesity. J. Cell. Physiol. 2019, 234, 3336–3346. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rochette, L.; Dogon, G.; Zeller, M.; Cottin, Y.; Vergely, C. GDF15 and Cardiac Cells: Current Concepts and New Insights. Int. J. Mol. Sci. 2021, 22, 8889. https://doi.org/10.3390/ijms22168889
Rochette L, Dogon G, Zeller M, Cottin Y, Vergely C. GDF15 and Cardiac Cells: Current Concepts and New Insights. International Journal of Molecular Sciences. 2021; 22(16):8889. https://doi.org/10.3390/ijms22168889
Chicago/Turabian StyleRochette, Luc, Geoffrey Dogon, Marianne Zeller, Yves Cottin, and Catherine Vergely. 2021. "GDF15 and Cardiac Cells: Current Concepts and New Insights" International Journal of Molecular Sciences 22, no. 16: 8889. https://doi.org/10.3390/ijms22168889
APA StyleRochette, L., Dogon, G., Zeller, M., Cottin, Y., & Vergely, C. (2021). GDF15 and Cardiac Cells: Current Concepts and New Insights. International Journal of Molecular Sciences, 22(16), 8889. https://doi.org/10.3390/ijms22168889