

The Role of P2X7 Purinoceptors in the Pathogenesis and Treatment of Muscular Dystrophies



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sterile Inflammation Is a Hallmark of Dystrophic Muscles
3. Purinergic Signaling and Its Alterations in Dystrophic Muscles
4. Altered P2X7 Expression and Function in Dystrophic Cells
5. P2X7 Up-Regulation: A Dystrophic Abnormality or Compensatory Adaptation?
6. Therapeutic Effects of P2X7 Blockade in Dystrophino- and Sarcoglycanopathies
7. P2X7 in Dystrophic Brains
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers. 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet. J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef]
- Massouridès, E.; Polentes, J.; Mangeot, P.E.; Mournetas, V.; Nectoux, J.; Deburgrave, N.; Nusbaum, P.; Leturcq, F.; Popplewell, L.; Dickson, G.; et al. Dp412e: A novel human embryonic dystrophin isoform induced by BMP4 in early differentiated cells. Skelet Muscle 2015, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Chesshyre, M.; Ridout, D.; Hashimoto, Y.; Ookubo, Y.; Torelli, S.; Maresh, K.; Ricotti, V.; Abbott, L.; Gupta, V.A.; Main, M.; et al. Investigating the role of dystrophin isoform deficiency in motor function in Duchenne muscular dystrophy. J. Cachexia Sarcopenia Muscle 2022, 13, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Desguerre, I.; Christov, C.; Mayer, M.; Zeller, R.; Becane, H.M.; Bastuji-Garin, S.; Leturcq, F.; Chiron, C.; Chelly, J.; Gherardi, R.K. Clinical heterogeneity of duchenne muscular dystrophy (DMD): Definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE 2009, 4, e4347. [Google Scholar] [CrossRef]
- Young, C.N.J.; Gosselin, M.R.F.; Rumney, R.; Oksiejuk, A.; Chira, N.; Bozycki, L.; Matryba, P.; Łukasiewicz, K.; Kao, A.P.; Dunlop, J.; et al. Total Absence of Dystrophin Expression Exacerbates Ectopic Myofiber Calcification and Fibrosis and Alters Macrophage Infiltration Patterns. Am. J. Pathol. 2020, 190, 190–205. [Google Scholar] [CrossRef]
- Daoud, F.; Angeard, N.; Demerre, B.; Martie, I.; Benyaou, R.; Leturcq, F.; Cossée, M.; Deburgrave, N.; Saillour, Y.; Tuffery, S.; et al. Analysis of Dp71 contribution in the severity of mental retardation through comparison of Duchenne and Becker patients differing by mutation consequences on Dp71 expression. Hum. Mol. Genet. 2009, 18, 3779–3794. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Marsolier, J.; Laforet, P.; Pegoraro, E.; Vissing, J.; Richard, I.; Sarcoglycanopathies Working Group. 1st International Workshop on Clinical trial readiness for Sarcoglycanopathies 15–16 November 2016, Evry, France. Neuromuscul. Disord. 2017, 27, 683–692. [Google Scholar] [CrossRef]
- Straub, V.; Murphy, A.; Udd, B.; LGMD Workshop Study Group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, The Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef]
- Mournetas, V.; Massouridès, E.; Dupont, J.B.; Kornobis, E.; Polvèche, H.; Jarrige, M.; Dorval, A.R.L.; Gosselin, M.R.F.; Manousopoulou, A.; Garbis, S.D.; et al. Myogenesis modelled by human pluripotent stem cells: A multi-omic study of Duchenne myopathy early onset. J. Cachexia Sarcopenia Muscle 2021, 12, 209–232. [Google Scholar] [CrossRef]
- Chang, N.C.; Sincennes, M.C.; Chevalier, F.P.; Brun, C.E.; Lacaria, M.; Segalés, J.; Muñoz-Cánoves, P.; Ming, H.; Rudnicki, M.A. The Dystrophin Glycoprotein Complex Regulates the Epigenetic Activation of Muscle Stem Cell Commitment. Cell Stem Cell 2018, 22, 755–768.e6. [Google Scholar] [CrossRef]
- Gosselin, M.R.F.; Mournetas, V.; Borczyk, M.; Verma, S.; Occhipinti, A.; Róg, J.; Bozycki, L.; Korostynski, M.; Robson, S.C.; Angione, C.; et al. Loss of full-length dystrophin expression results in major cell-autonomous abnormalities in proliferating myoblasts. Elife 2022, 11, e75521. [Google Scholar] [CrossRef]
- Shoji, E.; Sakurai, H.; Nishino, T.; Nakahata, T.; Heike, T.; Awaya, T.; Fujii, N.; Manabe, Y.; Matsuo, M.; Sehara-Fujisawa, A. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 2015, 5, 12831. [Google Scholar] [CrossRef]
- Panci, G.; Chazaud, B. Inflammation during post-injury skeletal muscle regeneration. Semin. Cell Dev. Biol. 2021, 119, 32–38. [Google Scholar] [CrossRef]
- Chazaud, B. Inflammation and Skeletal Muscle Regeneration: Leave It to the Macrophages! Trends Immunol. 2020, 41, 481–492. [Google Scholar] [CrossRef]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [CrossRef]
- Hartigan-O’Connor, D.; Kirk, C.J.; Crawford, R.; Mulé, J.J.; Chamberlain, J.S. Immune evasion by muscle-specific gene expression in dystrophic muscle. Mol. Ther. 2001, 4, 525–533. [Google Scholar] [CrossRef]
- Gibertini, S.; Zanotti, S.; Savadori, P.; Curcio, M.; Saredi, S.; Salerno, F.; Andreetta, F.; Bernasconi, P.; Mantegazza, R.; Mora, M. Fibrosis and inflammation are greater in muscles of beta-sarcoglycan-null mouse than mdx mouse. Cell Tissue Res. 2014, 356, 427–443. [Google Scholar] [CrossRef]
- Haslett, J.N.; Sanoudou, D.; Kho, A.T.; Bennett, R.R.; Greenberg, S.A.; Kohane, I.S.; Beggs, A.H.; Kunkel, L.M. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc. Natl. Acad. Sci. USA 2002, 99, 15000–15005. [Google Scholar] [CrossRef] [PubMed]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.; Li, Q.; Ding, J.; Liang, F.; Gusev, E.; Lapohos, O.; Fonseca, G.J.; Kaufmann, E.; Divangahi, M.; Petrof, B.J. TLR4 is a regulator of trained immunity in a murine model of Duchenne muscular dystrophy. Nat. Commun. 2022, 13, 879. [Google Scholar] [CrossRef] [PubMed]
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. Downregulation of the NLRP3 inflammasome by adiponectin rescues Duchenne muscular dystrophy. BMC Biol. 2018, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Schroder, K.; Pelegrín, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 2022, 43, 653–668. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Chang, L.; Niu, F.; Chen, J.; Cao, X.; Liu, Z.; Bao, X.; Xu, Y. Ghrelin improves muscle function in dystrophin-deficient mdx mice by inhibiting NLRP3 inflammasome activation. Life Sci. 2019, 232, 116654. [Google Scholar] [CrossRef]
- Dubuisson, N.; Davis-López de Carrizosa, M.A.; Versele, R.; Selvais, C.M.; Noel, L.; Van den Bergh, P.Y.D.; Brichard, S.M.; Abou-Samra, M. Inhibiting the inflam-masome with MCC950 counteracts muscle pyroptosis and improves Du-chenne muscular dystrophy. Front. Immunol. 2022, 13, 1049076. [Google Scholar] [CrossRef]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef]
- Nalbandian, A.; Khan, A.A.; Srivastava, R.; Llewellyn, K.J.; Tan, B.; Shukr, N.; Fazli, Y.; Kimonis, V.E.; BenMohamed, L. Activation of the NLRP3 Inflammasome Is associated with Valosin-Containing Protein Myopathy. Inflammation 2017, 40, 21–41. [Google Scholar] [CrossRef]
- Lau, Y.S.; Zhao, L.; Zhang, C.; Li, H.; Han, R. Genetic disruption of the inflammasome adaptor ASC has minimal impact on the pathogenesis of Duchenne muscular dystrophy in mdx mice. Life Sci. 2020, 257, 118069. [Google Scholar] [CrossRef]
- Accogli, T.; Hibos, C.; Vegran, F. Canonical and non-canonical functions of NLRP3. J. Adv. Res. 2023, in press. [CrossRef]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Farini, A.; Meregalli, M.; Belicchi, M.; Battistelli, M.; Parolini, D.; D’Antona, G.; Gavina, M.; Ottoboni, L.; Constantin, G.; Bottinelli, R.; et al. T and B lymphocyte depletion has a marked effect on the fibrosis of dystrophic skeletal muscles in the scid/mdx mouse. J. Pathol. 2007, 213, 229–238. [Google Scholar] [CrossRef]
- Mojumdar, K.; Liang, F.; Giordano, C.; Lemaire, C.; Danialou, G.; Okazaki, T.; Bourdon, J.; Rafei, M.; Galipeau, J.; Divangahi, M.; et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol. Med. 2014, 6, 1476–1492. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Zelikovich, A.S.; Salamone, I.M.; Fischer, J.A.; McNally, E.M. Mechanisms and Clinical Applications of Glucocorticoid Steroids in Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, 39–52. [Google Scholar] [CrossRef]
- Hedemann, A. Severe murine limb-girdle muscular dystrophy type 2C pathology is diminished by FTY720 treatment. Muscle Nerve. 2017, 56, 486–494. [Google Scholar] [CrossRef]
- Careccia, G.; Saclier, M.; Tirone, M.; Ruggieri, E.; Principi, E.; Raffaghello, L.; Torchio, S.; Recchia, D.; Canepari, M.; Gorzanelli, A.; et al. Rebalancing expression of HMGB1 redox isoforms to counteract muscular dystrophy. Sci. Transl. Med. 2021, 13, eaay8416. [Google Scholar] [CrossRef]
- Pelegrin, P. P2X7 receptor and the NLRP3 inflammasome: Partners in crime. Biochem. Pharmacol. 2021, 187, 114385. [Google Scholar] [CrossRef]
- Yeung, D.; Zablocki, K.; Lien, C.F.; Jiang, T.; Arkle, S.; Brutkowski, W.; Brown, J.; Lochmuller, H.; Simon, J.; Barnard, E.A.; et al. Increased susceptibility to ATP via alteration of P2X receptor function in dystrophic mdx mouse muscle cells. FASEB J. 2006, 20, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Young, C.N.; Brutkowski, W.; Lien, C.F.; Arkle, S.; Lochmüller, H.; Zabłocki, K.; Górecki, D.C. P2X7 purinoceptor al-terations in dystrophic mdx mouse muscles: Relationship to pathology and potential target for treatment. J. Cell Mol. Med. 2012, 16, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Gazzerro, E.; Baldassari, S.; Assereto, S.; Fruscione, F.; Pistorio, A.; Panicucci, C.; Volpi, S.; Perruzza, L.; Fiorillo, C.; Minetti, C.; et al. Enhancement of Muscle T Regulatory Cells and Improvement of Muscular Dystrophic Process in mdx Mice by Blockade of Extracellular ATP/P2X Axis. Am. J. Pathol. 2015, 185, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- MacIntosh, B.R.; Holash, R.J.; Renaud, J.M. Skeletal muscle fatigue--regulation of excitation-contraction coupling to avoid metabolic catastrophe. J. Cell Sci. 2012, 125 Pt 9, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Ryten, M.; Hoebertz, A.; Burnstock, G. Sequential expression of three receptor subtypes for extracellular ATP in developing rat skeletal muscle. Dev. Dyn. 2001, 221, 331–341. [Google Scholar] [CrossRef]
- Collet, C.; Strube, C.; Csernoch, L.; Mallouk, N.; Ojeda, C.; Allard, B.; Jacquemond, V. Effects of extracellular ATP on freshly isolated mouse skeletal muscle cells during pre-natal and post-natal development. Pflugers. Arch. 2002, 443, 771–778. [Google Scholar] [CrossRef]
- Cseri, J.; Szappanos, H.; Szigeti, G.P.; Csernátony, Z.; Kovács, L.; Csernoch, L. A purinergic signal transduction path-way in mammalian skeletal muscle cells in culture. Pflugers. Arch. 2002, 443, 731–738. [Google Scholar] [CrossRef]
- Araya, R.; Riquelme, M.A.; Brandan, E.; Sáez, J.C. The formation of skeletal muscle myotubes requires functional membrane receptors activated by extracellular ATP. Brain Res. Brain Res. Rev. 2004, 47, 174–188. [Google Scholar] [CrossRef]
- Deli, T.; Szappanos, H.; Szigeti, G.P.; Cseri, J.; Kovács, L.; Csernoch, L. Contribution from P2X and P2Y purinoreceptors to ATP-evoked changes in intracellular calcium concentration on cultured myotubes. Pflugers. Arch. 2007, 453, 519–529. [Google Scholar] [CrossRef]
- Tung, E.K.; Choi, R.C.; Siow, N.L.; Jiang, J.X.; Ling, K.K.; Simon, J.; Barnard, E.A.; Tsim, K.W. P2Y2 receptor activation regu-lates the expression of acetylcholinesterase and acetylcholine receptor genes at vertebrate neuromuscular junc-tions. Mol. Pharmacol. 2004, 66, 794–806. [Google Scholar] [CrossRef]
- Tsim, K.W.; Barnard, E.A. The signaling pathways mediated by P2Y nucleotide receptors in the formation and maintenance of the skeletal neuromuscular junction. Neurosignals 2002, 11, 58–64. [Google Scholar] [CrossRef]
- Silinsky, E.M. On the association between transmitter secretion and the release of adenine nucleotides from mammalian motor nerve terminals. J. Physiol. 1975, 247, 145–162. [Google Scholar] [CrossRef]
- Cheung, K.K.; Marques-da-Silva, C.; Vairo, L.; dos Santos, D.S.; Goldenberg, R.; Coutinho-Silva, R.; Burnstock, G. Pharmacological and molecular characterization of functional P2 receptors in rat embryonic cardiomyocytes. Purinergic Signal. 2015, 11, 127–138. [Google Scholar] [CrossRef]
- Woo, S.H.; Trinh, T.N. P2 Receptors in Cardiac Myocyte Pathophysiology and Mechanotransduction. Int. J. Mol. Sci. 2020, 22, 251. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Mazziotta, C.; Lanzillotti, C.; Stefani, C.; Badiale, G.; Campione, G.; Martini, F.; Tognon, M. The Role of Pu-rinergic P2X7 Receptor in Inflammation and Cancer: Novel Molecular Insights and Clinical Applications. Cancers 2022, 14, 1116. [Google Scholar] [CrossRef]
- Cheng, N.; Zhang, L.; Liu, L. Understanding the Role of Purinergic P2X7 Receptors in the Gastrointestinal Sysem: A Systematic Review. Front. Pharmacol. 2021, 12, 786579. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Schmalzing, G.; Markwardt, F. The Elusive P2X7 Macropore. Trends Cell Biol. 2018, 28, 392–404. [Google Scholar] [CrossRef]
- Zimmermann, H. Ectonucleoside triphosphate diphosphohydrolases and ecto-5’-nucleotidase in purinergic signaling: How the field developed and where we are now. Purinergic Signal. 2021, 17, 117–125. [Google Scholar] [CrossRef]
- Giuliani, A.L.; Sarti, A.C.; Di Virgilio, F. Ectonucleotidases in Acute and Chronic Inflammation. Front. Pharmacol. 2021, 11, 619458. [Google Scholar] [CrossRef]
- Zimmermann, H. History of ectonucleotidases and their role in purinergic signaling. Biochem. Pharmacol. 2021, 187, 114322. [Google Scholar] [CrossRef]
- Betto, R.; Senter, L.; Ceoldo, S.; Tarricone, E.; Biral, D.; Salviati, G. Ecto-ATPase activity of alpha-sarcoglycan (ad-halin). J. Biol. Chem. 1999, 274, 7907–7912. [Google Scholar] [CrossRef] [PubMed]
- Sandonà, D.; Gastaldello, S.; Martinello, T.; Betto, R. Characterization of the ATP-hydrolysing activity of alpha-sarcoglycan. Biochem. J. 2004, 381 Pt 1, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Zabłocka, B.; Górecki, D.C.; Zabłocki, K. Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. Int. J. Mol. Sci. 2021, 22, 11040. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Arnett, T.R.; Orriss, I.R. Purinergic signalling in the musculoskeletal system. Purinergic Signal. 2013, 9, 541–572. [Google Scholar] [CrossRef]
- Ryten, M.; Yang, S.Y.; Dunn, P.M.; Goldspink, G.; Burnstock, G. Purinoceptor expression in regenerating skeletal muscle in the mdx mouse model of muscular dystrophy and in satellite cell cultures. FASEB J. 2004, 18, 1404–1406. [Google Scholar] [CrossRef]
- Khairullin, A.E.; Grishin, S.N.; Ziganshin, A.U. P2 Receptor Signaling in Motor Units in Muscular Dystrophy. Int. J. Mol. Sci. 2023, 24, 1587. [Google Scholar] [CrossRef]
- Róg, J.; Oksiejuk, A.; Gosselin, M.R.F.; Brutkowski, W.; Dymkowska, D.; Nowak, N.; Robson, S.; Górecki, D.C.; Zabłocki, K. Dystrophic mdx mouse myoblasts exhibit elevated ATP/UTP-evoked metabotropic purinergic responses and alterations in calcium signalling. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1138–1151. [Google Scholar] [CrossRef]
- Róg, J.; Oksiejuk, A.; Górecki, D.C.; Zabłocki, K. Metabotropic purinergic receptor profiles and calcium signalling in primary mice myoblasts differ depending on their muscle origin and are altered in cells with mutated dystrophin gene (mdx mice). BioRxiv 2022. [Google Scholar] [CrossRef]
- Benzi, A.; Baratto, S.; Astigiano, C.; Sturla, L.; Panicucci, C.; Mamchaoui, K.; Raffaghello, L.; Bruzzone, S.; Gazzerro, E.; Bruno, C. Aberrant Adenosine Triphosphate Release and Impairment of P2Y2-Mediated Signaling in Sarcoglycanopathies. Lab. Investig. 2023, 103, 100037. [Google Scholar] [CrossRef]
- Gazzerro, E.; Baratto, S.; Assereto, S.; Baldassari, S.; Panicucci, C.; Raffaghello, L.; Scudieri, P.; De Battista, D.; Fiorillo, C.; Volpi, S.; et al. The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan-Deficient Muscular Dystrophy. Am. J. Pathol. 2019, 189, 354–369. [Google Scholar] [CrossRef]
- Ferrari, D.; Munerati, M.; Melchiorri, L.; Hanau, S.; di Virgilio, F.; Baricordi, O.R. Responses to extracellular ATP of lymphoblastoid cell lines from Duchenne muscular dystrophy patients. Am. J. Physiol. 1994, 267 Pt 1, C886–C892. [Google Scholar] [CrossRef]
- Young, C.N.; Sinadinos, A.; Lefebvre, A.; Chan, P.; Arkle, S.; Vaudry, D.; Gorecki, D.C. A novel mechanism of autophagic cell death in dystrophic muscle regulated by P2RX7 receptor large-pore formation and HSP90. Autophagy 2015, 11, 113–130. [Google Scholar] [CrossRef]
- Dubuisson, N.; Versele, R.; Davis-López de Carrizosa, M.A.; Selvais, C.M.; Brichard, S.M.; Abou-Samra, M. Walking down Skeletal Muscle Lane: From Inflammasome to Disease. Cells 2021, 10, 3023. [Google Scholar] [CrossRef]
- Morgan, J.E.; Prola, A.; Mariot, V.; Pini, V.; Meng, J.; Hourde, C.; Dumonceaux, J.; Conti, F.; Relaix, F.; Authier, F.J.; et al. Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat. Commun. 2018, 9, 3655. [Google Scholar] [CrossRef]
- Sinadinos, A.; Young, C.N.; Al-Khalidi, R.; Teti, A.; Kalinski, P.; Mohamad, S.; Floriot, L.; Henry, T.; Tozzi, G.; Jiang, T.; et al. P2RX7 purinoceptor: A therapeutic target for ameliorating the symptoms of duchenne muscular dystrophy. PLoS Med. 2015, 12, e1001888. [Google Scholar] [CrossRef]
- Raffaghello, L.; Principi, E.; Baratto, S.; Panicucci, C.; Pintus, S.; Antonini, F.; Del Zotto, G.; Benzi, A.; Bruzzone, S.; Scudieri, P.; et al. P2X7 Receptor Antagonist Reduces Fibrosis and Inflammation in a Mouse Model of Alpha-Sarcoglycan Muscular Dystrophy. Pharmaceuticals 2022, 15, 89. [Google Scholar] [CrossRef]
- Call, J.A.; Nichenko, A.S. Autophagy: An essential but limited cellular process for timely skeletal muscle recovery from injury. Autophagy 2020, 16, 1344–1347. [Google Scholar] [CrossRef]
- Yazid, M.D.; Hung-Chih, C. Perturbation of PI3K/Akt signaling affected autophagy modulation in dystrophin-deficient myoblasts. Cell Commun. Signal. 2021, 19, 105. [Google Scholar] [CrossRef]
- Pal, R.; Palmieri, M.; Loehr, J.A.; Li, S.; Abo-Zahrah, R.; Monroe, T.O.; Thakur, P.B.; Sardiello, M.; Rodney, G.G. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat. Commun. 2014, 5, 4425. [Google Scholar] [CrossRef]
- Adinolfi, E.; Kim, M.; Young, M.T.; Di Virgilio, F.; Surprenant, A. Tyrosine phosphorylation of HSP90 within the P2X7 receptor complex negatively regulates P2X7 receptors. J. Biol. Chem. 2003, 278, 37344–37351. [Google Scholar] [CrossRef]
- Kopp, R.; Krautloher, A.; Ramírez-Fernández, A.; Nicke, A. P2X7 Interactions and Signaling—Making Head or Tail of It. Front. Mol. Neurosci. 2019, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Vessey, D.A.; Li, L.; Kelley, M. Pannexin-I/P2X 7 purinergic receptor channels mediate the release of cardio-protectants induced by ischemic pre- and postconditioning. J. Cardiovasc. Pharmacol. Ther. 2010, 15, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Vessey, D.A.; Li, L.; Kelley, M. P2X7 receptor agonists pre- and postcondition the heart against ischemia-reperfusion injury by opening pannexin-1/P2X₇ channels. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H881–H887. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F. P2RX7: A receptor with a split personality in inflammation and cancer. Mol. Cell Oncol. 2015, 3, e1010937. [Google Scholar] [CrossRef] [PubMed]
- Fabbrizio, P.; Apolloni, S.; Bianchi, A.; Salvatori, I.; Valle, C.; Lanzuolo, C.; Bendotti, C.; Nardo, G.; Volonté, C. P2X7 activation enhances skeletal muscle metabolism and regeneration in SOD1G93A mouse model of amyotrophic lateral sclerosis. Brain Pathol. 2020, 30, 272–282. [Google Scholar] [CrossRef]
- Onopiuk, M.; Brutkowski, W.; Wierzbicka, K.; Wojciechowska, S.; Szczepanowska, J.; Fronk, J.; Lochmüller, H.; Górecki, D.C.; Zabłocki, K. Mutation in dystrophin-encoding gene affects energy metabolism in mouse myoblasts. Biochem. Biophys. Res. Commun. 2009, 386, 463–466. [Google Scholar] [CrossRef]
- Matre, P.R.; Mu, X.; Wu, J.; Danila, D.; Hall, M.A.; Kolonin, M.G.; Darabi, R.; Huard, J. CRISPR/Cas9-Based Dystro-phin Restoration Reveals a Novel Role for Dystrophin in Bioenergetics and Stress Resistance of Muscle Progenitors. Stem Cells 2019, 37, 1615–1628. [Google Scholar] [CrossRef]
- Bellissimo, C.A.; Garibotti, M.C.; Perry, C.G.R. Mitochondrial stress responses in Duchenne muscular dystrophy: Metabolic dysfunction or adaptive reprogramming? Am. J. Physiol. Cell Physiol. 2022, 323, C718–C730. [Google Scholar] [CrossRef]
- Rumney, R.M.H.; Róg, J.; Chira, N.; Kao, A.P.; Al-Khalidi, R.; Górecki, D.C. P2X7 Purinoceptor Affects Ectopic Calcification of Dystrophic Muscles. Front. Pharmacol. 2022, 13, 935804. [Google Scholar] [CrossRef]
- Young, C.N.J.; Chira, N.; Róg, J.; Al-Khalidi, R.; Benard, M.; Galas, L.; Chan, P.; Vaudry, D.; Zablocki, K.; Górecki, D.C. Sustained activation of P2X7 induces MMP-2-evoked cleavage and functional purinoceptor inhibition. J. Mol. Cell Biol. 2018, 10, 229–242. [Google Scholar] [CrossRef]
- Villalta, S.A.; Rosenthal, W.; Martinez, L.; Kaur, A.; Sparwasser, T.; Tidball, J.G.; Margeta, M.; Spencer, M.J.; Bluestone, J.A. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med. 2014, 6, 258ra142. [Google Scholar] [CrossRef]
- Boumechache, M.; Masin, M.; Edwardson, J.M.; Górecki, D.C.; Murrell-Lagnado, R. Analysis of assembly and trafficking of native P2X4 and P2X7 receptor complexes in rodent immune cells. J. Biol. Chem. 2009, 284, 13446–13454. [Google Scholar] [CrossRef]
- Keystone, E.C.; Wang, M.M.; Layton, M.; Hollis, S.; McInnes, I.B.; D1520C00001 Study Team. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann. Rheum. Dis. 2012, 71, 1630–1635, Erratum in Ann. Rheum. Dis. 2012, 71, 2064. [Google Scholar] [CrossRef]
- Stock, T.C.; Bloom, B.J.; Wei, N.; Ishaq, S.; Park, W.; Wang, X.; Gupta, P.; Mebus, C.A. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J. Rheumatol. 2012, 39, 720–727. [Google Scholar] [CrossRef]
- Eser, A.; Colombel, J.F.; Rutgeerts, P.; Vermeire, S.; Vogelsang, H.; Braddock, M.; Persson, T.; Reinisch, W. Safety and Efficacy of an Oral Inhibitor of the Purinergic Receptor P2X7 in Adult Patients with Moderately to Severely Active Crohn’s Disease: A Randomized Placebo-controlled, Double-blind, Phase IIa Study. Inflamm. Bowel. Dis. 2015, 21, 2247–2253. [Google Scholar] [CrossRef]
- Karasawa, A.; Kawate, T. Structural basis for subtype-specific inhibition of the P2X7 receptor. Elife 2016, 5, e22153. [Google Scholar] [CrossRef]
- Khalidi, R.; Panicucci, C.; Cox, P.; Chira, N.; Róg, J.; Young, C.N.J.; McGeehan, R.E.; Ambati, K.; Ambati, J.; Zabłocki, K.; et al. Zidovudine ameliorates pathology in the mouse model of Duchenne muscular dystrophy via P2RX7 purinoceptor antagonism. Acta Neuropathol. Commun. 2018, 6, 27. [Google Scholar] [CrossRef]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hirano, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T.; et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef]
- Narendran, S.; Pereira, F.; Yerramothu, P.; Apicella, I.; Wang, S.B.; Ambati, K.; Hirahara, S.; Kim, Y.; Ambati, M.; Ambati, V.L.; et al. Nucleoside reverse transcriptase inhibitors and Kamuvudines inhibit amyloid-β induced retinal pigmented epithelium degeneration. Signal. Transduct. Target Ther. 2021, 6, 149. [Google Scholar] [CrossRef]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef]
- Rivas-Yáñez, E.; Barrera-Avalos, C.; Parra-Tello, B.; Briceño, P.; Rosemblatt, M.V.; Saavedra-Almarza, J.; Rosemblatt, M.; Acuña-Castillo, C.; Bono, M.R.; Sauma, D. P2X7 Receptor at the Crossroads of T Cell Fate. Int. J. Mol. Sci. 2020, 21, 4937. [Google Scholar] [CrossRef] [PubMed]
- Rissiek, B.; Haag, F.; Boyer, O.; Koch-Nolte, F.; Adriouch, S. P2X7 on Mouse T Cells: One Channel, Many Functions. Front. Immunol. 2015, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, E.; Orioli, E.; Pegoraro, A.; Sangaletti, S.; Portararo, P.; Curti, A.; Colombo, M.P.; Di Virgilio, F.; Adinolfi, E. The P2X7 receptor modulates immune cells infiltration, ectonucleotidases expression and extracellular ATP levels in the tumor microenvironment. Oncogene 2019, 38, 3636–3650. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhao, Y.; Xiao, X.; Fan, Y.; Kloc, M.; Liu, W.; Ghobrial, R.M.; Lan, P.; He, X.; Li, X.C. Graft-Infiltrating Macrophages Adopt an M2 Phenotype and Are Inhibited by Purinergic Receptor P2X7 Antagonist in Chronic Rejection. Am. J. Transplant. 2016, 16, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Vergani, A.; Tezza, S.; D’Addio, F.; Fotino, C.; Liu, K.; Niewczas, M.; Bassi, R.; Molano, R.D.; Kleffel, S.; Petrelli, A.; et al. Long-term heart transplant survival by targeting the ionotropic purinergic receptor P2X7. Circulation 2013, 127, 463–475. [Google Scholar] [CrossRef]
- Jackson, T.; Seifi, M.; Górecki, D.C.; Swinny, J.D. Specific Dystrophins Selectively associate with Inhibitory and Excitatory Synapses of the Mouse Cerebellum and their Loss Alters Expression of P2X7 Puri-noceptors and Pro-Inflammatory Mediators. Cell Mol. Neurobiol. 2022, 42, 2357–2377. [Google Scholar] [CrossRef]
- Cuthbertson, P.; Geraghty, N.J.; Adhikary, S.R.; Casolin, S.; Watson, D.; Sluyter, R. P2X7 receptor antagonism increases regulatory T cells and reduces clinical and histological graft-versus-host disease in a humanised mouse model. Clin. Sci. 2021, 135, 495–513. [Google Scholar] [CrossRef]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Corsi, P.; Ribatti, D.; Quondamatteo, F.; Herken, R.; Girolamo, F.; Marzullo, A.; Svelto, M.; et al. Severe alterations of endothelial and glial cells in the blood-brain barrier of dystrophic mdx mice. Glia 2003, 42, 235–251. [Google Scholar] [CrossRef]
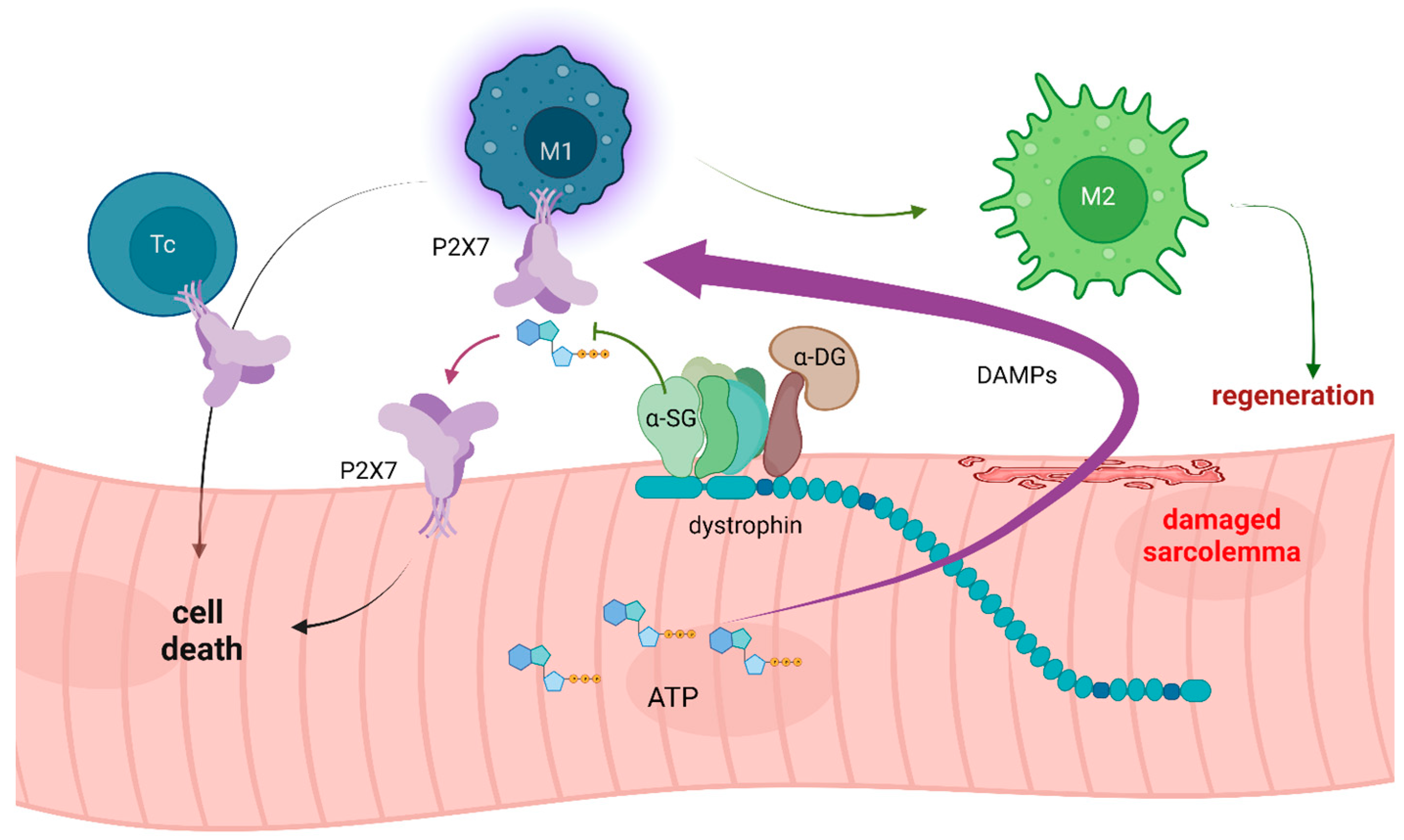
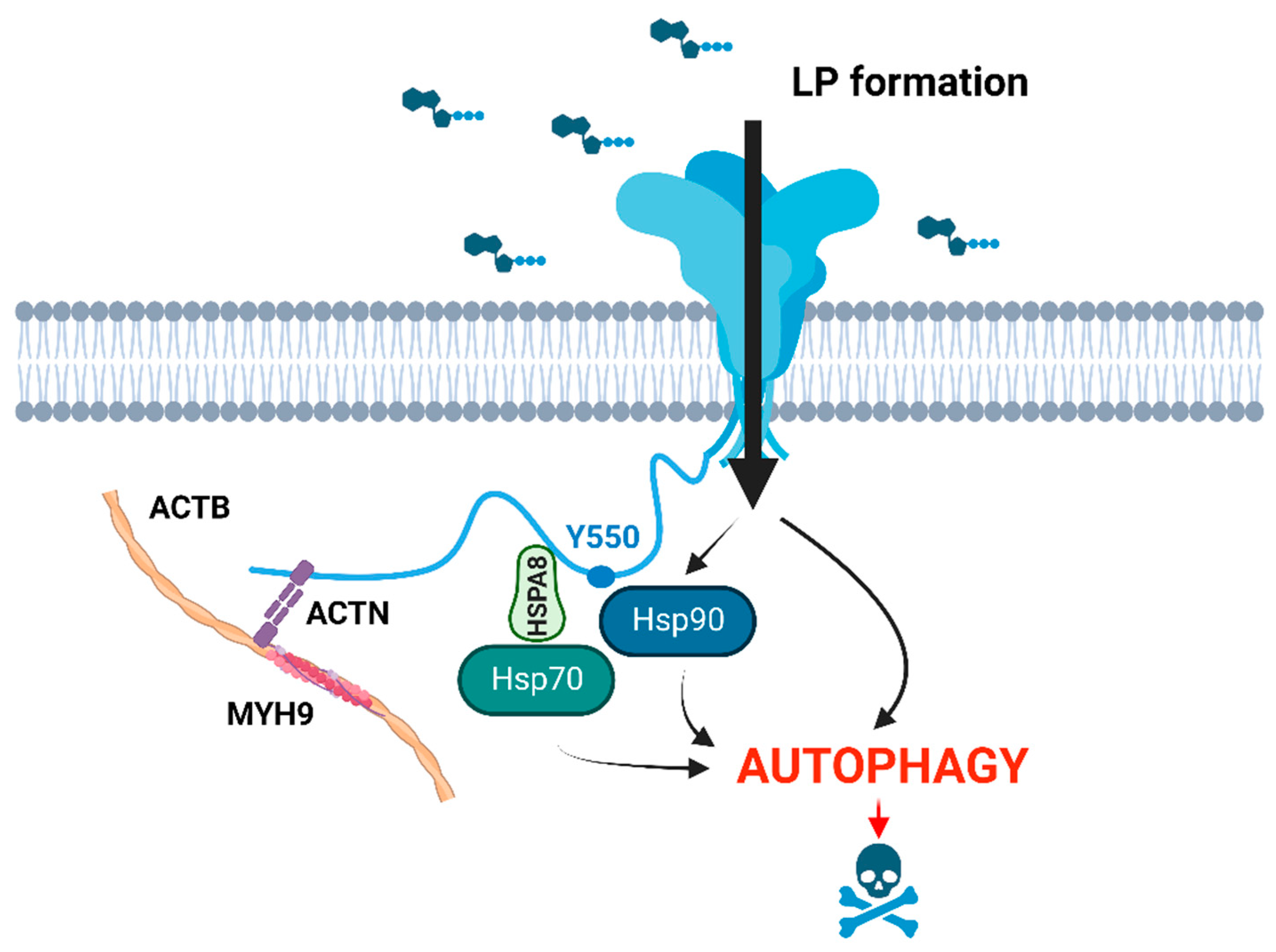

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zabłocki, K.; Górecki, D.C. The Role of P2X7 Purinoceptors in the Pathogenesis and Treatment of Muscular Dystrophies. Int. J. Mol. Sci. 2023, 24, 9434. https://doi.org/10.3390/ijms24119434
Zabłocki K, Górecki DC. The Role of P2X7 Purinoceptors in the Pathogenesis and Treatment of Muscular Dystrophies. International Journal of Molecular Sciences. 2023; 24(11):9434. https://doi.org/10.3390/ijms24119434
Chicago/Turabian StyleZabłocki, Krzysztof, and Dariusz C. Górecki. 2023. "The Role of P2X7 Purinoceptors in the Pathogenesis and Treatment of Muscular Dystrophies" International Journal of Molecular Sciences 24, no. 11: 9434. https://doi.org/10.3390/ijms24119434