
The Role of Interleukin-17 in Juvenile Idiopathic Arthritis: From Pathogenesis to Treatment

 , and
, and 
Abstract
:1. Introduction
2. Molecular Features and Signaling of IL-17
3. The Pro-Inflammatory Function of IL-17
4. Classification of JIA Subtypes
5. The Current Treatment of JIA
6. The Role of IL-17 in Oligoarticular, Polyarticular, and Enthesitis-Related Arthritis Subtypes
7. The Potential Role of IL-17 in Systemic JIA
8. The IL-23–IL-17 Axis: The Blockade of IL-23 in the Therapy of JIA
9. The Role of Anti-IL-17A Blocking Antibodies in JPsA and ERA Subtypes of JIA
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Martini, A.; Lovell, D.J.; Albani, S.; Brunner, H.I.; Hyrich, K.L.; Thompson, S.D.; Ruperto, N. Juvenile idiopathic arthritis. Nat. Rev. Dis. Prim. 2022, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Rypdal, V.; Arnstad, E.D.; Aalto, K.; Berntson, L.; Ekelund, M.; Fasth, A.; Glerup, M.; Herlin, T.; Nielsen, S.; Peltoniemi, S.; et al. Predicting unfavorable long-term outcome in juvenile idiopathic arthritis: Results from the Nordic cohort study. Arthritis Res. Ther. 2018, 20, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaag, A.M.; Kirkhus, E.; Tornqvist, L.; Lilleby, V.; Aulie, H.A.; Flato, B. Radiographic damage in hands and wrists of patients with juvenile idiopathic arthritis after 29 years of disease duration. Pediatr. Rheumatol. Online J. 2017, 15, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giancane, G.; Muratore, V.; Marzetti, V.; Quilis, N.; Benavente, B.S.; Bagnasco, F.; Alongi, A.; Civino, A.; Quartulli, L.; Consolaro, A.; et al. Disease activity and damage in juvenile idiopathic arthritis: Methotrexate era versus biologic era. Arthritis Res. Ther. 2019, 21, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onel, K.B.; Horton, D.B.; Lovell, D.J.; Shenoi, S.; Cuello, C.A.; Angeles-Han, S.T.; Becker, M.L.; Cron, R.Q.; Feldman, B.M.; Ferguson, P.J.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Juvenile Idiopathic Arthritis: Therapeutic Approaches for Oligoarthritis, Temporomandibular Joint Arthritis, and Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2022, 74, 553–569. [Google Scholar] [CrossRef]
- Rouvier, E.; Luciani, M.F.; Mattei, M.G.; Denizot, F.; Golstein, P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J. Immunol. 1993, 150, 5445–5456. [Google Scholar]
- Yao, Z.; Painter, S.L.; Fanslow, W.C.; Ulrich, D.; Macduff, B.M.; Spriggs, M.K.; Armitage, R.J. Human IL-17: A novel cytokine derived from T cells. J. Immunol. 1995, 155, 5483–5486. [Google Scholar]
- Hymowitz, S.G.; Filvaroff, E.H.; Yin, J.P.; Lee, J.; Cai, L.; Risser, P.; Maruoka, M.; Mao, W.; Foster, J.; Kelley, R.F. IL-17s adopt a cystine knot fold: Structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001, 20, 5332–5341. [Google Scholar] [CrossRef] [Green Version]
- Novatchkova, M.; Leibbrandt, A.; Werzowa, J.; Neubuser, A.; Eisenhaber, F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem. Sci. 2003, 28, 226–229. [Google Scholar] [CrossRef]
- Chen, X.; Cai, G.; Liu, C.; Zhao, J.; Gu, C.; Wu, L.; Hamilton, T.A.; Zhang, C.J.; Ko, J.; Zhu, L.; et al. IL-17R-EGFR axis links wound healing to tumorigenesis in Lrig1(+) stem cells. J. Exp. Med. 2019, 216, 195–214. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Fanslow, W.C.; Seldin, M.F.; Rousseau, A.M.; Painter, S.L.; Comeau, M.R.; Cohen, J.I.; Spriggs, M.K. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity 1995, 3, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Gaffen, S.L. Life before seventeen: Cloning of the IL-17 receptor. J. Immunol. 2011, 187, 4389–4391. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.F.; Bennett, F.; Li, B.; Brooks, J.; Luxenberg, D.P.; Whitters, M.J.; Tomkinson, K.N.; Fitz, L.J.; Wolfman, N.M.; Collins, M.; et al. The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. J. Immunol. 2008, 181, 2799–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.H.; Dong, C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 2007, 17, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.C.; Long, A.J.; Bennett, F.; Whitters, M.J.; Karim, R.; Collins, M.; Goldman, S.J.; Dunussi-Joannopoulos, K.; Williams, C.M.; Wright, J.F.; et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J. Immunol. 2007, 179, 7791–7799. [Google Scholar] [CrossRef] [Green Version]
- Toy, D.; Kugler, D.; Wolfson, M.; Vanden Bos, T.; Gurgel, J.; Derry, J.; Tocker, J.; Peschon, J. Cutting edge: Interleukin 17 signals through a heteromeric receptor complex. J. Immunol. 2006, 177, 36–39. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Ota, N.; Peng, I.; Refino, C.J.; Danilenko, D.M.; Caplazi, P.; Ouyang, W. IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. J. Immunol. 2010, 184, 4307–4316. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.-H.; Ye, X.-Q.; Iwakura, Y. Interleukin-17 family members in health and disease. Int. Immunol. 2021, 33, 723–729. [Google Scholar] [CrossRef]
- Qian, Y.; Liu, C.; Hartupee, J.; Altuntas, C.Z.; Gulen, M.F.; Jane-Wit, D.; Xiao, J.; Lu, Y.; Giltiay, N.; Liu, J.; et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 2007, 8, 247–256. [Google Scholar] [CrossRef]
- Sonder, S.U.; Saret, S.; Tang, W.; Sturdevant, D.E.; Porcella, S.F.; Siebenlist, U. IL-17-induced NF-kappaB activation via CIKS/Act1: Physiologic significance and signaling mechanisms. J. Biol. Chem. 2011, 286, 12881–12890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonder, S.U.; Paun, A.; Ha, H.L.; Johnson, P.F.; Siebenlist, U. CIKS/Act1-mediated signaling by IL-17 cytokines in context: Implications for how a CIKS gene variant may predispose to psoriasis. J. Immunol. 2012, 188, 5906–5914. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Gaffen, S.L. IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida albicans. J. Immunol. 2015, 195, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Vinh, D.C.; Casanova, J.L.; Puel, A. Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr. Opin. Microbiol. 2017, 40, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008, 29, 628–636. [Google Scholar] [CrossRef] [Green Version]
- Ruddy, M.J.; Wong, G.C.; Liu, X.K.; Yamamoto, H.; Kasayama, S.; Kirkwood, K.L.; Gaffen, S.L. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J. Biol. Chem. 2004, 279, 2559–2567. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Dai, D.; He, X.; Zhu, S.; Yao, Y.; Gao, H.; Wang, J.; Qu, F.; Qiu, J.; Wang, H.; et al. Growth Factor FGF2 Cooperates with Interleukin-17 to Repair Intestinal Epithelial Damage. Immunity 2015, 43, 488–501. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.H.; Richardson, J.P.; Zhou, C.; Coleman, B.M.; Moyes, D.L.; Ho, J.; Huppler, A.R.; Ramani, K.; McGeachy, M.J.; Mufazalov, I.A.; et al. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci. Immunol. 2017, 2, eaam8834. [Google Scholar] [CrossRef] [Green Version]
- Drummond, R.A.; Lionakis, M.S. Organ-specific mechanisms linking innate and adaptive antifungal immunity. Semin. Cell Dev. Biol. 2019, 89, 78–90. [Google Scholar] [CrossRef]
- Li, J.; Casanova, J.-L.; Puel, A. Mucocutaneous IL-17 immunity in mice and humans: Host defense vs. excessive inflammation. Mucosal Immunol. 2018, 11, 581–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Omenetti, S. The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef]
- Ishigame, H.; Kakuta, S.; Nagai, T.; Kadoki, M.; Nambu, A.; Komiyama, Y.; Fujikado, N.; Tanahashi, Y.; Akitsu, A.; Kotaki, H.; et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009, 30, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.H.; Reynolds, J.M.; Pappu, B.P.; Chen, G.; Martinez, G.J.; Dong, C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity 2011, 35, 611–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166. [Google Scholar] [CrossRef]
- Riether, C.; Radpour, R.; Kallen, N.M.; Burgin, D.T.; Bachmann, C.; Schurch, C.M.; Luthi, U.; Arambasic, M.; Hoppe, S.; Albers, C.E.; et al. Metoclopramide treatment blocks CD93-signaling-mediated self-renewal of chronic myeloid leukemia stem cells. Cell Rep. 2021, 34, 108663. [Google Scholar] [CrossRef]
- Veldhoen, M. Interleukin 17 is a chief orchestrator of immunity. Nat. Immunol. 2017, 18, 612–621. [Google Scholar] [CrossRef]
- Kolls, J.K.; McCray, P.B.; Chan, Y.R. Cytokine-mediated regulation of antimicrobial proteins. Nat. Rev. Immunol. 2008, 8, 829–835. [Google Scholar] [CrossRef] [Green Version]
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 2010, 129, 311–321. [Google Scholar] [CrossRef]
- Huang, W.; Na, L.; Fidel, P.L.; Schwarzenberger, P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 2004, 190, 624–631. [Google Scholar] [CrossRef] [Green Version]
- Kisand, K.; Boe Wolff, A.S.; Podkrajsek, K.T.; Tserel, L.; Link, M.; Kisand, K.V.; Ersvaer, E.; Perheentupa, J.; Erichsen, M.M.; Bratanic, N.; et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 2010, 207, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Monin, L.; Gordon, R.; Aggor, F.E.Y.; Bechara, R.; Edwards, T.N.; Kaplan, D.H.; Gingras, S.; Gaffen, S.L. An IL-17F.S65L Knock-In Mouse Reveals Similarities and Differences in IL-17F Function in Oral Candidiasis: A New Tool to Understand IL-17F. J. Immunol. 2020, 205, 720–730. [Google Scholar] [CrossRef]
- Albanesi, C.; Cavani, A.; Girolomoni, G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: Synergistic or antagonist effects with IFN-gamma and TNF-alpha. J. Immunol. 1999, 162, 494–502. [Google Scholar] [PubMed]
- Antonysamy, M.A.; Fanslow, W.C.; Fu, F.; Li, W.; Qian, S.; Troutt, A.B.; Thomson, A.W. Evidence for a role of IL-17 in organ allograft rejection: IL-17 promotes the functional differentiation of dendritic cell progenitors. J. Immunol. 1999, 162, 577–584. [Google Scholar] [PubMed]
- Infante-Duarte, C.; Horton, H.F.; Byrne, M.C.; Kamradt, T. Microbial lipopeptides induce the production of IL-17 in Th cells. J. Immunol. 2000, 165, 6107–6115. [Google Scholar] [CrossRef] [Green Version]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Ciofani, M.; Madar, A.; Galan, C.; Sellars, M.; Mace, K.; Pauli, F.; Agarwal, A.; Huang, W.; Parkhurst, C.N.; Muratet, M.; et al. A validated regulatory network for Th17 cell specification. Cell 2012, 151, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; O’Shea, J.J. Th17 cells: A new fate for differentiating helper T cells. Immunol. Res. 2008, 41, 87–102. [Google Scholar] [CrossRef]
- Martini, S.; Pozzi, G.; Carubbi, C.; Masselli, E.; Galli, D.; Di Nuzzo, S.; Banchini, A.; Gobbi, G.; Vitale, M.; Mirandola, P. PKCepsilon promotes human Th17 differentiation: Implications in the pathophysiology of psoriasis. Eur. J. Immunol. 2018, 48, 644–654. [Google Scholar] [CrossRef]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- He, Y.W.; Deftos, M.L.; Ojala, E.W.; Bevan, M.J. RORgamma t, a novel isoform of an orphan receptor, negatively regulates Fas ligand expression and IL-2 production in T cells. Immunity 1998, 9, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 562. [Google Scholar] [CrossRef] [Green Version]
- Lückel, C.; Picard, F.S.R.; Huber, M. Tc17 biology and function: Novel concepts. Eur. J. Immunol. 2020, 50, 1257–1267. [Google Scholar] [CrossRef]
- Srenathan, U.; Steel, K.; Taams, L.S. IL-17+ CD8+ T cells: Differentiation, phenotype and role in inflammatory disease. Immunol. Lett. 2016, 178, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Papotto, P.H.; Ribot, J.C.; Silva-Santos, B. IL-17(+) gammadelta T cells as kick-starters of inflammation. Nat. Immunol. 2017, 18, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, M.D.; Spits, H. Human innate lymphoid cells. Blood 2014, 124, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Cao, A.; Yao, S.; Evans-Marin, H.L.; Liu, H.; Wu, W.; Carlsen, E.D.; Dann, S.M.; Soong, L.; Sun, J.; et al. mTOR Mediates IL-23 Induction of Neutrophil IL-17 and IL-22 Production. J. Immunol. 2016, 196, 4390–4399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamassia, N.; Arruda-Silva, F.; Calzetti, F.; Lonardi, S.; Gasperini, S.; Gardiman, E.; Bianchetto-Aguilera, F.; Gatta, L.B.; Girolomoni, G.; Mantovani, A.; et al. A Reappraisal on the Potential Ability of Human Neutrophils to Express and Produce IL-17 Family Members In Vitro: Failure to Reproducibly Detect It. Front. Immunol. 2018, 9, 795. [Google Scholar] [CrossRef]
- Taylor, P.R.; Leal, S.M., Jr.; Sun, Y.; Pearlman, E. Aspergillus and Fusarium corneal infections are regulated by Th17 cells and IL-17-producing neutrophils. J. Immunol. 2014, 192, 3319–3327. [Google Scholar] [CrossRef] [Green Version]
- Werner, J.L.; Gessner, M.A.; Lilly, L.M.; Nelson, M.P.; Metz, A.E.; Horn, D.; Dunaway, C.W.; Deshane, J.; Chaplin, D.D.; Weaver, C.T.; et al. Neutrophils produce interleukin 17A (IL-17A) in a dectin-1- and IL-23-dependent manner during invasive fungal infection. Infect. Immun. 2011, 79, 3966–3977. [Google Scholar] [CrossRef] [Green Version]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Genovese, M.C.; Greenwald, M.; Cho, C.S.; Berman, A.; Jin, L.; Cameron, G.S.; Benichou, O.; Xie, L.; Braun, D.; Berclaz, P.Y.; et al. A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol. 2014, 66, 1693–1704. [Google Scholar] [CrossRef]
- Pavelka, K.; Chon, Y.; Newmark, R.; Lin, S.L.; Baumgartner, S.; Erondu, N. A study to evaluate the safety, tolerability, and efficacy of brodalumab in subjects with rheumatoid arthritis and an inadequate response to methotrexate. J. Rheumatol. 2015, 42, 912–919. [Google Scholar] [CrossRef]
- McInnes, I.B.; Sieper, J.; Braun, J.; Emery, P.; van der Heijde, D.; Isaacs, J.D.; Dahmen, G.; Wollenhaupt, J.; Schulze-Koops, H.; Kogan, J.; et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: A 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann. Rheum. Dis. 2014, 73, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; McInnes, I.B. Secukinumab: A New Treatment Option for Psoriatic Arthritis. Rheumatol. Ther. 2016, 3, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M.; et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Mens, L.J.J.; van de Sande, M.G.H.; Menegatti, S.; Chen, S.; Blijdorp, I.C.J.; de Jong, H.M.; Fluri, I.A.; Latuhihin, T.E.; van Kuijk, A.W.R.; Rogge, L.; et al. Brief Report: Interleukin-17 Blockade with Secukinumab in Peripheral Spondyloarthritis Impacts Synovial Immunopathology Without Compromising Systemic Immune Responses. Arthritis Rheumatol. 2018, 70, 1994–2002. [Google Scholar] [CrossRef] [Green Version]
- Taams, L.S. Interleukin-17 in rheumatoid arthritis: Trials and tribulations. J. Exp. Med. 2020, 217, e20192048. [Google Scholar] [CrossRef]
- Messina, G.; Forni, S.; Rosadini, D.; Falcone, M.; Collini, F.; Nante, N. Risk adjusted mortality after hip replacement surgery: A retrospective study. Ann. Dell’istituto Super. Sanita 2017, 53, 40–45. [Google Scholar]
- Messina, G.; Rasimelli, L.; Bonavita, C.; Ceriale, E.; Quercioli, C.; Nante, N. Which factors influence functional patients improvements during rehabilitation? Glob. J. Health Sci. 2014, 6, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Abd Almonaem, E.R.; Shaheen, A.M.; Abdelrahman, A.M.N.; Hassan, W.A.; Daay El Khair, N.M.; Abdel Haie, O.M. Association between Interleukin-17F 7488A/G and 7383A/G polymorphisms and susceptibility to juvenile idiopathic arthritis. Pediatr. Res. 2022. [Google Scholar] [CrossRef]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.M.; et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar]
- Behrens, E.M.; Beukelman, T.; Gallo, L.; Spangler, J.; Rosenkranz, M.; Arkachaisri, T.; Ayala, R.; Groh, B.; Finkel, T.H.; Cron, R.Q. Evaluation of the presentation of systemic onset juvenile rheumatoid arthritis: Data from the Pennsylvania Systemic Onset Juvenile Arthritis Registry (PASOJAR). J. Rheumatol. 2008, 35, 343–348. [Google Scholar]
- Singh-Grewal, D.; Schneider, R.; Bayer, N.; Feldman, B.M. Predictors of disease course and remission in systemic juvenile idiopathic arthritis: Significance of early clinical and laboratory features. Arthritis Rheumatol. 2006, 54, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Lomater, C.; Gerloni, V.; Gattinara, M.; Mazzotti, J.; Cimaz, R.; Fantini, F. Systemic onset juvenile idiopathic arthritis: A retrospective study of 80 consecutive patients followed for 10 years. J. Rheumatol. 2000, 27, 491–496. [Google Scholar] [PubMed]
- Janow, G.; Schanberg, L.E.; Setoguchi, S.; Hasselblad, V.; Mellins, E.D.; Schneider, R.; Kimura, Y. The Systemic Juvenile Idiopathic Arthritis Cohort of the Childhood Arthritis and Rheumatology Research Alliance Registry: 2010–2013. J. Rheumatol. 2016, 43, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Packham, J.C.; Hall, M.A.; Pimm, T.J. Long-term follow-up of 246 adults with juvenile idiopathic arthritis: Predictive factors for mood and pain. Rheumatology 2002, 41, 1444–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellins, E.D.; Macaubas, C.; Grom, A.A. Pathogenesis of systemic juvenile idiopathic arthritis: Some answers, more questions. Nat. Rev. Rheumatol. 2011, 7, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Martini, A.; Ravelli, A.; Avcin, T.; Beresford, M.W.; Burgos-Vargas, R.; Cuttica, R.; Ilowite, N.T.; Khubchandani, R.; Laxer, R.M.; Lovell, D.J.; et al. Toward New Classification Criteria for Juvenile Idiopathic Arthritis: First Steps, Pediatric Rheumatology International Trials Organization International Consensus. J. Rheumatol. 2019, 46, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, A.; Davi, S.; Bracciolini, G.; Pistorio, A.; Consolaro, A.; van Dijkhuizen, E.H.P.; Lattanzi, B.; Filocamo, G.; Verazza, S.; Gerloni, V.; et al. Intra-articular corticosteroids versus intra-articular corticosteroids plus methotrexate in oligoarticular juvenile idiopathic arthritis: A multicentre, prospective, randomised, open-label trial. Lancet 2017, 389, 909–916. [Google Scholar] [CrossRef]
- Giannini, E.H.; Brewer, E.J.; Kuzmina, N.; Shaikov, A.; Maximov, A.; Vorontsov, I.; Fink, C.W.; Newman, A.J.; Cassidy, J.T.; Zemel, L.S. Methotrexate in resistant juvenile rheumatoid arthritis. Results of the U.S.A.-U.S.S.R. double-blind, placebo-controlled trial. The Pediatric Rheumatology Collaborative Study Group and The Cooperative Children’s Study Group. N. Engl. J. Med. 1992, 326, 1043–1049. [Google Scholar] [CrossRef]
- Ruperto, N.; Murray, K.J.; Gerloni, V.; Wulffraat, N.; de Oliveira, S.K.; Falcini, F.; Dolezalov, P.; Alessio, M.; Burgos-Vargas, R.; Corona, F.; et al. A randomized trial of parenteral methotrexate comparing an intermediate dose with a higher dose in children with juvenile idiopathic arthritis who failed to respond to standard doses of methotrexate. Arthritis Rheumatol. 2004, 50, 2191–2201. [Google Scholar] [CrossRef]
- Perisano, C.; Vitiello, R.; Sgambato, A.; Greco, T.; Cianni, L.; Ragonesi, G.; Malara, T.; Maccauro, G.; Martini, M. Evaluation of PD1 and PD-L1 expression in high-grade sarcomas of the limbs in the adults: Possible implications of immunotherapy. J. Biol. Regul. Homeost. Agents 2020, 34, 289–294. [Google Scholar]
- de Jager, W.; Hoppenreijs, E.P.; Wulffraat, N.M.; Wedderburn, L.R.; Kuis, W.; Prakken, B.J. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: A cross-sectional study. Ann. Rheum. Dis. 2007, 66, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, I.D.; Griffin, P.; Michel, J.J.; Yano, H.; Gaffen, S.L.; Mueller, R.G.; Dvergsten, J.A.; Piganelli, J.D.; Rosenkranz, M.E.; Kietz, D.A.; et al. T Cell Receptor-Independent, CD31/IL-17A-Driven Inflammatory Axis Shapes Synovitis in Juvenile Idiopathic Arthritis. Front. Immunol. 2018, 9, 1802. [Google Scholar] [CrossRef] [PubMed]
- Martini, A. It is time to rethink juvenile idiopathic arthritis classification and nomenclature. Ann. Rheum. Dis. 2012, 71, 1437–1439. [Google Scholar] [CrossRef] [PubMed]
- Nistala, K.; Moncrieffe, H.; Newton, K.R.; Varsani, H.; Hunter, P.; Wedderburn, L.R. Interleukin-17-producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheumatol. 2008, 58, 875–887. [Google Scholar] [CrossRef] [PubMed]
- de Kleer, I.M.; Wedderburn, L.R.; Taams, L.S.; Patel, A.; Varsani, H.; Klein, M.; de Jager, W.; Pugayung, G.; Giannoni, F.; Rijkers, G.; et al. CD4+CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J. Immunol. 2004, 172, 6435–6443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Misra, R.; Aggarwal, A. Interleukin 17 levels are increased in juvenile idiopathic arthritis synovial fluid and induce synovial fibroblasts to produce proinflammatory cytokines and matrix metalloproteinases. J. Rheumatol. 2008, 35, 515–519. [Google Scholar]
- Mahendra, A.; Misra, R.; Aggarwal, A. Th1 and Th17 Predominance in the Enthesitis-related Arthritis Form of Juvenile Idiopathic Arthritis. J. Rheumatol. 2009, 36, 1730–1736. [Google Scholar] [CrossRef]
- Olivito, B.; Simonini, G.; Ciullini, S.; Moriondo, M.; Betti, L.; Gambineri, E.; Cantarini, L.; De Martino, M.; Azzari, C.; Cimaz, R. Th17 transcription factor RORC2 is inversely correlated with FOXP3 expression in the joints of children with juvenile idiopathic arthritis. J. Rheumatol. 2009, 36, 2017–2024. [Google Scholar] [CrossRef] [Green Version]
- Cosmi, L.; Cimaz, R.; Maggi, L.; Santarlasci, V.; Capone, M.; Borriello, F.; Frosali, F.; Querci, V.; Simonini, G.; Barra, G.; et al. Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheumatol. 2011, 63, 2504–2515. [Google Scholar] [CrossRef]
- Miossec, P. Interleukin-17 and Th17 cells: From adult to juvenile arthritis—Now it is serious! Arthritis Rheumatol. 2011, 63, 2168–2171. [Google Scholar] [CrossRef]
- Wu, S.A.; Yeh, K.W.; Lee, W.I.; Yao, T.C.; Huang, J.L. Persistent improper upregulation of Th17 and TReg cells in patients with juvenile idiopathic arthritis. J. Microbiol. Immunol. Infect. 2016, 49, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosser, E.C.; Lom, H.; Bending, D.; Duurland, C.L.; Bajaj-Elliott, M.; Wedderburn, L.R. Innate Lymphoid Cells and T Cells Contribute to the Interleukin-17A Signature Detected in the Synovial Fluid of Patients with Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2019, 71, 460–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omoyinmi, E.; Hamaoui, R.; Pesenacker, A.; Nistala, K.; Moncrieffe, H.; Ursu, S.; Wedderburn, L.R.; Woo, P. Th1 and Th17 cell subpopulations are enriched in the peripheral blood of patients with systemic juvenile idiopathic arthritis. Rheumatology 2012, 51, 1881–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessel, C.; Lippitz, K.; Weinhage, T.; Hinze, C.; Wittkowski, H.; Holzinger, D.; Fall, N.; Grom, A.A.; Gruen, N.; Foell, D. Proinflammatory Cytokine Environments Can Drive Interleukin-17 Overexpression by gamma/delta T Cells in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2017, 69, 1480–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, L.A.; Hoyt, K.J.; Lee, P.Y.; Rao, D.A.; Jonsson, A.H.; Nguyen, J.P.; Rutherford, K.; Jule, A.M.; Charbonnier, L.M.; Case, S.; et al. Th17 reprogramming of T cells in systemic juvenile idiopathic arthritis. JCI Insight 2020, 5, e132508. [Google Scholar] [CrossRef] [Green Version]
- Cingoz, O. Ustekinumab. MAbs 2009, 1, 216–221. [Google Scholar] [CrossRef]
- Dobbin-Sears, I.; Roberts, J.; O’Rielly, D.D.; Rahman, P. Ustekinumab in psoriatic arthritis and related phenotypes. Ther. Adv. Chronic Dis. 2018, 9, 191–198. [Google Scholar] [CrossRef]
- Yiu, Z.Z.; Warren, R.B. Ustekinumab for the treatment of psoriasis: An evidence update. Semin. Cutan. Med. Surg. 2018, 37, 143–147. [Google Scholar] [CrossRef]
- Gisbert, J.P.; Chaparro, M. Ustekinumab to treat Crohn’s disease. Gastroenterol. Hepatol. 2017, 40, 688–698. [Google Scholar] [CrossRef]
- Mannion, M.L.; McAllister, L.; Cron, R.Q.; Stoll, M.L. Ustekinumab as a Therapeutic Option for Children with Refractory Enthesitis-Related Arthritis. J. Clin. Rheumatol. 2016, 22, 282–284. [Google Scholar] [CrossRef]
- Sanford, M.; McKeage, K. Secukinumab: First global approval. Drugs 2015, 75, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, A.; Blanco, R.; Dokoupilova, E.; Hall, S.; Kameda, H.; Kivitz, A.J.; Poddubnyy, D.; van de Sande, M.; Wiksten, A.S.; Porter, B.O.; et al. Improvement of Signs and Symptoms of Nonradiographic Axial Spondyloarthritis in Patients Treated With Secukinumab: Primary Results of a Randomized, Placebo-Controlled Phase III Study. Arthritis Rheumatol. 2021, 73, 110–120. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Mease, P.J.; Kirkham, B.; Kavanaugh, A.; Ritchlin, C.T.; Rahman, P.; van der Heijde, D.; Landewe, R.; Conaghan, P.G.; Gottlieb, A.B.; et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 386, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Brunello, F.; Tirelli, F.; Pegoraro, L.; Dell’Apa, F.; Alfisi, A.; Calzamatta, G.; Folisi, C.; Zulian, F. New Insights on Juvenile Psoriatic Arthritis. Front. Pediatr. 2022, 10, 884727. [Google Scholar] [CrossRef] [PubMed]
- Weiss, P.F.; Klink, A.J.; Behrens, E.M.; Sherry, D.D.; Finkel, T.H.; Feudtner, C.; Keren, R. Enthesitis in an inception cohort of enthesitis-related arthritis. Arthritis Care Res. (Hoboken) 2011, 63, 1307–1312. [Google Scholar] [CrossRef]
- Weiss, P.F.; Fuhlbrigge, R.C.; von Scheven, E.; Lovell, D.J.; Colbert, R.A.; Brunner, H.I.; Council, P.A. Children With Enthesitis-Related Arthritis and Possible Benefits From Treatments for Adults With Spondyloarthritis. Arthritis Care Res. (Hoboken) 2022, 74, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Ringold, S.; Angeles-Han, S.T.; Beukelman, T.; Lovell, D.; Cuello, C.A.; Becker, M.L.; Colbert, R.A.; Feldman, B.M.; Ferguson, P.J.; Gewanter, H.; et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Treatment of Juvenile Idiopathic Arthritis: Therapeutic Approaches for Non-Systemic Polyarthritis, Sacroiliitis, and Enthesitis. Arthritis Rheumatol. 2019, 71, 846–863. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.; Henrey, A.; Loughin, T.; Berard, R.A.; Shiff, N.J.; Jurencak, R.; Benseler, S.M.; Tucker, L.B. Predicting Which Children with Juvenile Idiopathic Arthritis Will Have a Severe Disease Course: Results from the ReACCh-Out Cohort. J. Rheumatol. 2017, 44, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, A.; Consolaro, A.; Horneff, G.; Laxer, R.M.; Lovell, D.J.; Wulffraat, N.M.; Akikusa, J.D.; Al-Mayouf, S.M.; Anton, J.; Avcin, T.; et al. Treating juvenile idiopathic arthritis to target: Recommendations of an international task force. Ann. Rheum. Dis. 2018, 77, 819–828. [Google Scholar] [CrossRef]
- Burgos-Vargas, R.; Tse, S.M.; Horneff, G.; Pangan, A.L.; Kalabic, J.; Goss, S.; Unnebrink, K.; Anderson, J.K. A Randomized, Double-Blind, Placebo-Controlled Multicenter Study of Adalimumab in Pediatric Patients With Enthesitis-Related Arthritis. Arthritis Care Res. (Hoboken) 2015, 67, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Brunner, H.I.; Schanberg, L.E.; Kimura, Y.; Dennos, A.; Co, D.O.; Colbert, R.A.; Fuhlbrigge, R.C.; Goldmuntz, E.; Kingsbury, D.J.; Patty-Resk, C.; et al. New Medications Are Needed for Children With Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2020, 72, 1945–1951. [Google Scholar] [CrossRef] [PubMed]
- Weiss, P.F.; Beukelman, T.; Schanberg, L.E.; Kimura, Y.; Colbert, R.A. Enthesitis-related arthritis is associated with higher pain intensity and poorer health status in comparison with other categories of juvenile idiopathic arthritis: The Childhood Arthritis and Rheumatology Research Alliance Registry. J. Rheumatol. 2012, 39, 2341–2351. [Google Scholar] [CrossRef] [Green Version]
- Baer, J.; Klotsche, J.; Foeldvari, I. Secukinumab in the treatment for patients with juvenile enthesitis related arthritis non-responsive to anti-TNF treatment according the Juvenile Spondyloarthritis Disease Activity Index. Clin. Exp. Rheumatol. 2022, 40, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.I.; Foeldvari, I.; Alexeeva, E.; Ayaz, N.A.; Calvo Penades, I.; Kasapcopur, O.; Chasnyk, V.G.; Hufnagel, M.; Zuber, Z.; Schulert, G.; et al. Secukinumab in enthesitis-related arthritis and juvenile psoriatic arthritis: A randomised, double-blind, placebo-controlled, treatment withdrawal, phase 3 trial. Ann. Rheum. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
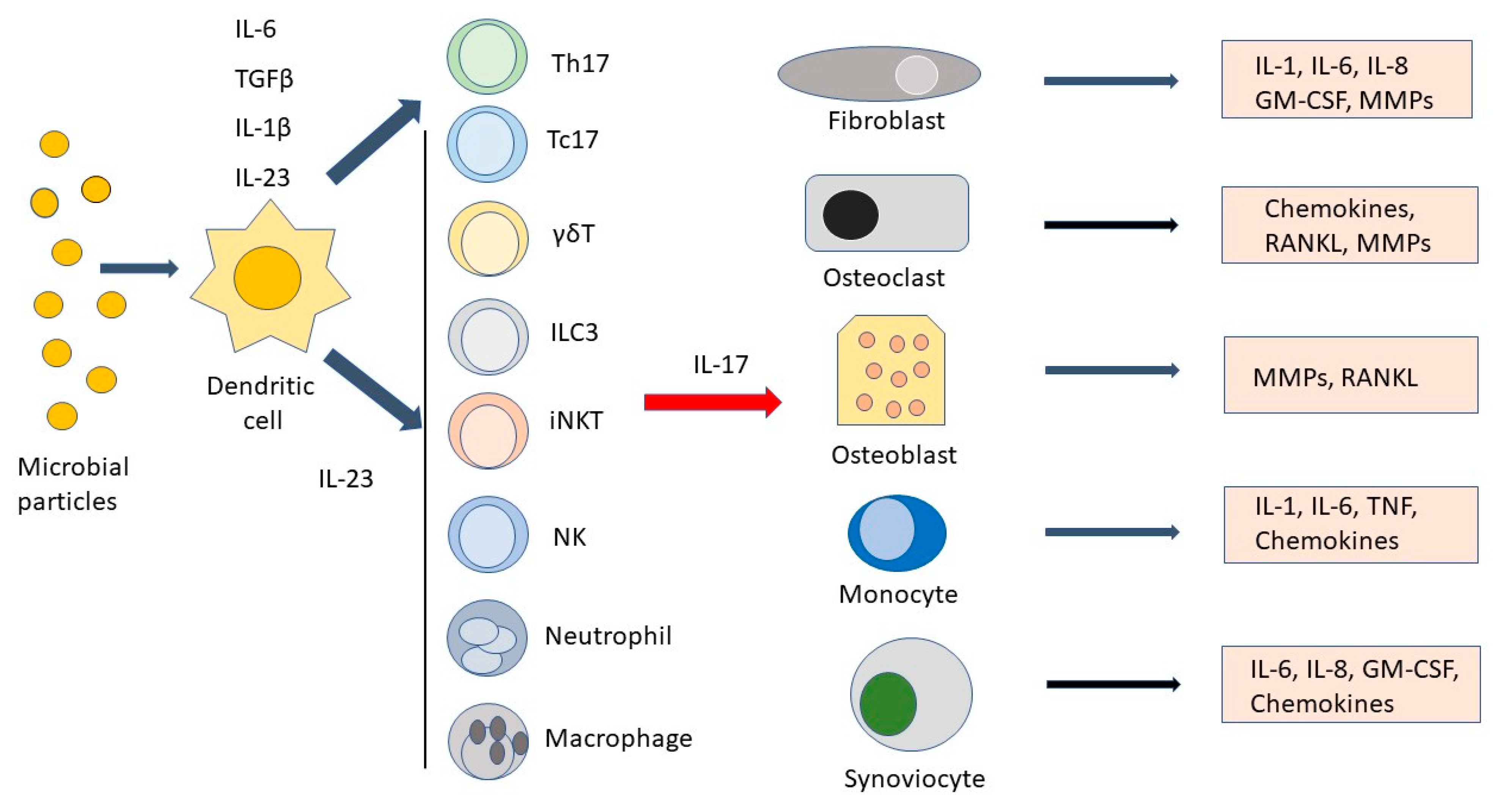

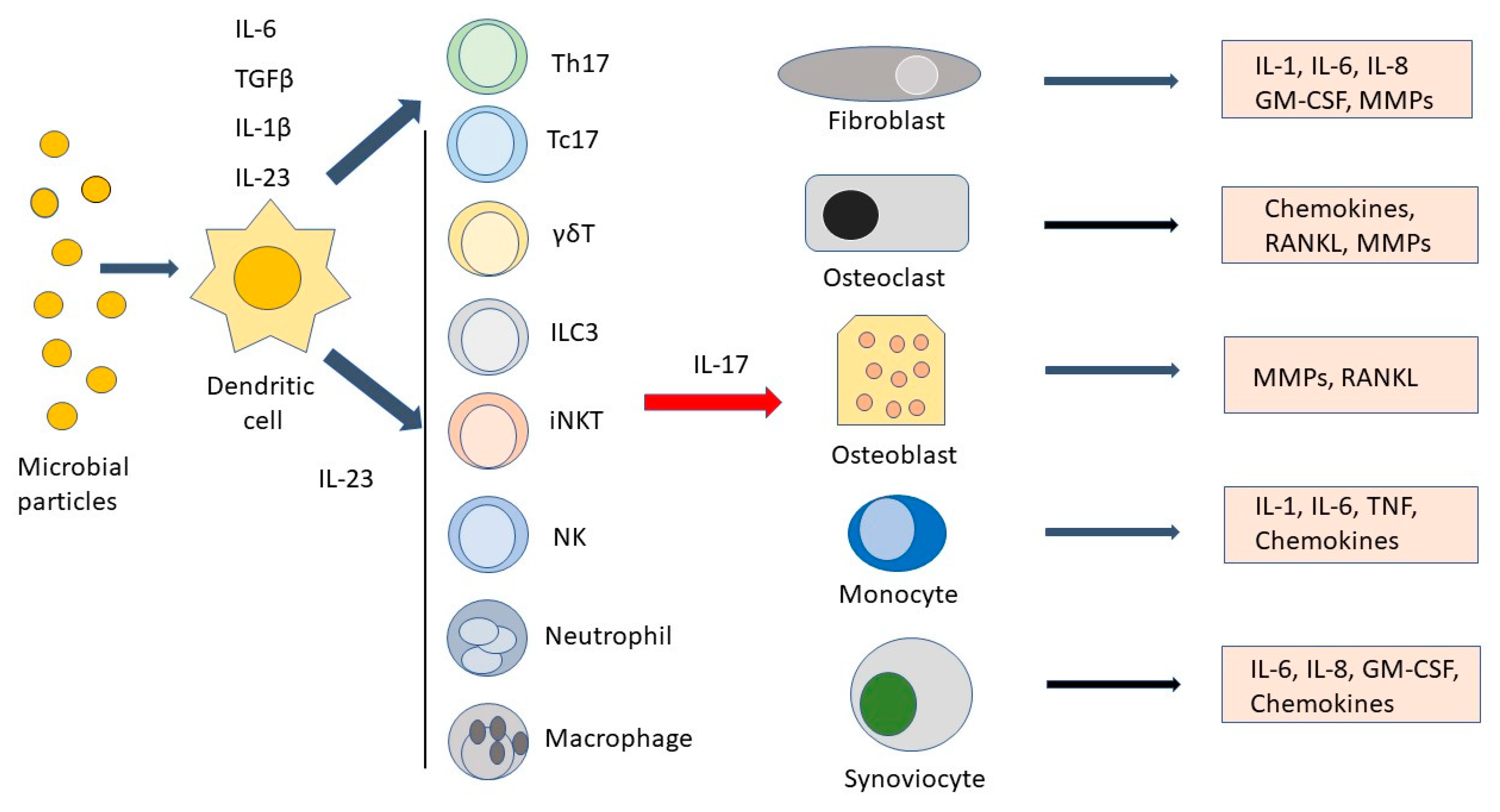{kind=link}
| Cytokine | Receptor | Activated Transcription Factor |
|---|---|---|
| IL-17 A/A | IL-17RA/IL-17RC IL-17RC/IL-17RC IL-17RA/IL-17RD | C/EPBβ, AP-1, NF-κB (NF-κB) (NF-κB) |
| IL-17 F/F | IL-17RA/IL-17RC IL-17RC/IL-17RC | NF-κB (NF-κB) |
| IL-17A/F | IL-17RA/IL-17RC IL-17RC/IL-17RC | NF-κB (NF-κB) |
| IL-17B/B | IL-17RA/IL-17RB | C/EPBβ, AP-1, NF-κB |
| IL-17E/E (IL-25) | IL-17RA/IL-17RB | C/EPBβ, AP-1, NF-κB |
| IL-17C/C | IL-17RA/IL-17RE | NF-κBζ |
| IL-17D | CD93 | Intra-cellular CD93 domain |
| Biologic | Mechanism of Action | JIA Subtype | Approving Agency |
|---|---|---|---|
| Etanercept | Binding to TNFα | pJIA, JPsA, ERA | FDA/EMA |
| Adalimumab | Binding to TNFα | pJIA, ERA | FDA/EMA |
| Golimumab | Binding to TNFα | pJIA | FDA/EMA |
| Tocilizumab | Binding to IL6R | sJIA, pJIA | FDA/EMA |
| Anakinra | Binding to IL-1Ra | sJIA | EMA |
| Canakinumab | Binding to IL-1β | sJIA | FDA/EMA |
| Abatacept | Binding to CD80/CD86 | pJIA | FDA/EMA |
| Ustekinumab | Binding to IL-23/IL-12 | JPsA | FDA |
| Secukinumab | Binding to IL-17A | JPsA, ERA | FDA/EMA |
| Biologic | Mechanism of Action | Approved Indication |
|---|---|---|
| Ustekinumab | Binding to IL-23/IL-12 | PsO, PsA, CD, UC |
| Guselkumab | Binding to IL-23 | PsO, PsA |
| Tildrakizumab | Binding to IL-23 | PsO |
| Risankizumab | Binding to IL-23 | PsO, PsA, CD |
| Secukinumab | Binding to IL-17A | PsO, PsA, AS, nr-axSpA |
| Ixekizumab | Binding to IL-17A | PsO, PsA |
| Brodalumab | Binding to IL-17RA | PsO |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paroli, M.; Spadea, L.; Caccavale, R.; Spadea, L.; Paroli, M.P.; Nante, N. The Role of Interleukin-17 in Juvenile Idiopathic Arthritis: From Pathogenesis to Treatment. Medicina 2022, 58, 1552. https://doi.org/10.3390/medicina58111552
Paroli M, Spadea L, Caccavale R, Spadea L, Paroli MP, Nante N. The Role of Interleukin-17 in Juvenile Idiopathic Arthritis: From Pathogenesis to Treatment. Medicina. 2022; 58(11):1552. https://doi.org/10.3390/medicina58111552
Chicago/Turabian StyleParoli, Marino, Luca Spadea, Rosalba Caccavale, Leopoldo Spadea, Maria Pia Paroli, and Nicola Nante. 2022. "The Role of Interleukin-17 in Juvenile Idiopathic Arthritis: From Pathogenesis to Treatment" Medicina 58, no. 11: 1552. https://doi.org/10.3390/medicina58111552
APA StyleParoli, M., Spadea, L., Caccavale, R., Spadea, L., Paroli, M. P., & Nante, N. (2022). The Role of Interleukin-17 in Juvenile Idiopathic Arthritis: From Pathogenesis to Treatment. Medicina, 58(11), 1552. https://doi.org/10.3390/medicina58111552