
New Hybrid Compounds Combining Fragments of Usnic Acid and Thioether Are Inhibitors of Human Enzymes TDP1, TDP2 and PARP1

 , , , , ,
, , , , ,  ,
, 
Abstract
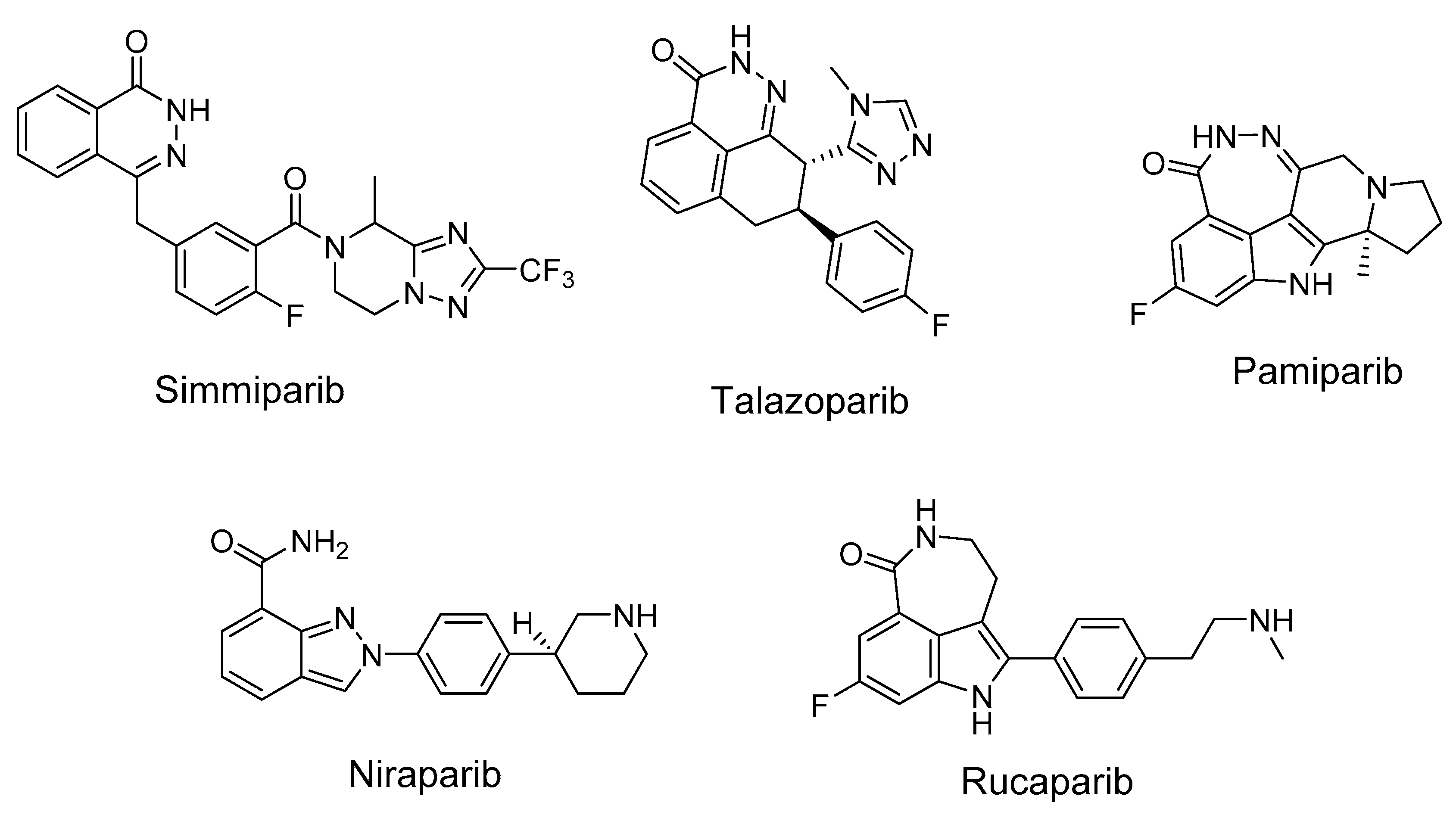
:1. Introduction
2. Results and Discussion
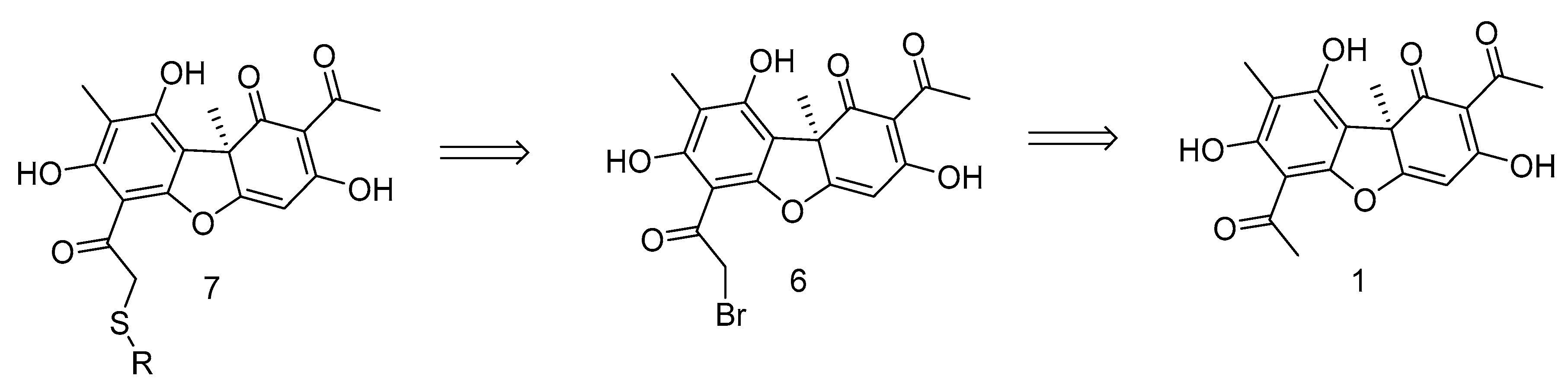
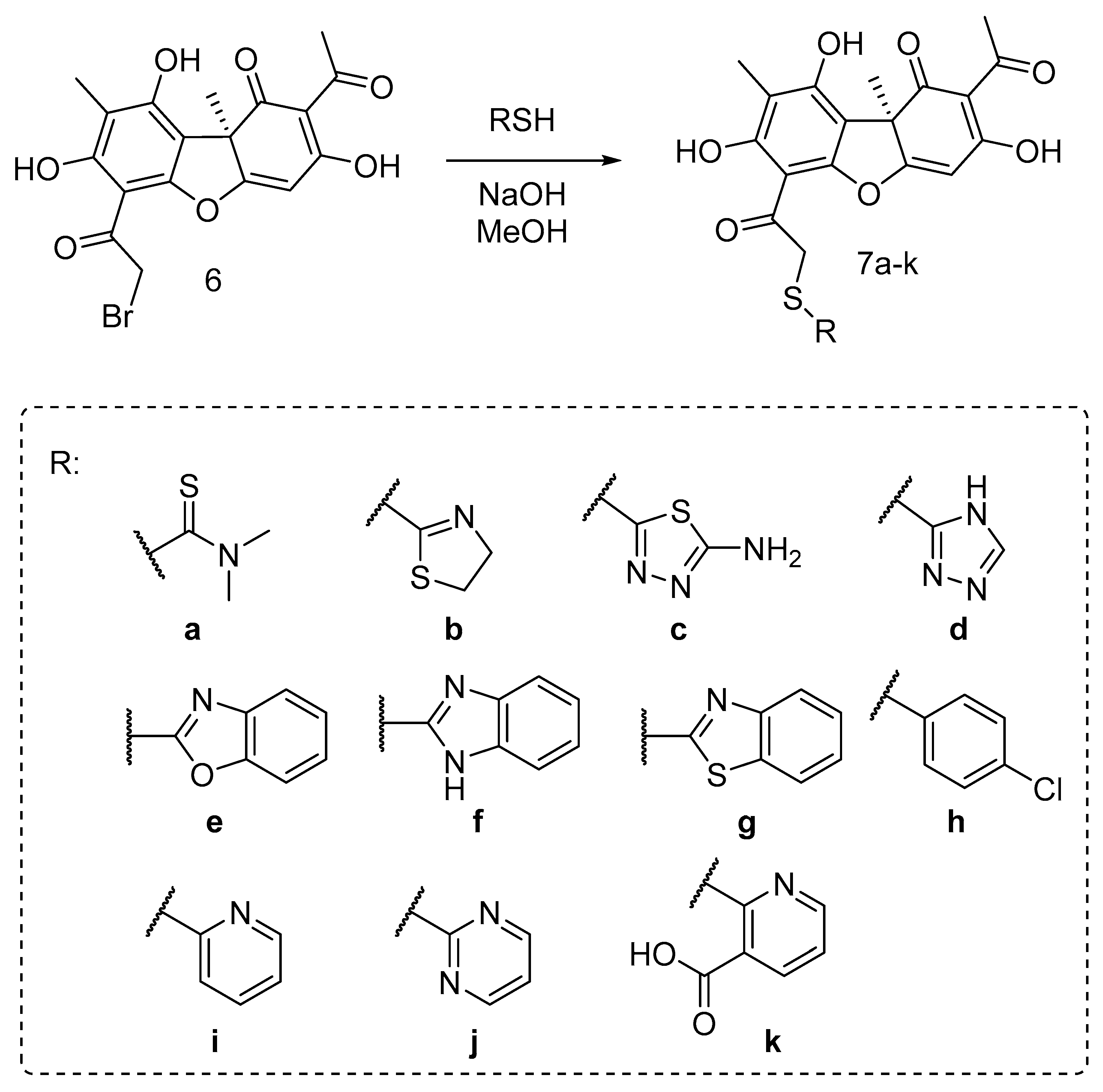
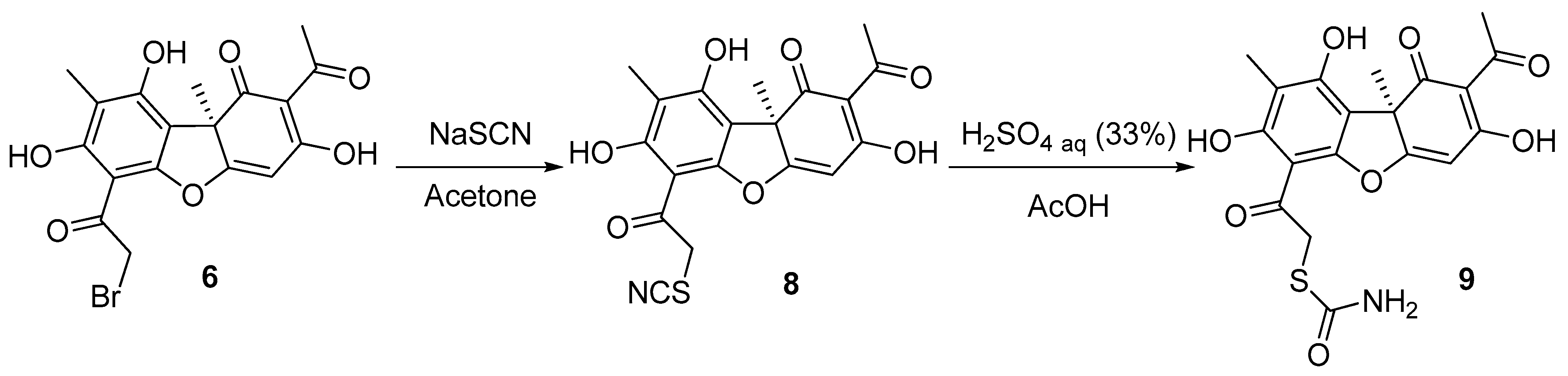
2.1. Chemistry
2.2. Biology
2.2.1. Real-Time Fluorescence Assay of TDP1 Activity
2.2.2. Type of Inhibition of TDP1 Enzyme Reaction for the Most Effective Compounds
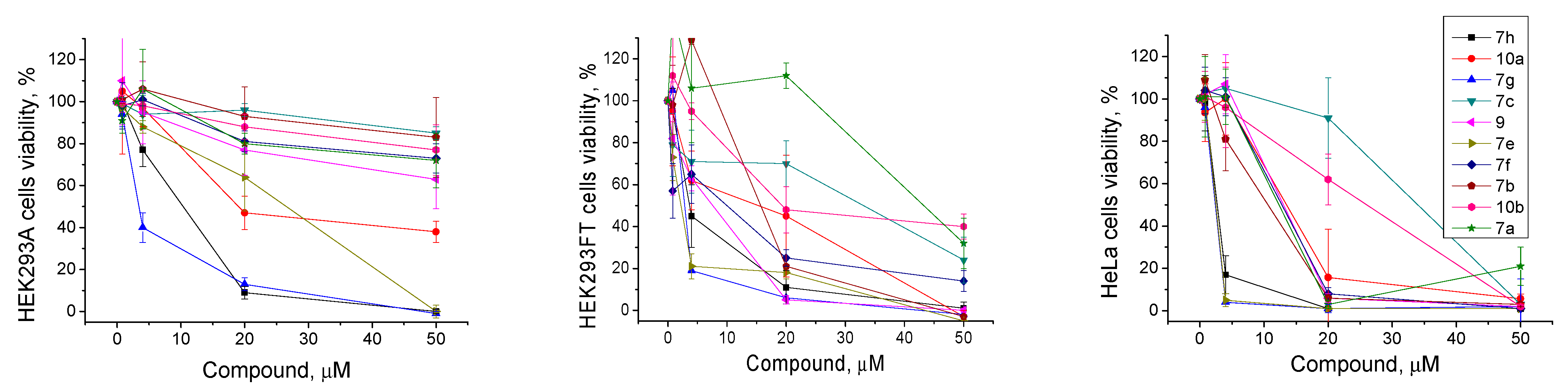
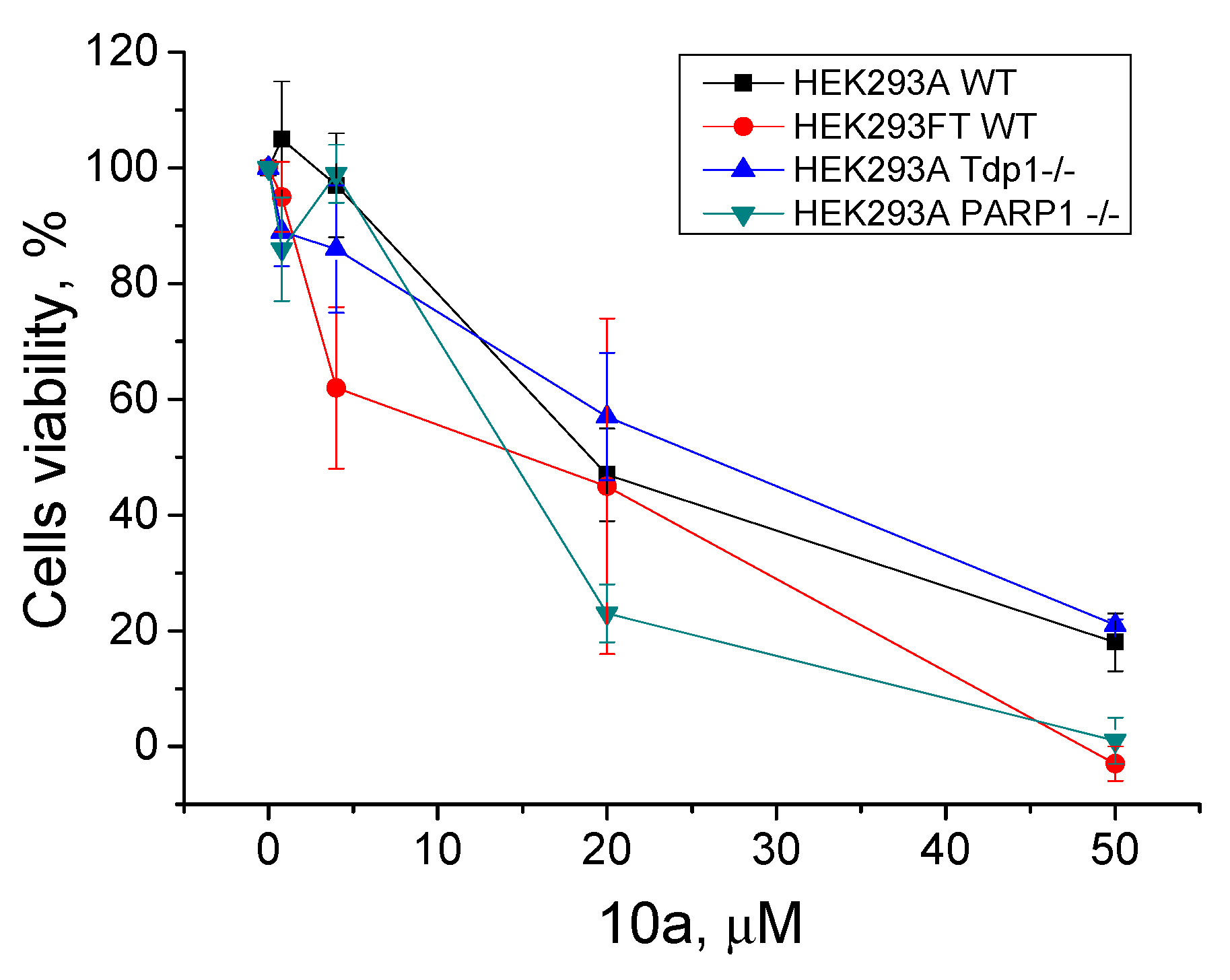
2.2.3. The Effect of TDP1 Inhibitors on Cells Viability
2.2.4. PARP1 and PARP2 Activity in the Presence of TDP1 Inhibitors
2.2.5. TDP2 Activity in the Presence of TDP1 Inhibitors
2.2.6. The Effect of TDP1 Inhibitors on Cell Viability in Combination with Topotecan
3. Materials and Methods
3.1. Chemistry
3.1.1. Reaction of 2 with Thiols (General Method)
3.1.2. Hydrolysis of Compound 8
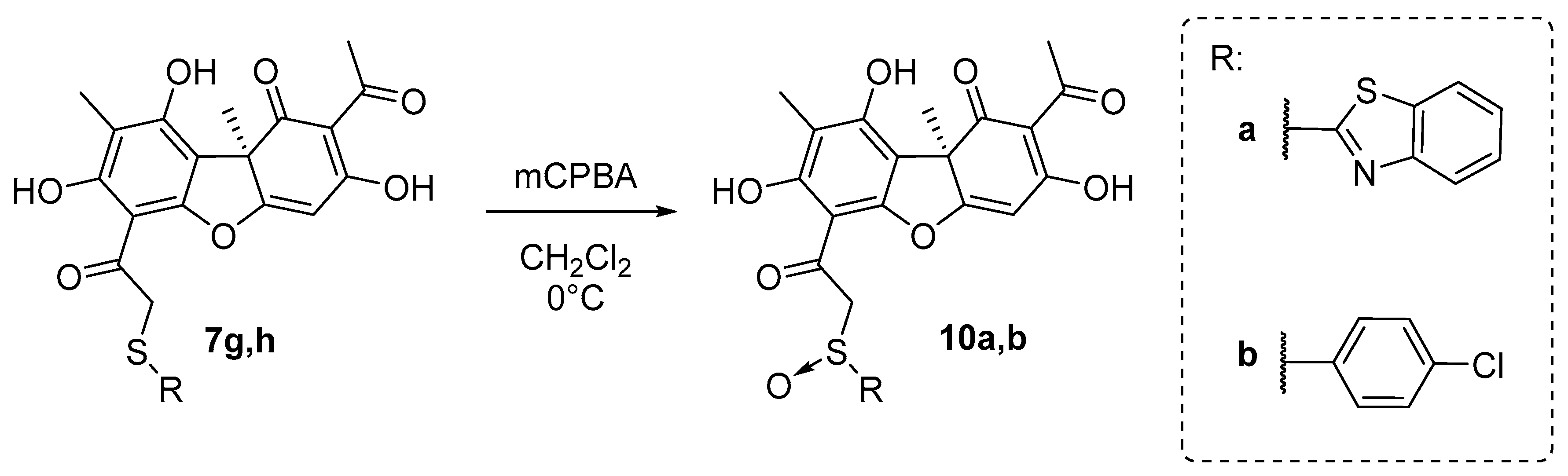
3.1.3. Oxidation of Compounds 7g,h
3.2. Biology
3.2.1. Preparation of Labeled Oligonucleotides
3.2.2. Expression and Purification of Human Recombinant Tyrosyl-DNA Phosphodiesterase 2 (TDP2)
3.2.3. Real-Time Detection of TDP1 Activity
3.2.4. Gel-Based TDP2 Activity Assay
3.2.5. PARP1 and PARP2 Enzyme Assay
3.2.6. Cell Culture Cytotoxicity Assay
3.2.7. Plasmid Construction for Human PARP1 Gene Knockout
3.2.8. Knockout HEK293A Clones’ Generation
3.2.9. Analysis of CRISPR/Cas9-Mediated Deletions in PARP1 Gene
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Zakharenko, A.; Dyrkheeva, N.; Lavrik, O. Dual DNA topoisomerase 1 and tyrosyl-DNA phosphodiesterase 1 inhibition for improved anticancer activity. Med. Res. Rev. 2019, 39, 1427–1441. [Google Scholar] [CrossRef] [PubMed]
- Kawale, A.S.; Povirk, L.F. Tyrosyl-DNA phosphodiesterases: Rescuing the genome from the risks of relaxation. Nucleic Acids Res. 2018, 46, 520–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagoz, M.; Gilbert, D.C.; El-Khamisy, S.; Chalmers, A.J. DNA repair and resistance to topoisomerase I inhibitors: Mechanisms, biomarkers and therapeutic targets. Curr. Med. Chem. 2012, 19, 3874–3885. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Huang, S.Y.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 2014, 19, 114–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Cortés-Ledesma, F.; El Khamisy, S.F.; Caldecott, K.W. TDP2/TTRAP is the major 5’-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 2011, 286, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Herreros, F.; Romero-Granados, R.; Zeng, Z.; Alvarez-Quilón, A.; Quintero, C.; Ju, L.; Umans, L.; Vermeire, L.; Huylebroeck, D.; Caldecott, K.W.; et al. TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo. PLoS Genet. 2013, 9, e1003226. [Google Scholar] [CrossRef]
- Marchand, C.; Abdelmalak, M.; Kankanala, J.; Huang, S.Y.; Kiselev, E.; Fesen, K.; Kurahashi, K.; Sasanuma, H.; Takeda, S.; Aihara, H.; et al. Deazaflavin Inhibitors of Tyrosyl-DNA Phosphodiesterase 2 (TDP2) Specific for the Human Enzyme and Active against Cellular TDP2. ACS Chem. Biol. 2016, 11, 1925–1933. [Google Scholar] [CrossRef]
- Raoof, A.; Depledge, P.; Hamilton, N.M.; Hamilton, N.S.; Hitchin, J.R.; Hopkins, G.V.; Jordan, A.M.; Maguire, L.A.; McGonagle, A.E.; Mould, D.P.; et al. Toxoflavins and deazaflavins as the first reported selective small molecule inhibitors of tyrosyl-DNA phosphodiesterase II. J. Med. Chem. 2013, 56, 6352–6370. [Google Scholar] [CrossRef]
- Laev, S.S.; Salakhutdinov, N.F.; Lavrik, O.I. Tyrosyl-DNA phosphodiesterase inhibitors: Progress and potential. Bioorg. Med. Chem. 2016, 24, 5017–5027. [Google Scholar] [CrossRef]
- Zhang, Y.; He, X.Z.; Yang, H.; Liu, H.Y.; An, L.K. Robustadial A and B from Eucalyptus globulus Labill. and their anticancer activity as selective tyrosyl-DNA phosphodiesterase 2 inhibitors. Phytother. Res. 2021, 35, 5282–5289. [Google Scholar] [CrossRef]
- Yang, H.; Zhu, X.Q.; Wang, W.; Chen, Y.; Hu, Z.; Zhang, Y.; Hu, D.X.; Yu, L.M.; Agama, K.; Pommier, Y.; et al. The synthesis of furoquinolinedione and isoxazoloquinolinedione derivatives as selective Tyrosyl-DNA phosphodiesterase 2 (TDP2) inhibitors. Bioorg. Chem. 2021, 111, 104881. [Google Scholar] [CrossRef]
- Senaweera, S.; He, T.; Cui, H.; Aihara, H.; Wang, Z. 4-Benzylideneisoquinoline-1,3(2H,4H)-diones as tyrosyl DNA phosphodiesterase 2 (TDP2) inhibitors. Med. Chem. Res. 2021, 30, 371–386. [Google Scholar] [CrossRef]
- Wang, P.; Elsayed, M.S.A.; Plescia, C.B.; Ravji, A.; Redon, C.E.; Kiselev, E.; Marchand, C.; Zeleznik, O.; Agama, K.; Pommier, Y.; et al. Synthesis and Biological Evaluation of the First Triple Inhibitors of Human Topoisomerase 1, Tyrosyl-DNA Phosphodiesterase 1 (Tdp1), and Tyrosyl-DNA Phosphodiesterase 2 (Tdp2). J. Med. Chem. 2017, 60, 3275–3288. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, O.I. PARPs’ impact on base excision DNA repair. DNA Repair 2020, 93, 102911. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Camacho, C.V.; Kraus, W.L. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp. Mol. Med. 2021, 53, 42–51. [Google Scholar] [CrossRef]
- Pacher, P.; Szabó, C. Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: The therapeutic potential of PARP inhibitors. Cardiovasc. Drug Rev. 2007, 25, 235–260. [Google Scholar] [CrossRef] [Green Version]
- Das, B.B.; Huang, S.Y.; Murai, J.; Rehman, I.; Amé, J.C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef] [PubMed]
- Moor, N.A.; Vasil’eva, I.A.; Anarbaev, R.O.; Antson, A.A.; Lavrik, O.I. Quantitative characterization of protein-protein complexes involved in base excision DNA repair. Nucleic Acids Res. 2015, 43, 6009–6022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebedeva, N.A.; Anarbaev, R.O.; Sukhanova, M.; Vasil’eva, I.A.; Rechkunova, N.I.; Lavrik, O.I. Poly(ADP-ribose)polymerase 1 stimulates the AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1. Biosci. Rep. 2015, 35, e00230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.K.; Stefanick, D.F.; Zhao, M.L.; Janoshazi, A.K.; Gassman, N.R.; Seddon, H.J.; Wilson, S.H. XRCC1-mediated repair of strand breaks independent of PNKP binding. DNA Repair 2017, 60, 52–63. [Google Scholar] [CrossRef]
- Prasad, R.; Horton, J.K.; Dai, D.P.; Wilson, S.H. Repair pathway for PARP-1 DNA-protein crosslinks. DNA Repair 2019, 73, 71–77. [Google Scholar] [CrossRef]
- Brettrager, E.J.; van Waardenburg, R.C.A.M. Targeting Tyrosyl-DNA phosphodiesterase I to enhance toxicity of phosphodiester linked DNA-adducts. Cancer Drug Resist. 2019, 2, 1153–1163. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, W.; El-Shafie, L.; Hassan, M.K.; Farag, M.A.; El-Khamisy, S.F. Isoeugenol is a selective potentiator of camptothecin cytotoxicity in vertebrate cells lacking TDP1. Sci. Rep. 2016, 6, 26626. [Google Scholar] [CrossRef]
- Al Abo, M.; Sasanuma, H.; Liu, X.; Rajapakse, V.N.; Huang, S.Y.; Kiselev, E.; Takeda, S.; Plunkett, W.; Pommier, Y. TDP1 is Critical for the Repair of DNA Breaks Induced by Sapacitabine, a Nucleoside also Targeting ATM- and BRCA-Deficient Tumors. Mol. Cancer Ther. 2017, 16, 2543–2551. [Google Scholar] [CrossRef] [Green Version]
- Fam, H.K.; Walton, C.; Mitra, S.A.; Chowdhury, M.; Osborne, N.; Choi, K.; Sun, G.; Wong, P.C.; O’Sullivan, M.J.; Turashvili, G.; et al. TDP1 and PARP1 deficiency are cytotoxic to rhabdomyosarcoma cells. Mol. Cancer Res. 2013, 11, 1179–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toots, M.; Ustav, M., Jr.; Männik, A.; Mumm, K.; Tämm, K.; Tamm, T.; Ustav, E.; Ustav, M. Identification of several high-risk HPV inhibitors and drug targets with a novel high-throughput screening assay. PLoS Pathog. 2017, 13, e1006168. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Ploner, A.; Elfström, K.M.; Wang, J.; Roth, A.; Fang, F.; Sundström, K.; Dillner, J.; Sparén, P. HPV Vaccination and the Risk of Invasive Cervical Cancer. N. Engl. J. Med. 2020, 383, 1340–1348. [Google Scholar] [CrossRef]
- Zakharenko, A.; Luzina, O.; Koval, O.; Nilov, D.; Gushchina, I.; Dyrkheeva, N.; Švedas, V.; Salakhutdinov, N.; Lavrik, O. Tyrosyl-DNA Phosphodiesterase 1 Inhibitors: Usnic Acid Enamines Enhance the Cytotoxic Effect of Camptothecin. J. Nat. Prod. 2016, 79, 2961–2967. [Google Scholar] [CrossRef]
- Zakharova, O.; Luzina, O.; Zakharenko, A.; Sokolov, D.; Filimonov, A.; Dyrkheeva, N.; Chepanova, A.; Ilina, E.; Ilyina, A.; Klabenkova, K.; et al. Synthesis and evaluation of aryliden- and hetarylidenfuranone derivatives of usnic acid as highly potent Tdp1 inhibitors. Bioorg. Med. Chem. 2018, 26, 4470–4480. [Google Scholar] [CrossRef]
- Filimonov, A.S.; Chepanova, A.A.; Luzina, O.A.; Zakharenko, A.L.; Zakharova, O.D.; Ilina, E.S.; Dyrkheeva, N.S.; Kuprushkin, M.S.; Kolotaev, A.V.; Khachatryan, D.S.; et al. New Hydrazinothiazole Derivatives of Usnic Acid as Potent Tdp1 Inhibitors. Molecules 2019, 24, 3711. [Google Scholar] [CrossRef] [Green Version]
- Zakharenko, O.; Luzina, O.; Sokolov, D.; Zakharova, O.; Rakhmanova, M.; Chepanova, A.; Dyrkheeva, N.; Lavrik, O.; Salakhutdinov, N. Usnic acid derivatives are effective inhibitors of tyrosyl-DNA phosphodiesterase 1//Rus. J. Bioorg. Chem. 2017, 43, 84–90. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Luzina, O.A.; Sokolov, D.N.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Patel, J.; Zakharova, O.D.; Chepanova, A.A.; Zafar, A.; et al. Novel tyrosyl-DNA phosphodiesterase 1 inhibitors enhance the therapeutic impact of topotecan on in vivo tumor models. Eur. J. Med. Chem. 2019, 161, 581–593. [Google Scholar] [CrossRef]
- Nikolin, V.P.; Popova, N.A.; Kaledin, V.I.; Luzina, O.A.; Zakharenko, A.L.; Salakhutdinov, N.F.; Lavrik, O.I. The influence of an enamine usnic acid derivative (a tyrosyl-DNA phosphodiesterase 1 inhibitor) on the therapeutic effect of topotecan against transplanted tumors in vivo. Clin. Exp. Metastasis. 2021, 38, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Dyrkheeva, N.S.; Zakharenko, A.L.; Novoselova, E.S.; Chepanova, A.A.; Popova, N.A.; Nikolin, V.P.; Luzina, O.A.; Salakhutdinov, N.F.; Ryabchikova, E.I.; Lavrik, O.I. Antitumor Activity of the Combination of Topotecan and Tyrosyl-DNA-Phosphodiesterase 1 Inhibitor on Model Krebs-2 Mouse Ascite Carcinoma. Mol. Biol. 2021, 55, 312–317. [Google Scholar] [CrossRef]
- Zakharenko, A.; Sokolov, D.; Luzina, O.; Sukhanova, M.; Khodyreva, S.; Zakharova, O.; Salakhutdinov, N.; Lavrik, O. Influence of usnic acid and its derivatives on the activity of mammalian poly(ADP-ribose)polymerase 1 and DNA polymerase beta. Med. Chem. 2012, 8, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, L.X.; Jiang, T.; Long, J.; Ma, Z.Y.; Lu, A.P.; Cheng, Y.; Cao, D.S. The ups and downs of Poly(ADP-ribose) Polymerase-1 inhibitors in cancer therapy-Current progress and future direction. Eur. J. Med. Chem. 2020, 203, 112570. [Google Scholar] [CrossRef]
- Shtro, A.A.; Zarubaev, V.V.; Luzina, O.A.; Sokolov, D.N.; Kiselev, O.I.; Salakhutdinov, N.F. Novel derivatives of usnic acid effectively inhibiting reproduction of influenza A virus. Bioorg Med. Chem. 2014, 22, 6826–6836. [Google Scholar] [CrossRef]
- Sokolov, D.N.; Luzina, O.A.; Salakhutdinov, N.F. Synthesis of Sulfones and Sulfoxides Based on (+)-usnic Acid. Chem. Nat. Compd. 2018, 54, 46–49. [Google Scholar] [CrossRef]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V.; et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef]
- Davies, D.R.; Interthal, H.; Champoux, J.J.; Hol, W.G. Insights into substrate binding and catalytic mechanism of human tyrosyl-DNA phosphodiesterase (Tdp1) from vanadate and tungstate-inhibited structures. J. Mol. Biol. 2002, 324, 917–932. [Google Scholar] [CrossRef]
- Davies, D.R.; Interthal, H.; Champoux, J.J.; Hol, W.G. Crystal structure of a transition state mimic for Tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived peptide. Chem. Biol. 2003, 10, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.R.; Interthal, H.; Champoux, J.J.; Hol, W.G. The crystal structure of human tyrosyl-DNA phosphodiesterase, Tdp1. Structure 2002, 10, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Interthal, H.; Chen, H.J.; Kehl-Fie, T.E.; Zotzmann, J.; Leppard, J.B.; Champoux, J.J. SCAN1 mutant Tdp1 accumulates the enzyme--DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005, 24, 2224–2233. [Google Scholar] [CrossRef] [Green Version]
- Il’ina, I.V.; Dyrkheeva, N.S.; Zakharenko, A.L.; Sidorenko, A.Y.; Li-Zhulanov, N.S.; Korchagina, D.V.; Chand, R.; Ayine-Tora, D.M.; Chepanova, A.A.; Zakharova, O.D.; et al. Design, Synthesis, and Biological Investigation of Novel Classes of 3-Carene-Derived Potent Inhibitors of TDP1. Molecules 2020, 25, 3496. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Filimonov, A.S.; Luzina, O.A.; Zakharenko, A.L.; Ilina, E.S.; Malakhova, A.A.; Medvedev, S.P.; Reynisson, J.; Volcho, K.P.; Zakian, S.M.; et al. New Hybrid Compounds Combining Fragments of Usnic Acid and Monoterpenoids for Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibition. Biomolecules 2021, 11, 973. [Google Scholar] [CrossRef]
- Polovinka, M.P.; Salakhutdinov, N.F.; Panchenko, M.Y. Method for Preparing Usnic Acid. Patent RU2317076, 20 February 2006. [Google Scholar]
- Luzina, O.; Filimonov, A.; Zakharenko, A.; Chepanova, A.; Zakharova, O.; Ilina, E.; Dyrkheeva, N.; Likhatskaya, G.; Salakhutdinov, N.; Lavrink, O. Usnic Acid Conjugates with Monoterpenoids as Potent Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. J. Nat. Prod. 2020, 83, 2320–2329. [Google Scholar] [CrossRef]
- Dyrkheeva, N.; Anarbaev, R.; Lebedeva, N.; Kuprushkin, M.; Kuznetsova, A.; Kuznetsov, N.; Rechkunova, N.; Lavrik, O. Human Tyrosyl-DNA Phosphodiesterase 1 Possesses Transphosphooligonucleotidation Activity With Primary Alcohols. Front. Cell Dev. Biol. 2020, 8, 604732. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, M.V.; Khodyreva, S.N.; Lavrik, O.I. Poly(ADP-ribose) polymerase-1 inhibits strand-displacement synthesis of DNA catalyzed by DNA polymerase beta. Biochemistry 2004, 69, 558–568. [Google Scholar] [CrossRef]
- Komarova, A.O.; Drenichev, M.S.; Dyrkheeva, N.S.; Kulikova, I.V.; Oslovsky, V.E.; Zakharova, O.D.; Zakharenko, A.L.; Mikhailov, S.N.; Lavrik, O.I. Novel group of tyrosyl-DNA-phosphodiesterase 1 inhibitors based on disaccharide nucleosides as drug prototypes for anti-cancer therapy. J. Enzym. Inhib. Med. Chem. 2018, 33, 1415–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherstyuk, Y.V.; Ivanisenko, N.V.; Zakharenko, A.L.; Sukhanova, M.V.; Peshkov, R.Y.; Eltsov, I.V.; Kutuzov, M.M.; Kurgina, T.A.; Belousova, E.A.; Ivanisenko, V.A.; et al. Design, Synthesis and Molecular Modeling Study of Conjugates of ADP and Morpholino Nucleosides as A Novel Class of Inhibitors of PARP-1, PARP-2 and PARP-3. Int. J. Mol. Sci. 2019, 21, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | IC50 (TDP1), µM | HEK293A CC50, µM | HEK293FT CC50, µM | HeLa CC50, µM | PARP1, 1 mM | TDP2, 1 mM | ||
|---|---|---|---|---|---|---|---|---|
| 1 | UA | - | >50 | ND | ND | 20 ± 10 | - | - |
| 2 | 9 |  | 6.6 ± 1.0 | >50 | 7 ± 2 | 13 ± 1 | + | + |
| 3 | 7a |  | 5.4 ± 2.9 | >50 | >50 | 13.0 ± 0.5 | + | + |
| 4 | 7b | 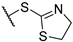 | 4.4 ± 1.0 | >50 | 16 ± 5 | 11.0 ± 0.5 | + | + |
| 5 | 7c | 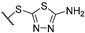 | 4.3 ± 0.5 | >50 | 35 ± 2 | 33 ± 2 | + | + |
| 6 | 7d |  | 25.2 ± 6.5 | ND | ND | ND | + | + |
| 7 | 7e | 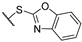 | 3.2 ± 0.5 | 26 ± 2 | 4.5 ± 1.0 | 2.9 ± 0.2 | - | + |
| 8 | 7f | 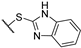 | 2.4 ± 1.0 | >50 | 10 ± 2 | 13 ± | - | + |
| 9 | 7g | 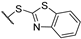 | 1.7 ± 0.6 | 3.5 ± 0.3 | 3 ± 1 | 2.0 ± 0.6 | - | + |
| 10 | 10a |  | 2.1 ± 0.2 | 20 ± 2 | 15 ± 2 | 15 ± 1 | - | + |
| 11 | 7h | 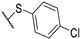 | 2.2 ± 0.5 | 10 ± 2 | 4 ± 1 | 2.5 ± 0.5 | + | + |
| 12 | 10b | 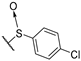 | 1.4 + 0.2 | >50 | 20 ± 3 | 27 ± 2 | - | + |
| 13 | 7i |  | 11.9 ± 0.4 | ND | ND | ND | - | + |
| 14 | 7j |  | 16.9 ± 2.4 | ND | ND | ND | + | + |
| 15 | 7k |  | 19.6 ± 2.8 | ND | ND | ND | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyrkheeva, N.S.; Filimonov, A.S.; Luzina, O.A.; Orlova, K.A.; Chernyshova, I.A.; Kornienko, T.E.; Malakhova, A.A.; Medvedev, S.P.; Zakharenko, A.L.; Ilina, E.S.; et al. New Hybrid Compounds Combining Fragments of Usnic Acid and Thioether Are Inhibitors of Human Enzymes TDP1, TDP2 and PARP1. Int. J. Mol. Sci. 2021, 22, 11336. https://doi.org/10.3390/ijms222111336
Dyrkheeva NS, Filimonov AS, Luzina OA, Orlova KA, Chernyshova IA, Kornienko TE, Malakhova AA, Medvedev SP, Zakharenko AL, Ilina ES, et al. New Hybrid Compounds Combining Fragments of Usnic Acid and Thioether Are Inhibitors of Human Enzymes TDP1, TDP2 and PARP1. International Journal of Molecular Sciences. 2021; 22(21):11336. https://doi.org/10.3390/ijms222111336
Chicago/Turabian StyleDyrkheeva, Nadezhda S., Aleksandr S. Filimonov, Olga A. Luzina, Kristina A. Orlova, Irina A. Chernyshova, Tatyana E. Kornienko, Anastasia A. Malakhova, Sergey P. Medvedev, Alexandra L. Zakharenko, Ekaterina S. Ilina, and et al. 2021. "New Hybrid Compounds Combining Fragments of Usnic Acid and Thioether Are Inhibitors of Human Enzymes TDP1, TDP2 and PARP1" International Journal of Molecular Sciences 22, no. 21: 11336. https://doi.org/10.3390/ijms222111336
APA StyleDyrkheeva, N. S., Filimonov, A. S., Luzina, O. A., Orlova, K. A., Chernyshova, I. A., Kornienko, T. E., Malakhova, A. A., Medvedev, S. P., Zakharenko, A. L., Ilina, E. S., Anarbaev, R. O., Naumenko, K. N., Klabenkova, K. V., Burakova, E. A., Stetsenko, D. A., Zakian, S. M., Salakhutdinov, N. F., & Lavrik, O. I. (2021). New Hybrid Compounds Combining Fragments of Usnic Acid and Thioether Are Inhibitors of Human Enzymes TDP1, TDP2 and PARP1. International Journal of Molecular Sciences, 22(21), 11336. https://doi.org/10.3390/ijms222111336