Abstract
Computational calculations have been used successfully to explain the reactivity of the >C=P- functionality in pyrido-annelated 1,3-azaphospholes. Theoretical investigation at the Density Functional Theory (DFT) level shows that the lone pair of the bridgehead nitrogen atoms is involved in extended conjugation, due to which electron density increases considerably in the five-membered azaphosphole ring. The electron density in the azaphosphole is further enhanced by the presence of an ester group at the 3-position making the >C=P- functionality electron-rich. Thus, 1,3-azaphospholo[5,1-a]pyridine, i.e., 2-phosphaindolizine having ester group at the 3-position only does not undergo Diels-Alder (DA) reaction with an electron rich diene, such as 2,3-dimethyl-1,3-butadiene (DMB). However, an ester group at 1-position acts as electron-sink, due to which transfer of the electron density to the >C=P- moiety is checked and DA reaction occurs across the >C=P- functionality. The coordination of the Lewis acid to the carbonyl group at the 3-position raises the activation barrier, while it is lowered remarkably when it is coordinated to the P atom. Furthermore, the attack of 1,3-butadiene on the Si face of the P-coordinated (o-menthoxy)aluminum dichloride complex is a lower activation energy path. Fukui functions could be used to explain relative reactivities of indolizine and 2-phosphaindolizine towards electrophilic substitution reactions.
1. Introduction
Almost a century ago, Paul Dirac [1] could foresee the possibility of applying quantum mechanical concepts to solving the experimental problems in chemistry when he remarked, “The underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known, and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble. It therefore becomes desirable that approximate practical methods of applying quantum mechanics should be developed, which can lead to an explanation of the main features of complex atomic systems without too much computation”. Dirac’s dream inched towards fruition when Kohn, Hohenberg, and Sham [2,3] developed the density functional theory (DFT). It was followed by dramatic progress when many DFT codes were made available commercially, which enabled even less experienced persons with modest computational facilities to do calculations with much accuracy and look into the electronic structures and many other properties of the substances of interest. During the last two decades, the availability of a variety of new and more accurate DFT functionals incorporating specific features has brought DFT computations to the stage where they are used advantageously as a tool not only to explain the experimental results but also to plan and modify experimental conditions for achieving the desired objectives [4,5,6,7,8,9]. In recent years, there has been a perceptible increase in the number of research papers in chemistry reporting both experimental and theoretical results together. Fristrup and co-workers in a recent article emphasized the synergy between experimental and theoretical methods in the exploration of homogeneous transitional metal catalysis [10]. In another interesting review article titled, “Towards the computational design of solid catalysts”, Norskov et al. [11] commented, “Theoretical methods can be used to describe surface chemical reactions in detail and to understand variations in catalytic activity from one catalyst to another.” In spite of many challenges in the field, they added “Computational approaches for the discovery and development of catalysts hold great promise for the future.” Similar views have been expressed in other reports [12,13,14].
In this perspective, we shall present mainly our own results highlighting the application of DFT calculations to plan and accomplish successfully some reactions of annelated 1,3-azaphospholes. It will also be illustrated how computational calculations helped to resolve an experimental log jam and paved the way for the use of catalysts, leading to a successful reaction and asymmetric synthesis.
2. Synthesis of Annelated 1,3-Azaphospholes
Before coming to the reactions, it will be appropriate to describe briefly two general methods of synthesis of annelated 1,3-azaphospholes that we developed.
2.1. [4+1] Cyclocondensation Method
The reaction of 2-ethyl-1-phenacylpyridinium bromide (1) with PCl3 in the presence of Et3N at room temperature (r.t.) afforded 3-benzoyl-1-methyl-1,3-azaphospholo[5,1-a]pyridine (2) in very good yield (Scheme 1). The general skeleton was named as 2-phosphaindolizine perceiving it to result from a CH/P exchange at the 2-position of indolizine nucleus [15].
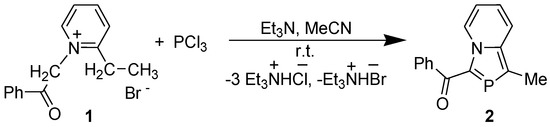
Scheme 1.
Synthesis of 2-phosphaindolizine derivative.
Later, this synthetic strategy made accessible not only variously substituted 2-phosphaindolizines [15,16,17], but also 1-aza-2-phosphaindolizines [18], 3-aza-2-phosphaindolizines [19], and 1,3-diaza-2-phosphaindolizines [20]. Besides, the method could be extended to the synthesis of 1,3-azaphospholes annelated to pyrazine [15], thiazole [21], benzothiazole [21], and oxazole [22] skeletons also (Scheme 2). These reactions have been compiled in a mini-review [23].
Scheme 2.
Synthesis of annelated 1,3-azaphospholes.
2.2. Synthesis Involving Tandem Pyridinium Ylide generation, Disproportionation, and 1,5-Electrocyclization
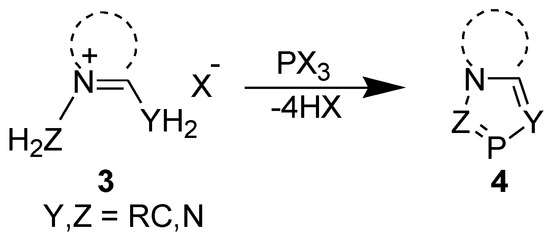
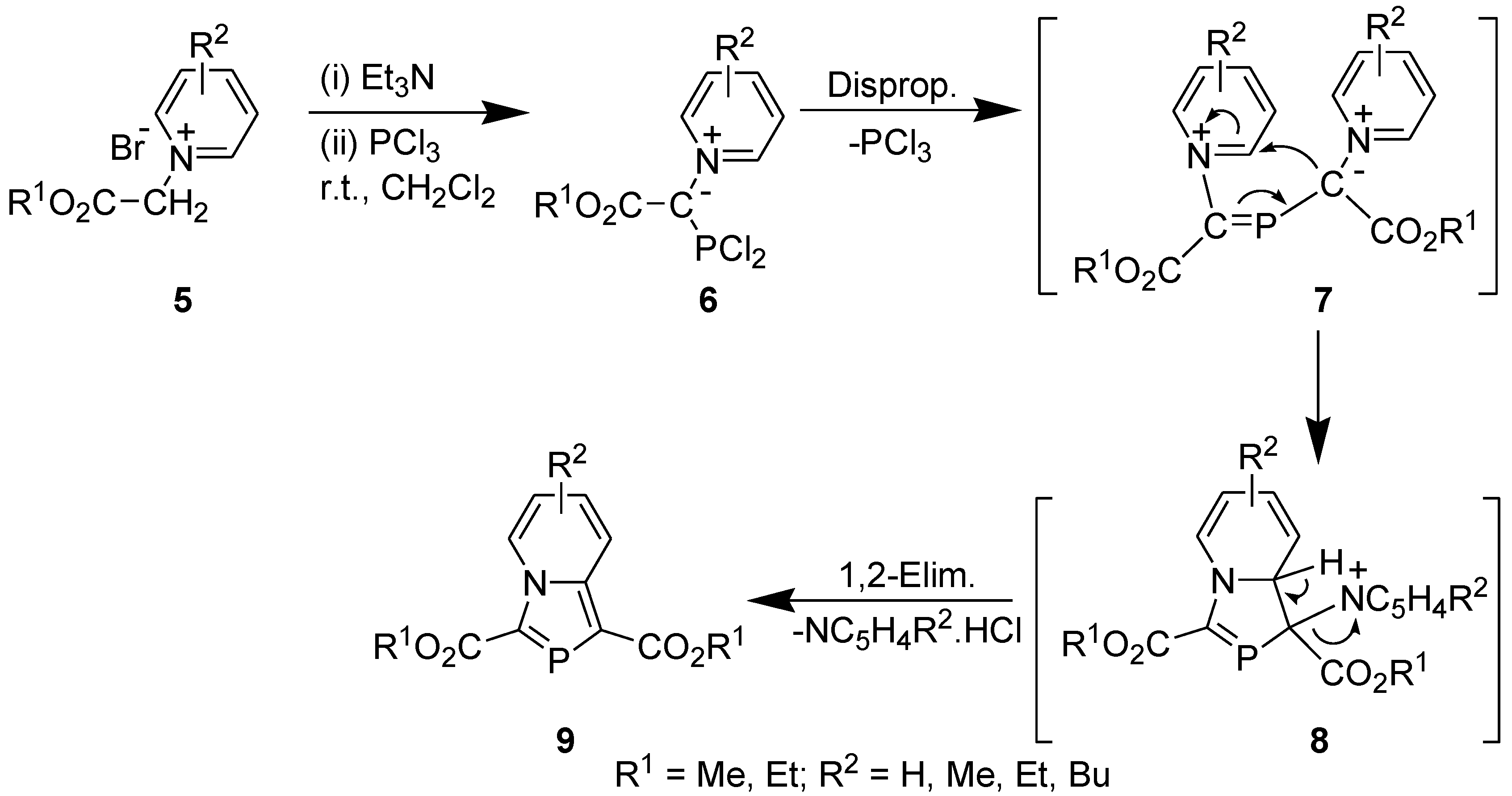
We obtained 1,3-bis(alkoxycarbonyl)-1,3-azaphospholo[5,1-a]pyridines (9) serendipitously on reacting (alkoxycarbonyl)methylpyridinium bromide with Et3N and PCl3. The reaction involved tandem pyridinium alkoxycarbonyl-dichlorophosphinomethylide (6) generation, disproportionation, 1,5-electrocyclization, and 1,2-elimination to furnish the final product (Scheme 3) [24,25].
Scheme 3.
Synthesis of 1,3-bis(alkoxycarbonyl)-1,3-azaphospholo[5,1-a]pyridines.
Subsequently, isoquinoline [26], and phenanthridine [27] analogues of 9 could also be prepared.
3. Reactions Response
3.1. Diels-Alder Reaction
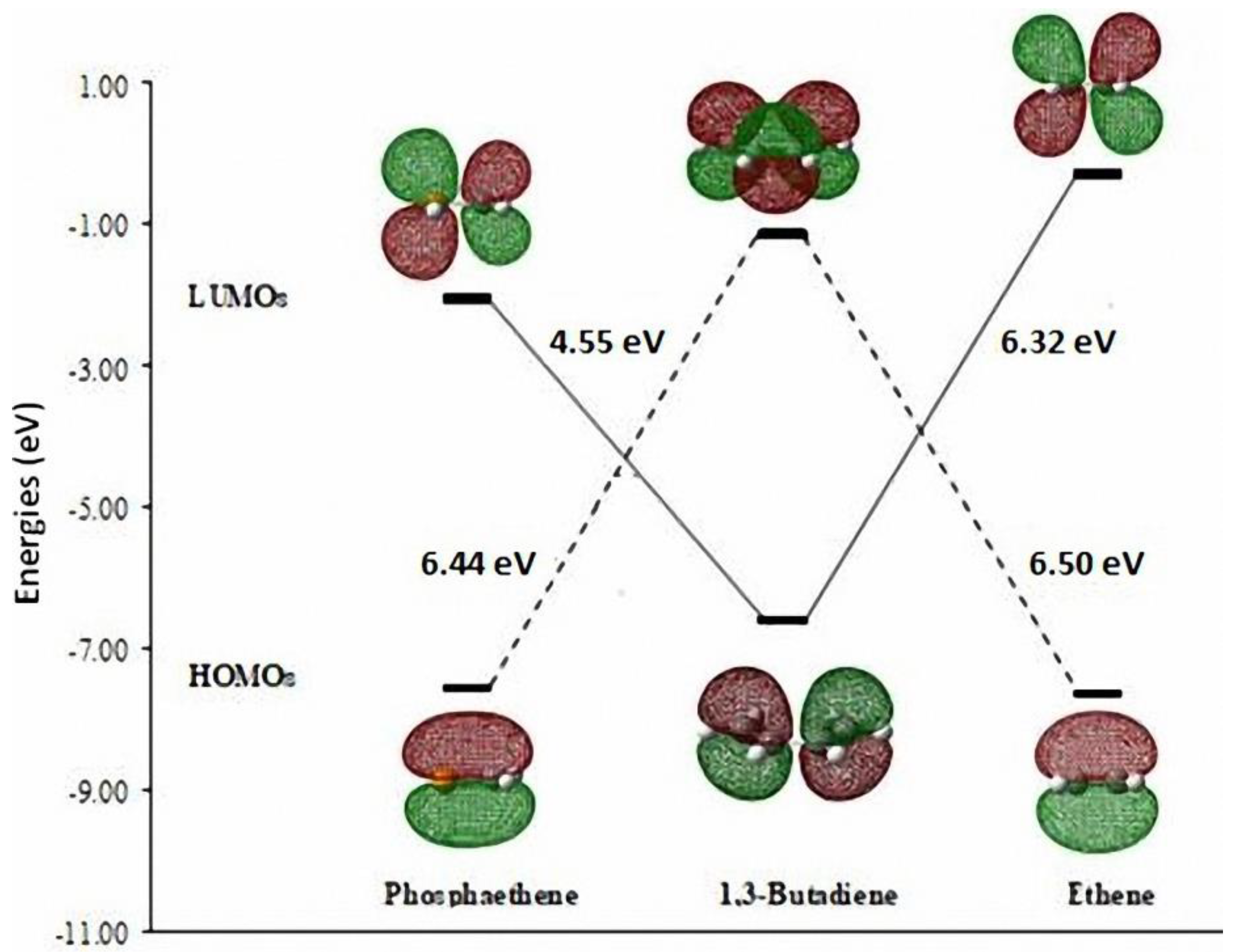
There have been earlier reports about the Diels-Alder (DA) reaction across the >C=P- functionality as dienophile [28,29]. We investigated theoretically the model DA reactions of ethene and phosphaethene with 1,3-butadiene at the DFT(B3LYP/6-31+G**) level [30]. The Frontier Molecular Orbitals (FMO) of the reactants are shown in Figure 1.
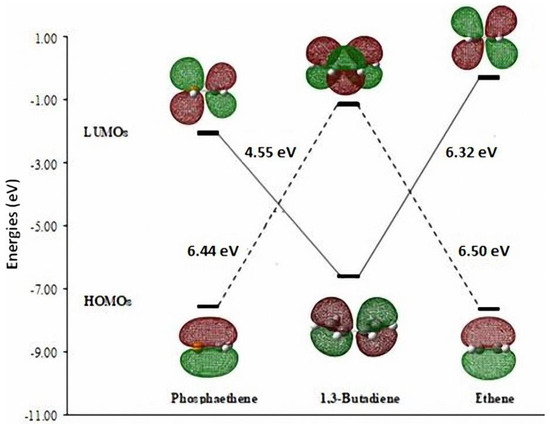
Figure 1.
The HOMO-LUMO energy differences between FMOs of phosphaethene, 1,3-butadiene, and ethene calculated at the B3LYP/6-31+G** level.
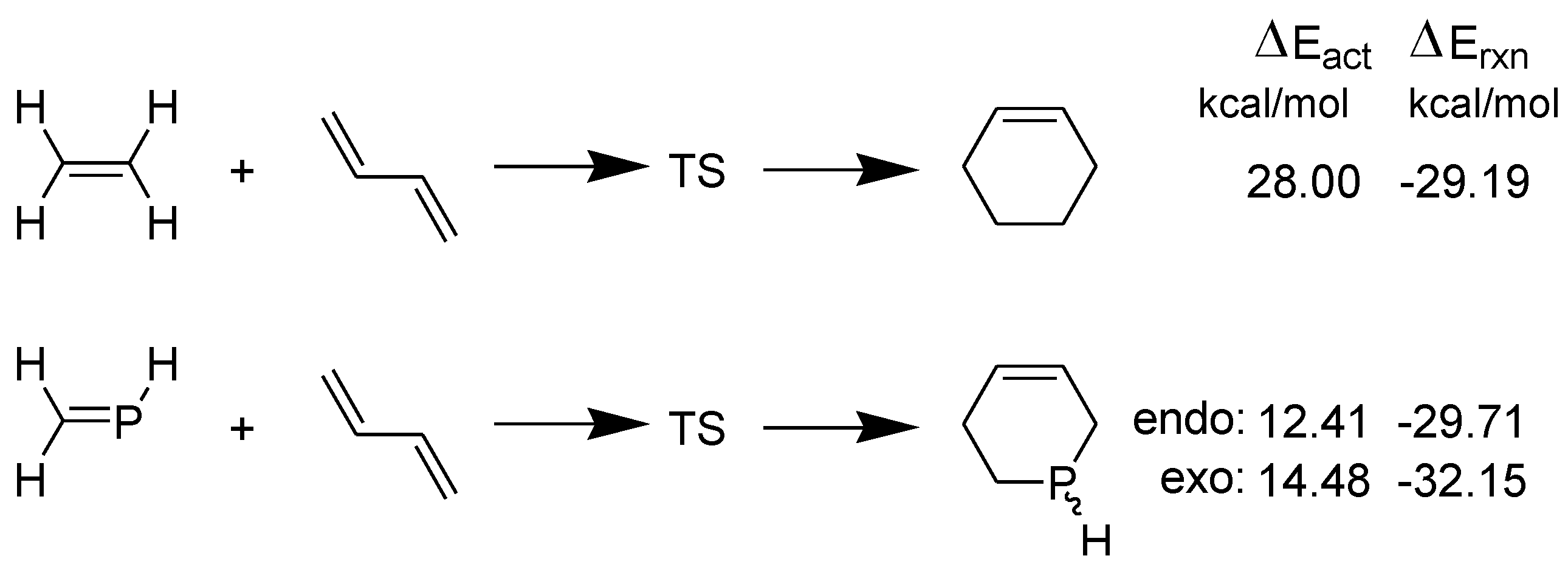
It may be noted that the HOMOdiene–LUMOphosphaethene gap (4.55 eV) is much lower than the gap HOMOdiene–LUMOethene (6.32 eV). Furthermore, the activation energy barriers (ΔEact.) for the two DA reactions are shown in Scheme 4 [30].
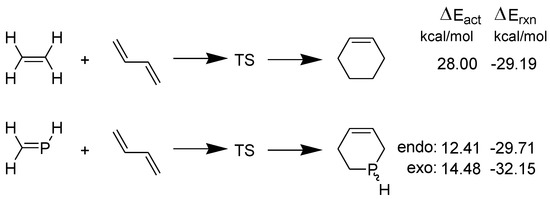
Scheme 4.
Model DA reactions of ethene and phosphaethene with 1,3-butadiene computed at B3LYP/6-31+G** in the gas phase.
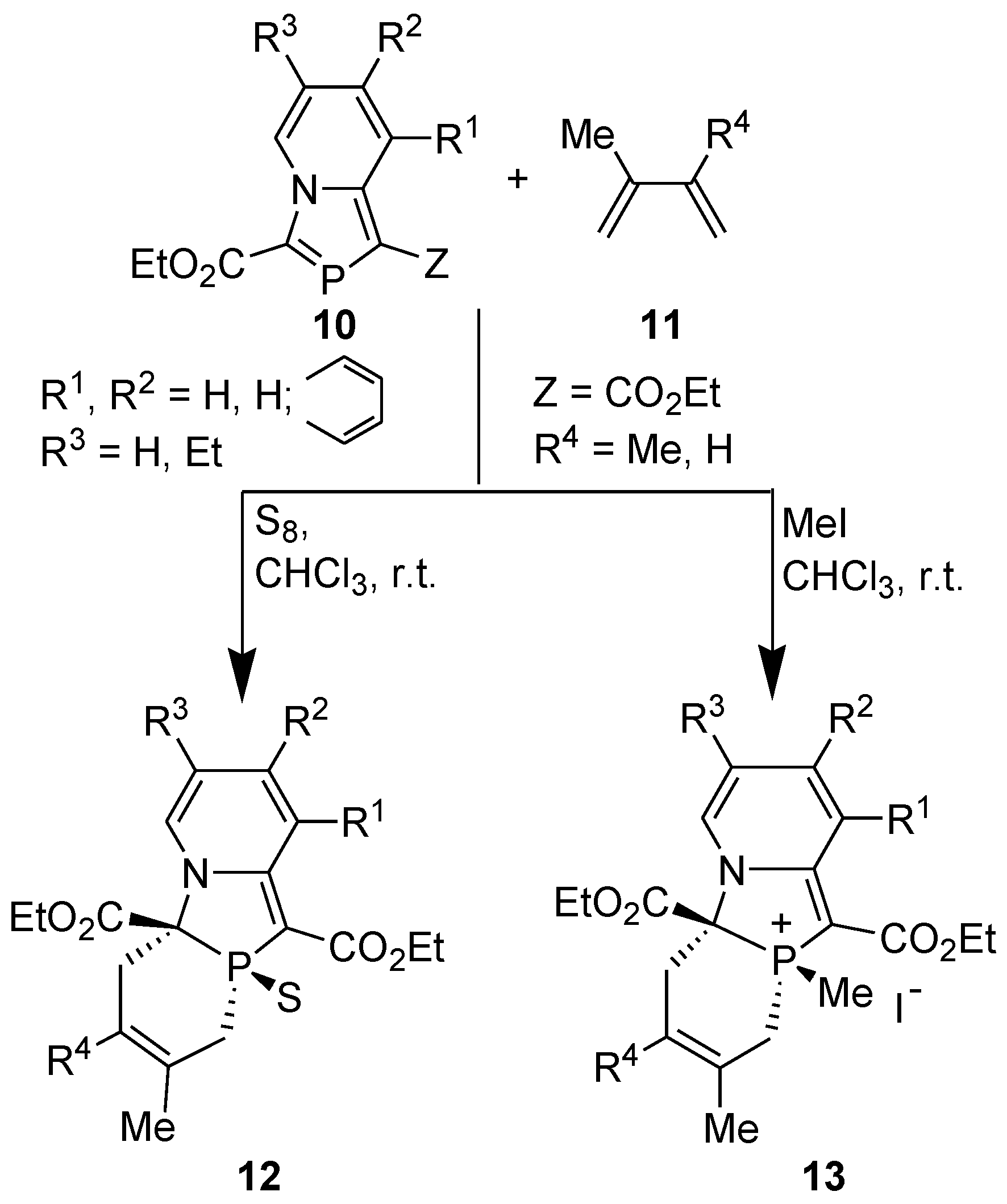
Encouraged by these theoretical results, we attempted DA reactions of 1,3-azaphospholo[5,1-a]pyridines, i.e., 2-phosphaindolizines prepared by two methods. However, to our great surprise, the compounds 3-ethoxycarbonyl-1-methyl-1,3-azaphospholo[5,1-a]pyridine 10a (10, Z=Me, R1=R2=R3=H) obtained through [4+1] cyclocondensation and 1,3-bis(ethoxylcarbonyl)-1,3-azaphospholo[5,1-a]pyridine 10b (10, Z=CO2Et, R1=R2=R3=H) showed remarkable difference in their reactivity with 2,3-dimethylbuta-1,3-diene (DMB). It was found that 3-(ethoxycarbonyl)-1-methyl-1,3-azaphospholo[5,1-a]pyridine (10a) did not undergo DA reaction with DMB, even upon boiling in toluene in the presence of sulfur (used to oxidize phosphorus atom of the initially formed cycloadduct to push the reaction in the forward direction, see later) [31]. But, 1,3-bis(ethoxylcarbonyl)-1,3-azaphospholo[5,1-a]pyridine (10b) underwent DA reaction with DMB at room temperature (r.t.). However, the reaction at room temperature (ca 25 °C) was very slow and could be completed (δ 31P NMR = 14.1 ppm) after heating at reflux temperature in chloroform for four days. On carrying out the reaction in the presence of sulfur or methyl iodide, it was complete at r.t. in 4 days [31]. Thus, the DA reactions of 1,3-bis(ethoxycarbonyl)-1,3-azaphospholo[5,1-a]pyridines and also of an isoquinoline analogue with DMB and isoprene could be accomplished successfully (Scheme 5) [31,32].
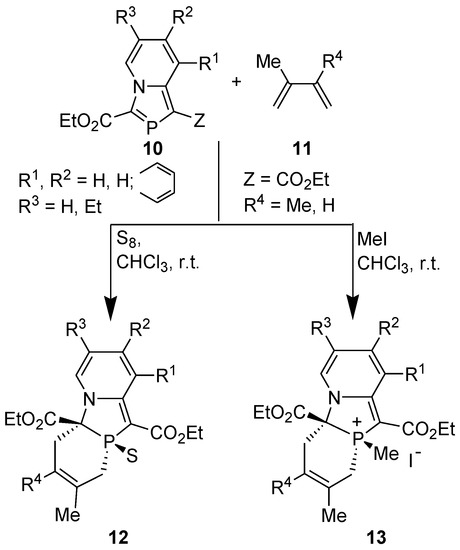
Scheme 5.
DA reactions of 1,3-bis-(ethoxycarbonyl)-1,3-azaphospholo[5,1-a]pyridines with 2,3-dimethyl-1,3-butadiene (DMB ) and isoprene.
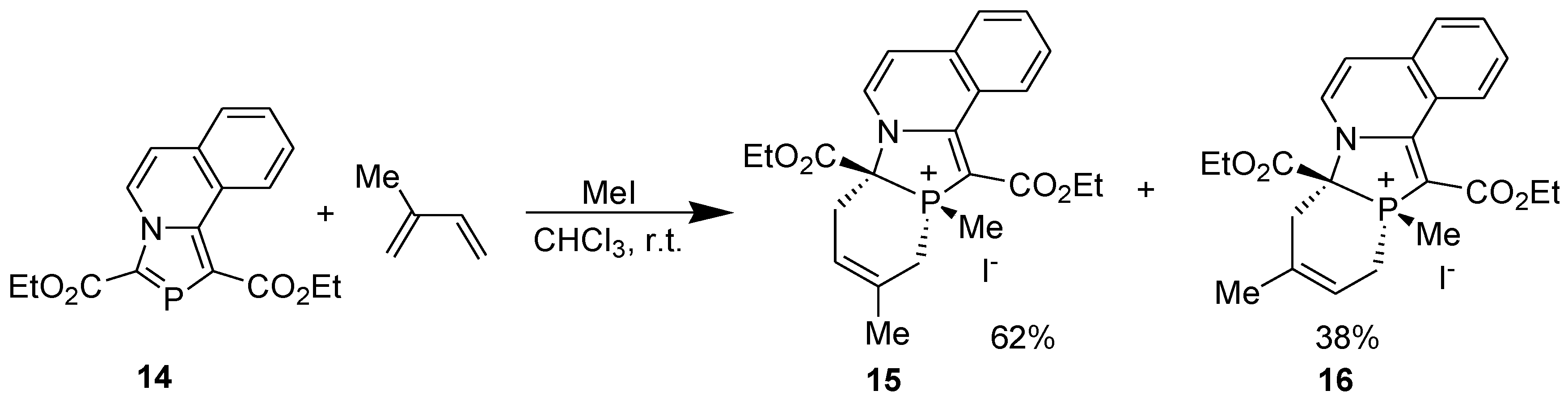
All the reactions occurred with complete stereo- and regioselectivity, except the reaction of 1,3-bis(ethoxycarbonyl)-1,3-azaphospholo[5,1-a]isoquinoline (14) with isoprene in the presence of methyl iodide which afforded two regioisomers, the major (62%) being 15 (Scheme 6) [32].
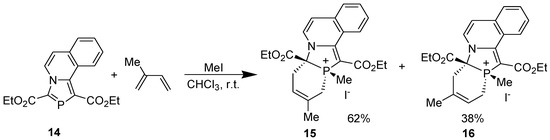
Scheme 6.
Regioselectivity in the DA reaction of 1,3-bis(ethoxycarbonyl)-1,3-azaphospholo[5,1-a]isoquinoline.
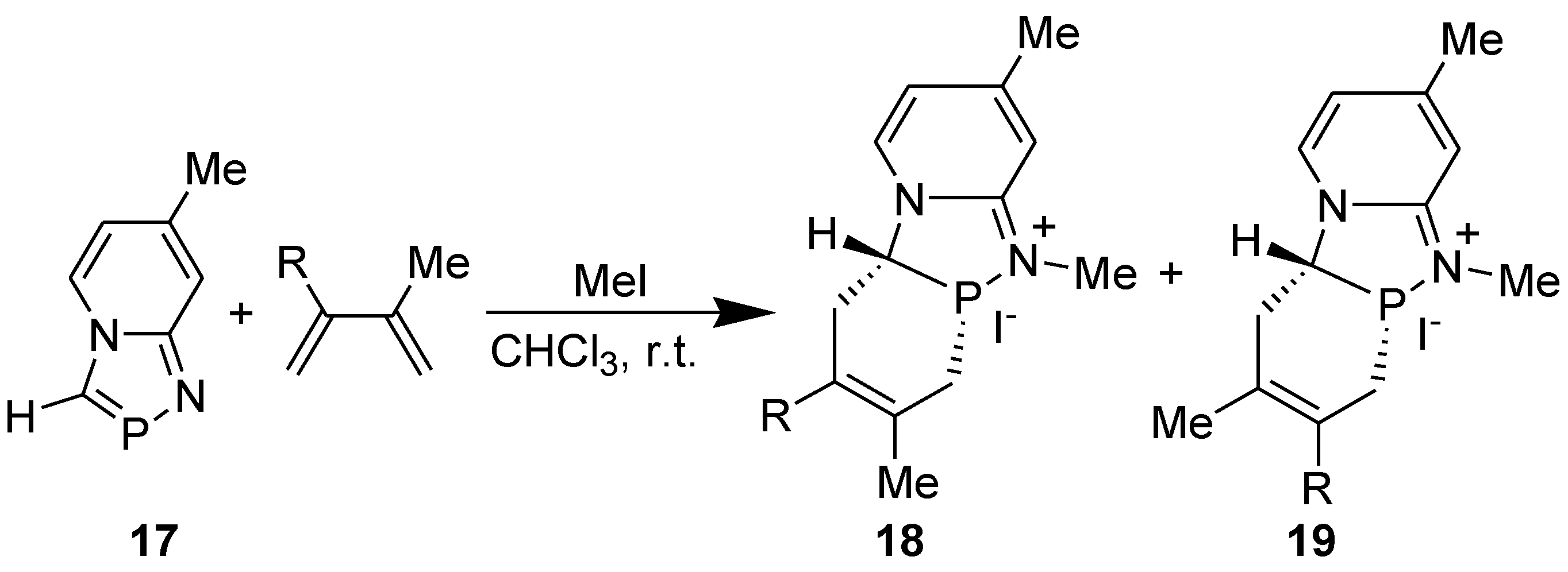
The structure of the cycloadduct 12 (R1R2= CH=CH-CH=CH-; R3=R4=H) was confirmed by single crystal X-ray diffraction studies [32]. That the [2+4]cycloaddition of annelated 1,3-azaphosphole with DMB precedes the oxidation of the phosphorus atom of the initially formed cycloadduct by sulfur or methyl iodide could be proved unambiguously by carrying out the DA reaction of 1,4,2-diazaphospholo[4-a]pyridine derivative 17 in the presence of methyl iodide, when N-methylated product 18 (R=Me) was formed (Scheme 7) [33]. It has been reported that [1,4,2]diazaphospholo[4-a]pyridines did not react with methyl iodide [34].
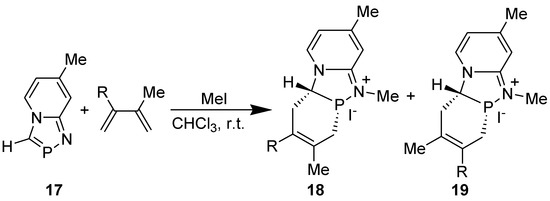
Scheme 7.
DA reaction of 7-methyl-1,4,2-diazaphospholo[4,5-a]pyridine in the presence of methyl iodide.
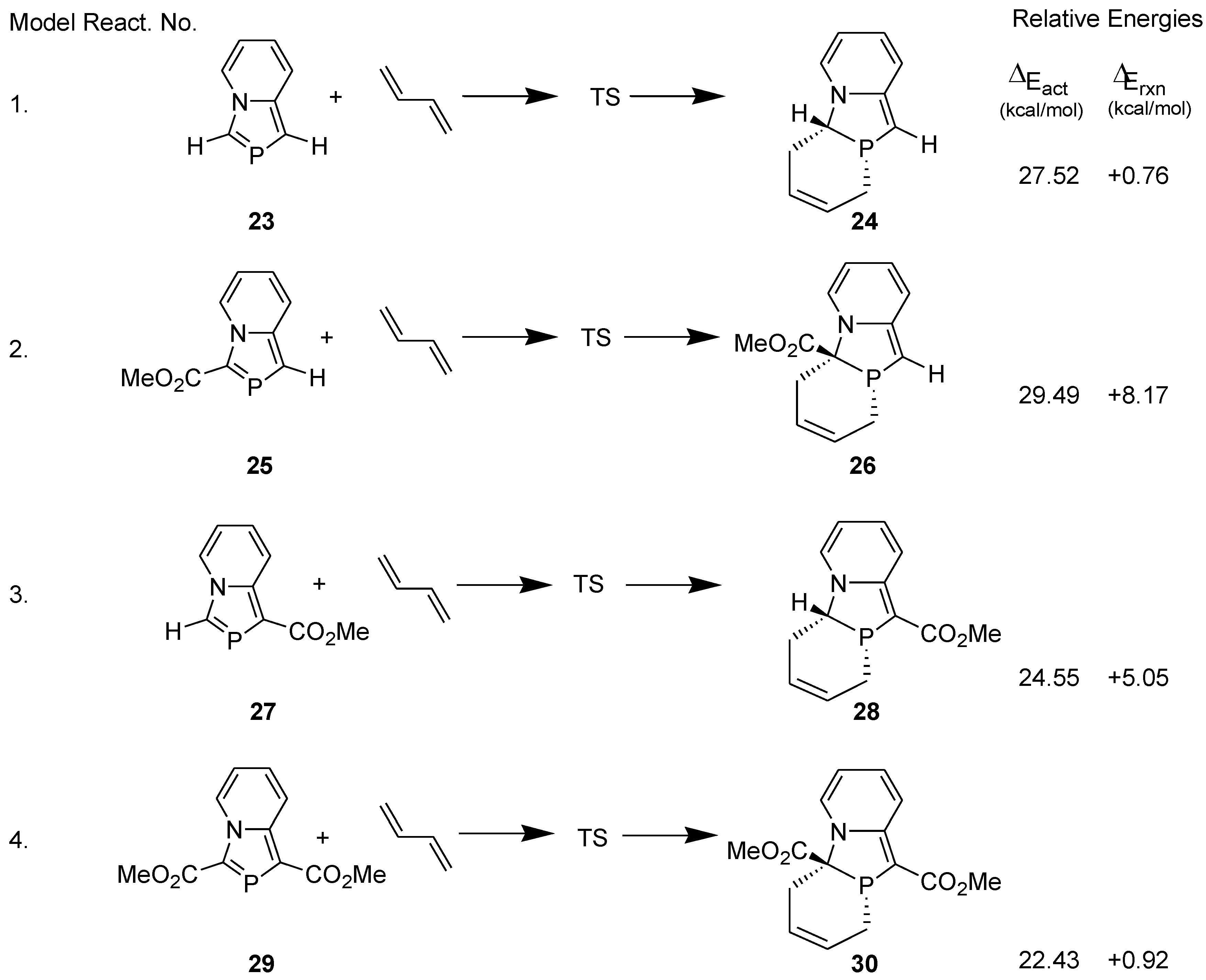
The regioselectivity observed in the reaction of 1,3-bis(ethoxycarbonyl)- 1,3-azaphospholo[5,1-a]pyridine with isoprene was investigated theoretically at the DFT (B3LYP/6-311++G**//B3LYP/6-31G**) level. Following model reactions were computed (Figure 2). For studying the solvent effect, single point energy was computed using gas phase-optimized geometry at the B3LYP/6-311++G** level with polarizable continuum model (PCM).
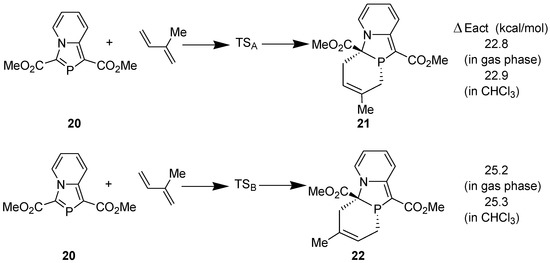
Figure 2.
Model DA reactions computed at B3LYP/6-311++G**//B3LYP/6-31G**.
The regioselectivity was calculated by using the formula, k1/k2 = e−∆E/RT [35]. A difference of 2.4 kcal/mol in the activation energies corresponds to only 2% regioselectivity. It is obvious that the observed regioselectivity of 100% cannot be accounted for at this theoretical level.
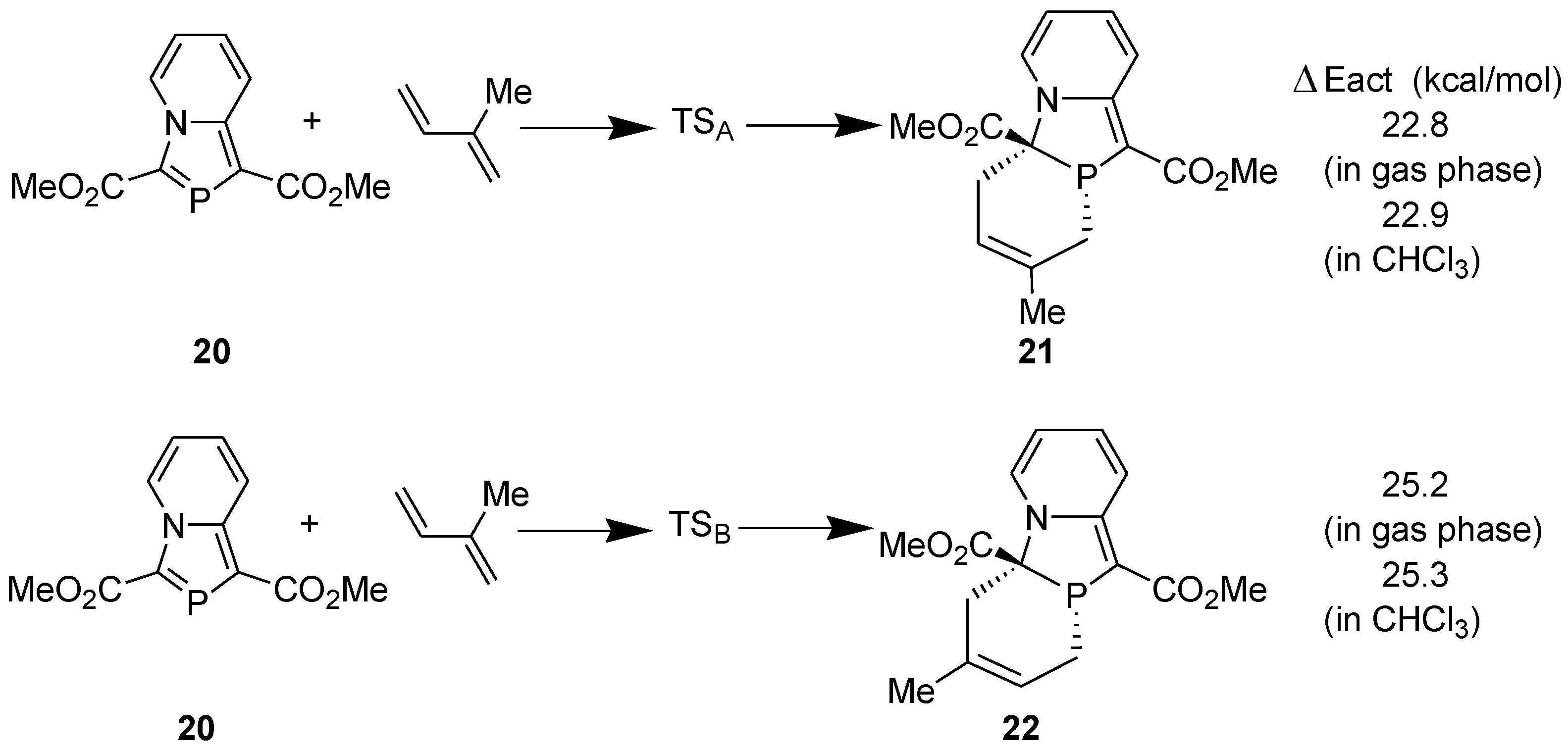
We investigated the reason for the difference in the dienophilic reactivities of 3-ethoxycarbonyl-1-methyl-1,3-azaphospholo[5,1-a]pyridine (10a) and 1,3-bis(ethoxycarbonyl)-1,3-azaphospholo[5,1-a]pyridine (10b), theoretically. For this purpose, following four model reactions were computed at the DFT (B3LYP/6-31G**) level (Scheme 8) [36].
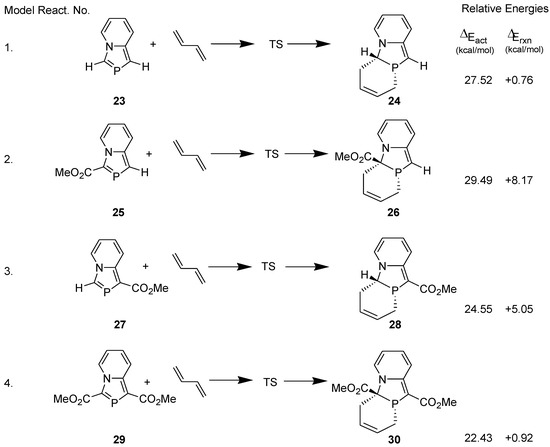
Scheme 8.
Model DA reactions computed at the B3LYP/6-31G** level.
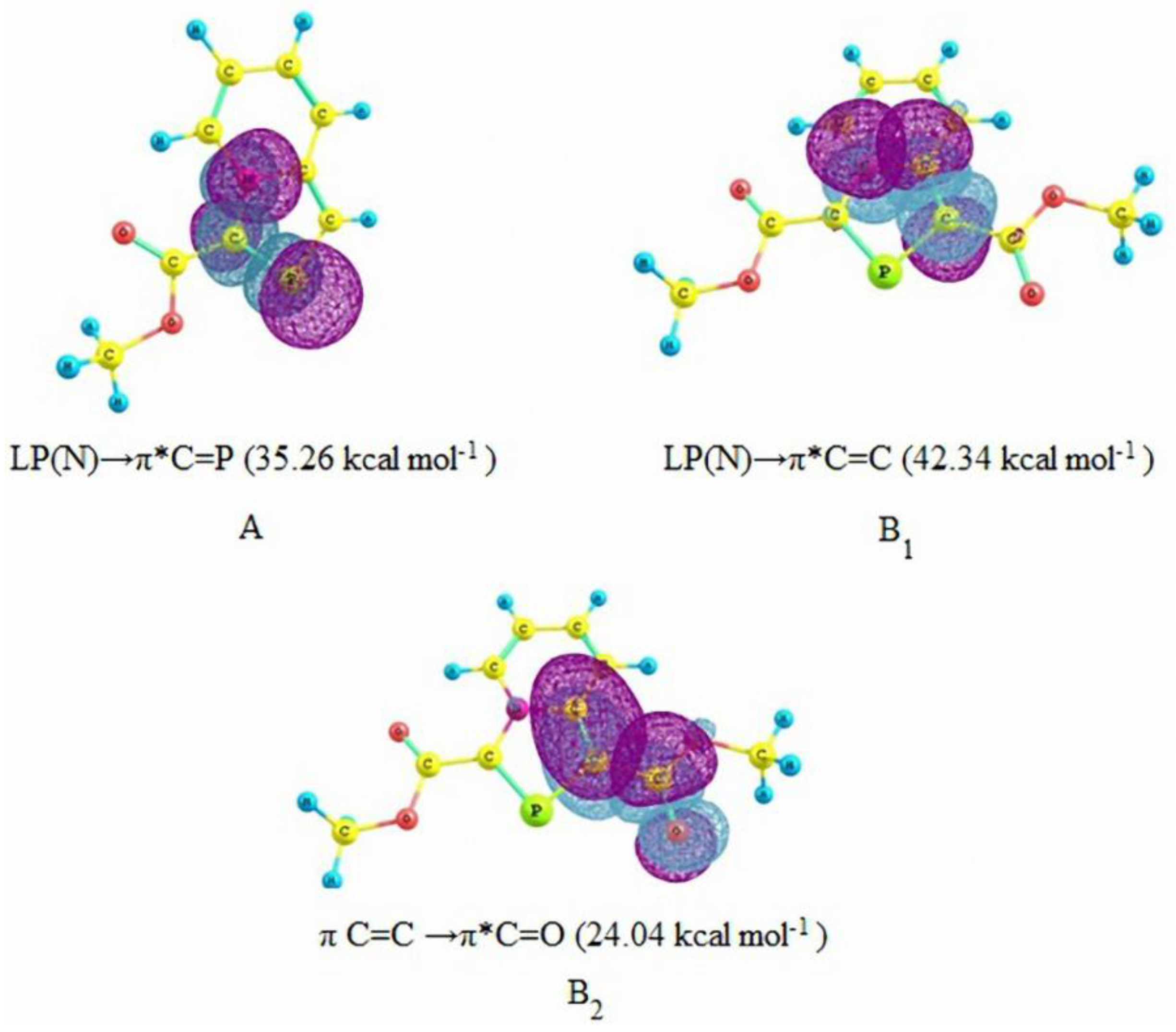
As expected, the activation energy barrier for the DA reaction with 1,3-bis(methoxycarbonyl)-1,3-azaphospholo[5,1-a]pyridine (29) is lowest. However, it is interesting to find that the activation barrier for the DA reaction with 3-methoxycarbonyl-1,3-azaphospholo[5,1-a]pyridine (25) is highest (29.49 kcal/mol), higher than even for the DA reaction with the unsubstituted 1,3-azaphospholo[5,1-a]pyridine (23). It is rather an unexpected effect of an electron-withdrawing group (EWG). To look into the real cause of this unusual effect, we carried out the natural bond orbital (NBO) analysis calculations, which revealed the existence of an interesting phenomenon: due to conjugation of the lone pair of the bridgehead nitrogen in 23, negative charge density is effectively transferred to the five-membered azaphosphole ring, which is further enhanced by the presence of an EWG at the 3-position. It is shown in Figure 3A. It makes the >C=P- functionality electron-rich, which fails to react with electron-rich diene, DMB. However, a CO2Me group at the 1-position acts as a trap for the negative charge, the latter being delocalized over to the carbonyl group. It is shown in Figure 3B. Thus in this case, the >C=P- functionality remains unaffected and is able to undergo the DA reaction with DMB.
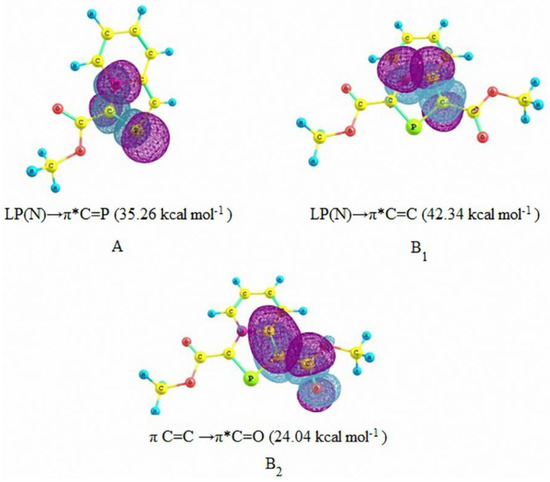
Figure 3.
Natural Bond Orbital (NBO) interactions in 3-methoxycarbonyl-1,3-azaphospholo[5,1-a]pyridine (A) and 1,3-bis(methoxycarbonyl)- 1,3-azaphospholo[5,1-a]pyridine (B1 and B2) with second order perturbation energies (in parenthesis).
3.2. Lewis Acid Catalyzed Diels-Alder Reaction
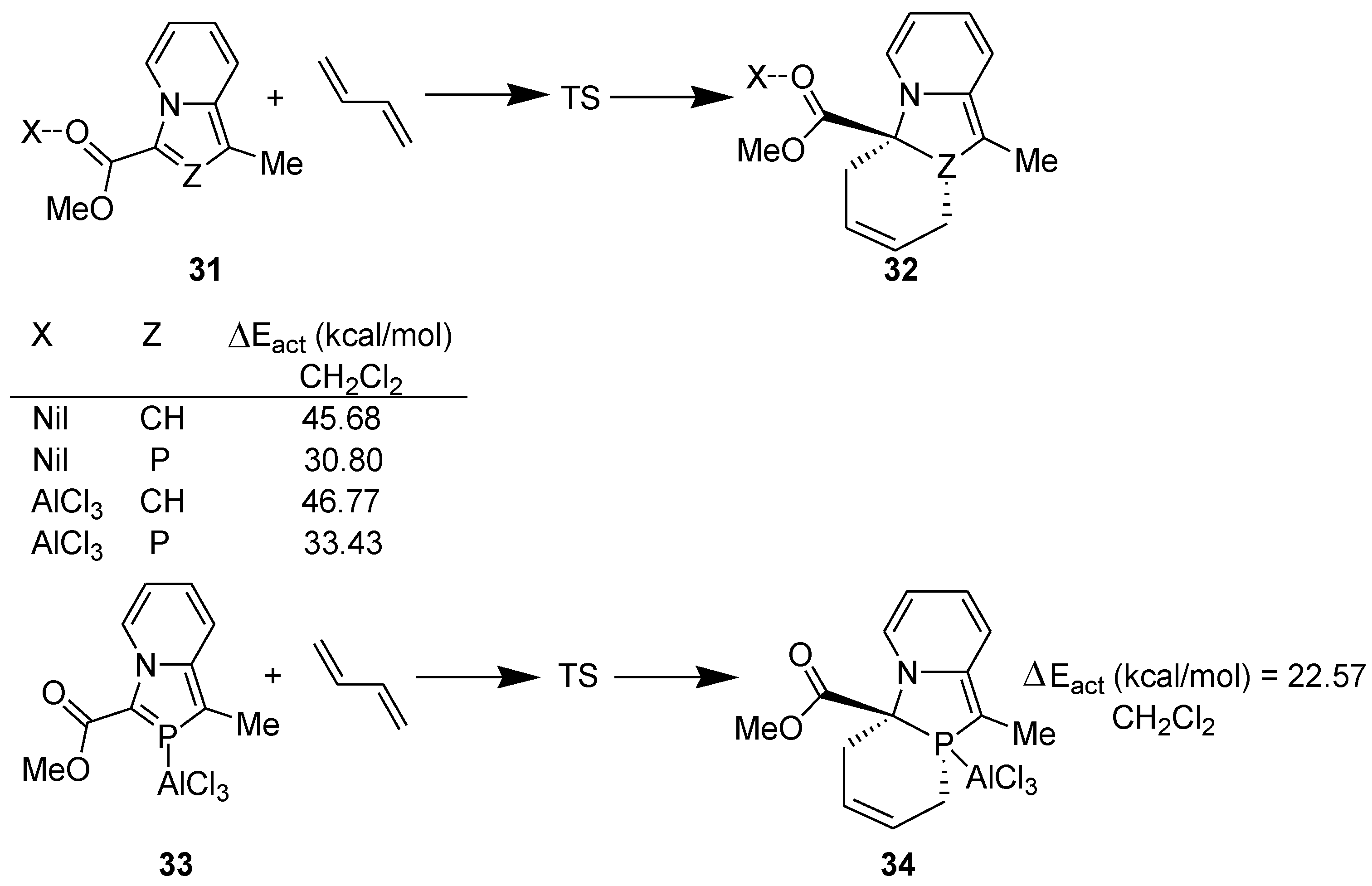
Then, we explored the possibility of using a Lewis acid as catalyst to accomplish DA reaction with 3-(alkoxycarbonyl)-1-methyl-1,3-azaphospholo[5,1-a]pyridines and computed the following model reactions at the DFT (B3LYP/6-31+G**) level (Scheme 9) [37]. For studying the solvent effect, single point energy was computed using gas phase-optimized geometry with PCM solvation model.
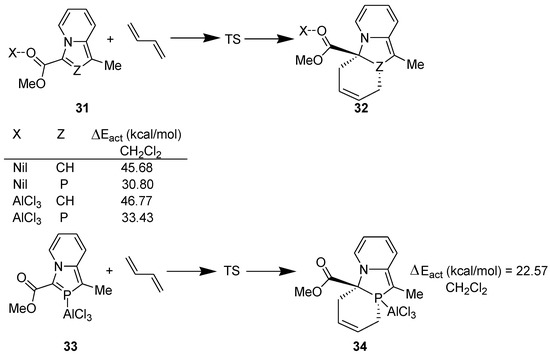
Scheme 9.
Model DA reactions computed at the B3LYP/6-31+G** level.
There was another surprise in store for us. When the catalyst, AlCl3, is coordinated to the carbonyl group of indolizine or 1,3-azaphospholo[5,1-a]pyridine, the activation energy barrier is increased further as compared with that in the absence of the catalyst. The NBO studies reveal that coordination of AlCl3 with the carbonyl group enhances transfer of the negative charge in these cases, making the >C=C< and >C=P- functionality in indolizine and 1,3-azaphospholo[5,1-a]pyridine, respectively, electron-rich. This appears to be the reason why no DA reaction across the >C=C< moiety of indolizine has been reported even in the presence of a catalyst.
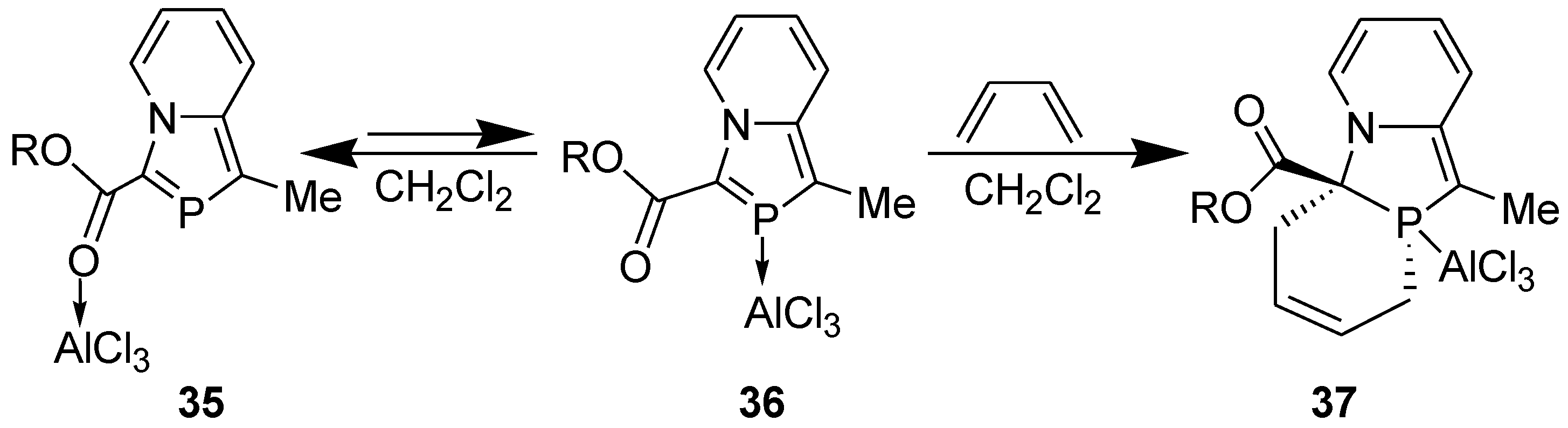
In 2-phosphaindolizine, however, there is another site for the coordination of AlCl3: the P atom. It may be noted that on coordination of the catalyst to the P atom, the activation energy barrier is lowered remarkably, to 22.57 kcal/mol (in CH2Cl2) only. These results indicated that in contrast to indolizine, DA reactions across the >C=P- moiety of 2-phosphaindolizine having EWG at 3-position only should be possible in the presence of a Lewis acid catalyst. However, in this context a question arises: if coordination of AlCl3 to the carbonyl group is thermodynamically more favorable than its coordination to the P atom by 3.43 kcal/mol, why should it coordinate to the P atom at all? In our opinion, in solution, an equilibrium may exist between the C=O- → AlCl3 and P → AlCl3 coordinated species, making a small concentration of the latter available that undergoes DA reaction with the diene to produce irreversibly the cycloadduct, thereby shifting the equilibrium to the right (Scheme 10).
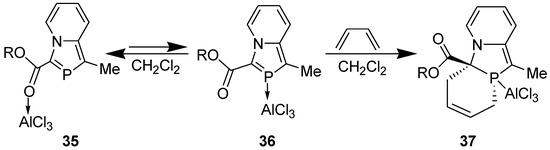
Scheme 10.
Equilibrium between C=O → and P → coordinated species, and DA reaction with the latter.
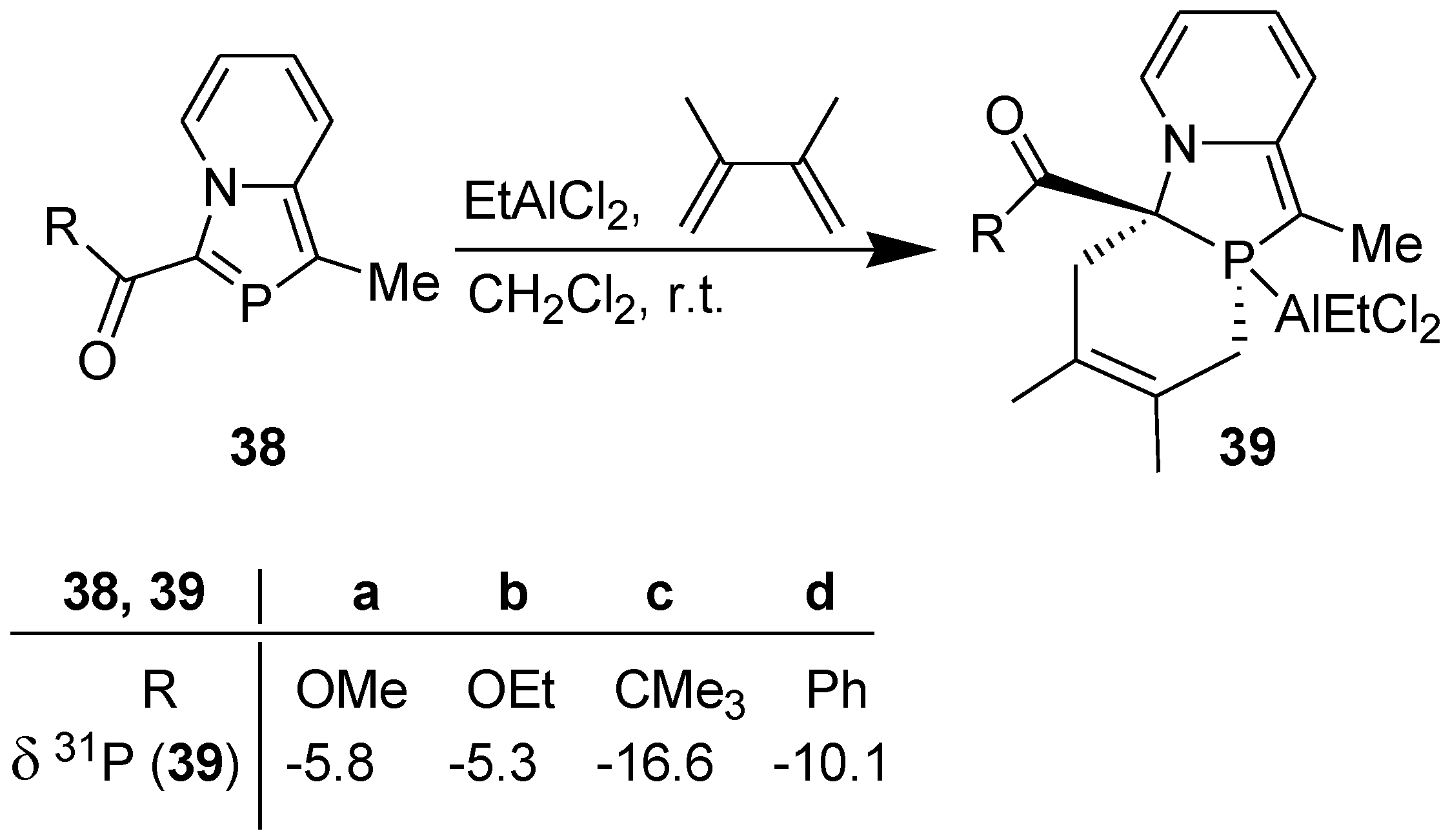
Guided by the above results, we accomplished successfully the DA reaction of 3-(alkoxycarbonyl/acyl)-1-methyl-2-phosphaindolizines (38) with DMB in the presence of ethylaluminium dichloride as a promoter (i.e., using its 1 equiv.) in dichloromethane at r.t. under nitrogen atmosphere (Scheme 11) [38]. The occurrence of a clean reaction was revealed by 31P NMR signal at δ = −16.6 to −5.3 ppm, which is in accordance with the 31P NMR chemical shifts reported for the [2+4] cycloadducts obtained from the DA reaction of P-W(Co)5 complexes of λ3-phosphinines [39].
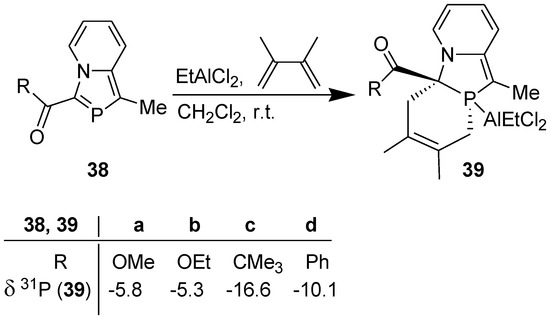
Scheme 11.
DA reaction of 3-(alkoxycarbonyl/acyl)-1-methyl-2-phosphaindolizines in the presence of EtAlCl2.
3.3. Asymmetric Diels-Alder Reaction with 2-Phosphaindolizines
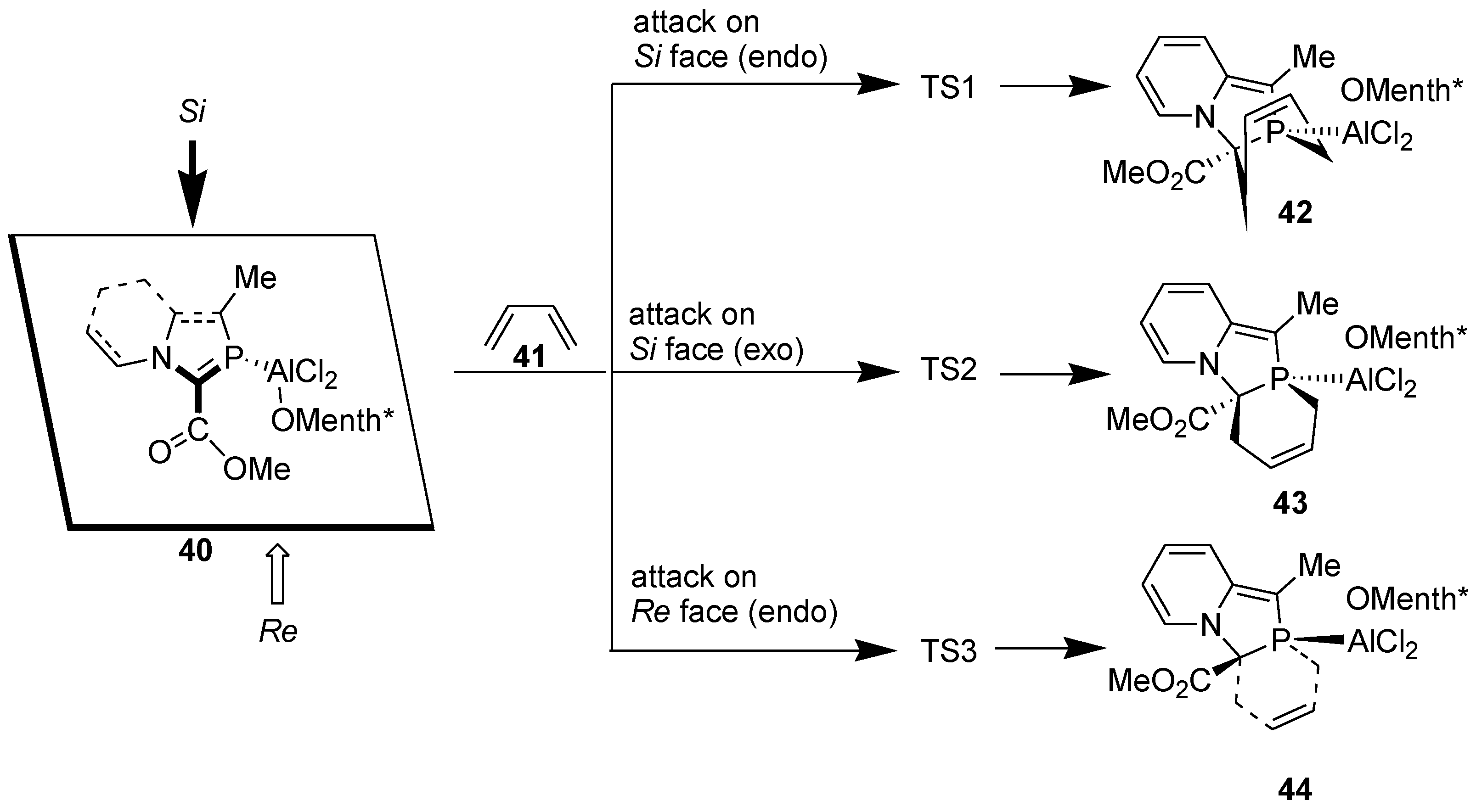
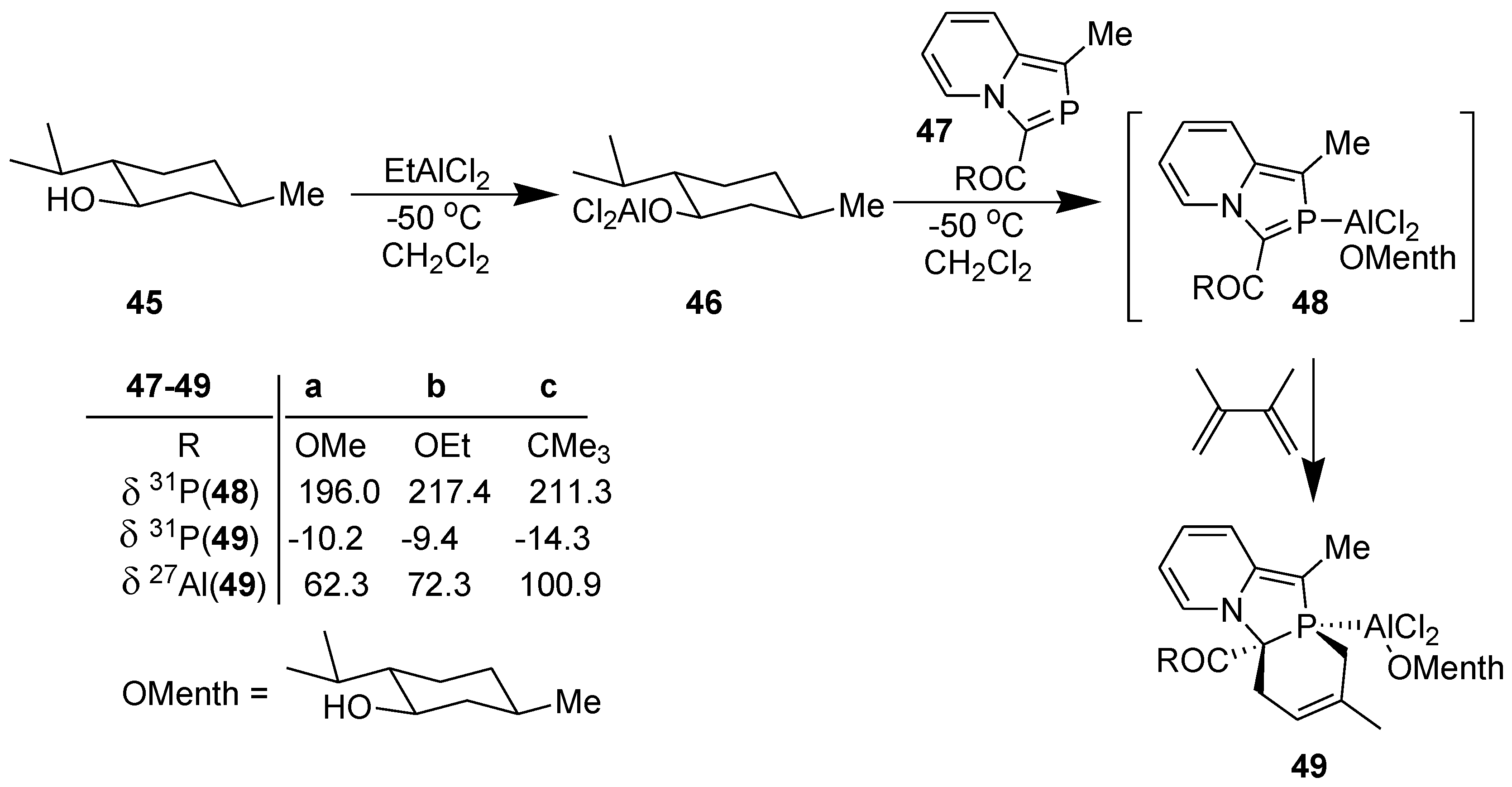
The successful results described above motivated us to investigate asymmetric DA reaction across the >C=P- moiety of 2-phosphaindolizines by using a chiral organoaluminium catalyst, namely, o-menthoxyaluminium dichloride [40].
We computed the following model reactions at the B3LYP/6-31+G* level (Scheme 12).
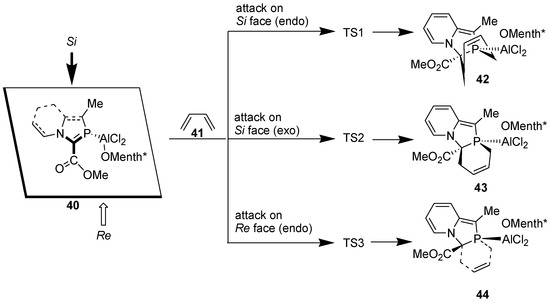
Scheme 12.
Attack of 1,3-butadiene on Si and Re faces of the >C=P- functionality of 2-phosphaindolizine complex computed at the B3LYP/6-31+G* level.
Computational calculations reveal that attack of the diene occurs preferentially from the Si face. Furthermore, during attack on the Si face, activation energy barrier for the exo approach is smaller than for the endo approach by ca. 0.3 kcal/mol. Presence of the bulky o-menthoxy group possibly makes the endo approach comparatively less accessible. All the reactions are moderately exothermic, but due to decrease in entropy, they are endergonic, Gibbs free energies values being +11.65 to +13.08 kcal/mol.
In accordance with the theoretical results, DA reaction of 2-phosphaindolizines (47) could be accomplished with complete diastereoselectivity, as indicated by 31P NMR, with a single product being formed in each case (Scheme 13) [40].
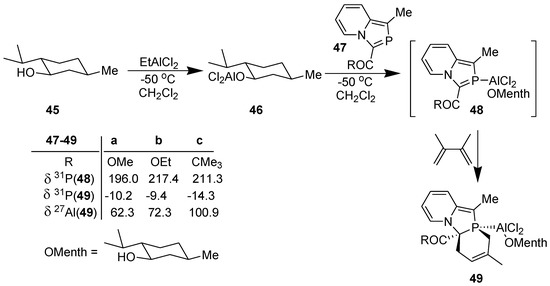
Scheme 13.
Diels-Alder reaction of 2-phosphaindolizine-n1-P-aluminium(o-menthoxy) dichloride with DMB.
3.4. Electrophilic Substitution
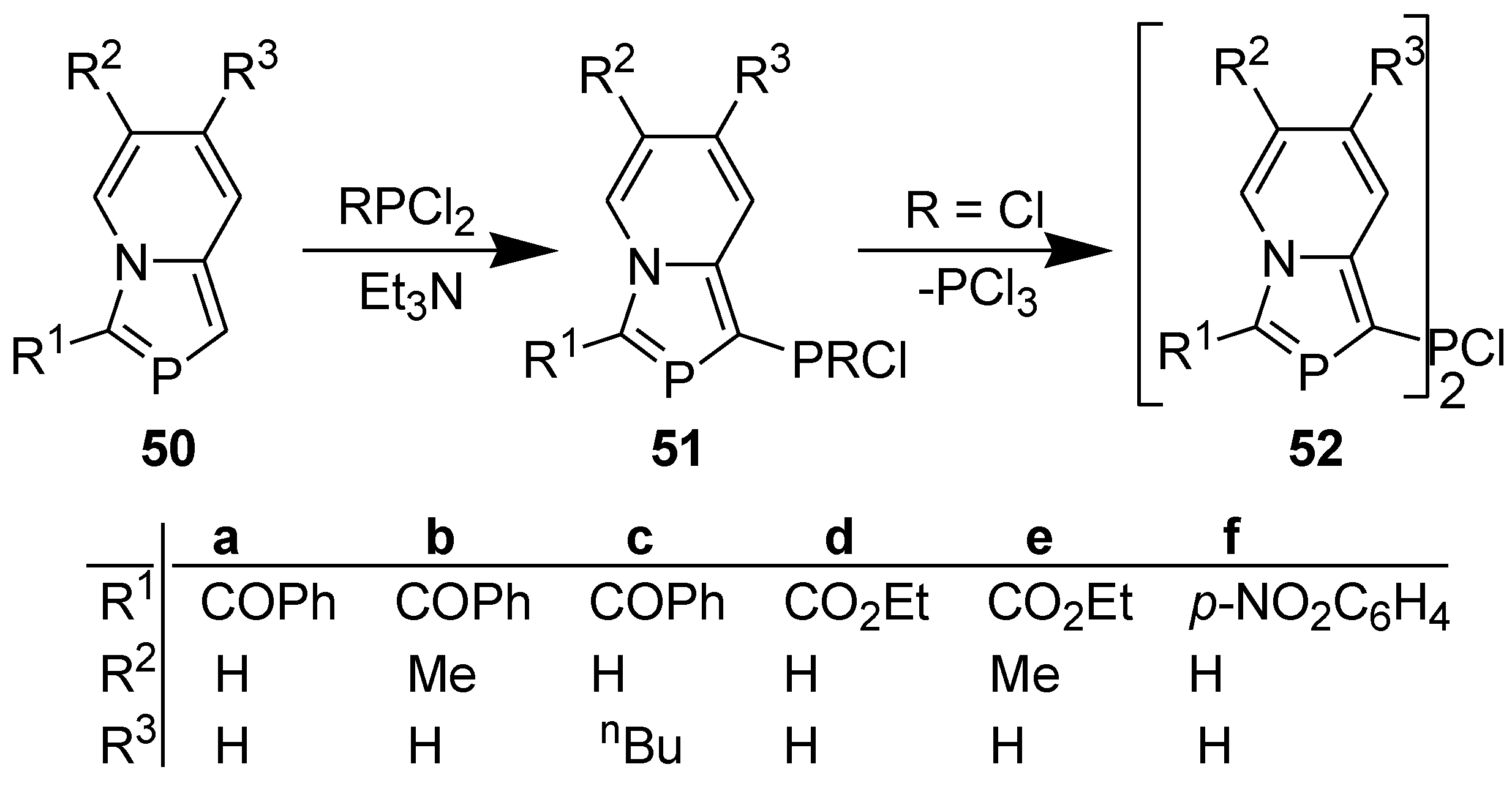
In contrast to indolizines [41], 1-unsubstituted 2-phosphaindolizines failed to react with MeCOCl, PhCOCl, or Me3SiCl, even on prolonged heating in the presence of Et3N [42]. However, like indolizines [43], 1-unsubstituted 2-phosphaindolizines reacted with PCl3 or PhPCl2 (but not with Ph2PCl) in the presence of Et3N to afford the phosphynated products (Scheme 14) [44]. It is noteworthy that on reacting with PCl3, the initially formed dichlorophosphino product (51, R=Cl) undergoes dismutation accompanied by the loss of PCl3 to afford a PCl bridged bis(2-phosphaindolizine) (52) as the final product.
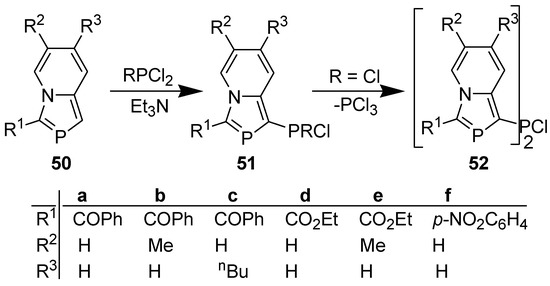
Scheme 14.
Reaction of 1-unsubstituted 2-phosphaindolizines with chlorophosphines.
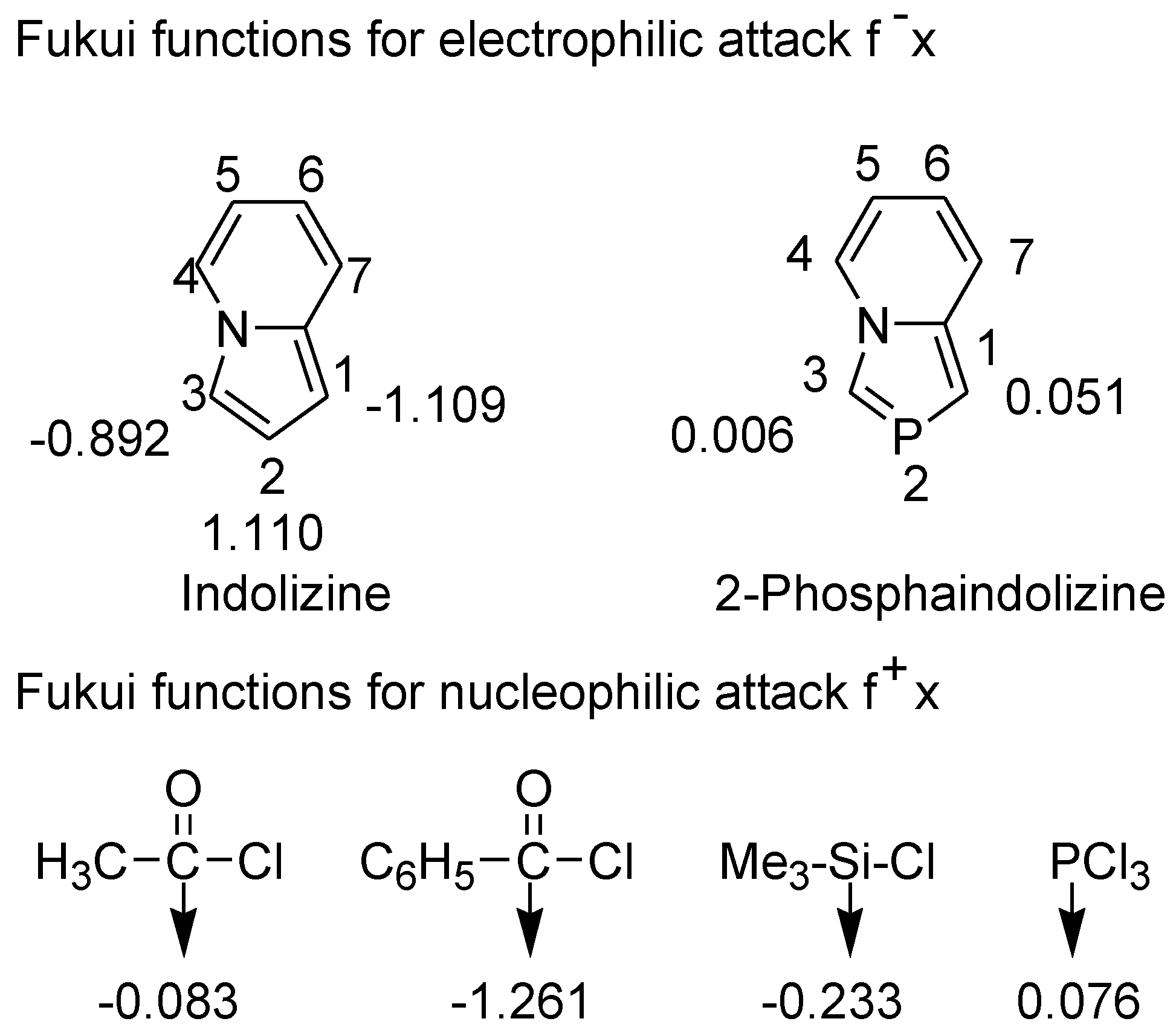
The concept of hard-soft acid-base (HSAB) theory was extended to explain the global and local reactivity of organic molecules towards nucleophilic and electrophilic reagents [45]. The chemical reactivity at a particular molecular site could be rationalized using a quantitative descriptor, the Fukui function (f(r)). We recently used Fukui function to rationalize 1,2- versus 1,4-addition of amines to maleic anhydride [46]. The difference in the behavior of indolizine and 2-phosphaindolizine towards electrophilic reagents can be rationalized on the basis of the Fukui functions calculated at the B3LYP/6-31+G* level (Figure 4) [47].
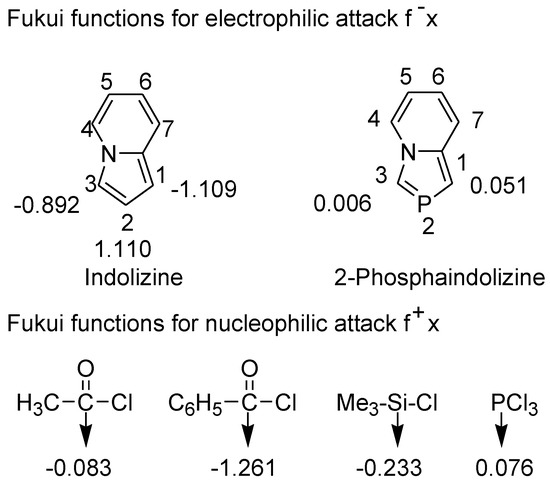
Figure 4.
Fukui functions calculated at the B3LYP/6-31+G* level.
Fukui functions reveal that C1 and C3 in indolizine are much harder than C1 and C3 in 2-phosphaindolizine. Thus, hard electrophilic reagents such as acyl chlorides and chlorotrimethylsilane attack at C1 and C3 positions of indolizine, but they fail to react with 2-phosphaindolizine. On the other hand, phosphorus trichloride is comparatively softer and hence reacts with 2-phosphaindolizine at C1 (C3 being substituted in our compounds). Being at the borderline, PCl3 reacts with indolizine also.
4. Conclusions
The computational calculations could be used as a useful tool to understand and solve problems encountered during reactions of annelated 1,3-azaphospholers. Furthermore, this strategy worked well while planning the reactions such as Lewis acid catalyzed and asymmetric DA reactions across the >C=P- functionality of 2-phosphaindolizines.
Author Contributions
All authors contributed equally in the compilation of this perspective.
Funding
This research received no external funding. APC was sponsored by MDPI.
Acknowledgments
We are thankful to the authorities of The IIS University, Jaipur (India) for the facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dirac, P.A.M. Quantum mechanics of many-electrons systems. Proc. R. Soc. Lond. Ser. A. 1929, 123, 714–733. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogenous electron gas. Phys. Rev. 1964, 136, B867–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Zangwill, A. A half century of density functional theory. Phys. Today 2015, 68, 34–39. [Google Scholar] [CrossRef]
- Jain, A.; Shin, Y.; Persson, K.A. Computational predictions of energy materials using density functional theory. Nat. Rev. Mater. 2016, 1, 1–13. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87, 897–923. [Google Scholar] [CrossRef]
- Tsipis, A.C. DFT flavor of coordination chemistry. Coord. Chem. Rev. 2014, 272, 1–29. [Google Scholar] [CrossRef]
- Cole, D.J.; Hine, N.D.M. Applications of large-scale density functional theory in biology. J. Phys. Condens. Matter 2016, 28, 393001. [Google Scholar] [CrossRef] [PubMed]
- Lupp, D.; Christensen, N.J.; Fristrup, P. Synergy between experimental and theoretical methods in the exploration of homogeneous transition metal catalysis. Dalton Trans. 2014, 43, 11093–11105. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lineberger, W.C.; Borden, W.T. The synergy between qualitative theory, quantitative calculations, and direct experiments in understanding, calculating, and measuring the energy differences between the lowest singlet and triplet states of organic diradicals. Phys. Chem. Chem. Phys. 2011, 13, 11792–11813. [Google Scholar] [CrossRef] [PubMed]
- Sierka, M. Synergy between theory and experiment in structure resolution of low-dimensional oxides. Prog. Surf. Sci. 2010, 85, 398–434. [Google Scholar] [CrossRef]
- Mcgrady, J. Guest Editor The themed collection, “The synergy between theory and experiment. Dalton Trans. 2009, 5805–6064. [Google Scholar] [CrossRef]
- Bansal, R.K.; Gupta, N.; Karaghiosoff, K.; Schmidpeter, A.; Spindler, C. 2-Phosphaindolizines. Chem. Ber. 1991, 124, 475–480. [Google Scholar] [CrossRef]
- Bansal, R.K.; Kabra, V.; Gupta, N.; Karaghiosoff, K. Synthesis of some new 2-phosphaindolizines. Indian J. Chem. 1992, 31, 254–256. [Google Scholar]
- Gupta, N.; Jain, C.B.; Heinicke, J.; Bharatiya, N.; Bansal, R.K.; Jones, P.G. 2-Phosphaindolizines. Heteroat. Chem. 1998, 9, 333–339. [Google Scholar] [CrossRef]
- Karaghioshoff, K.; Bansal, R.K.; Gupta, N. 1,4,2-Diazaphospholo[4,5-a]pyridines. Z. Naturforsch. B 1992, 47b, 373–378. [Google Scholar]
- Bansal, R.K.; Pandey, G.; Karaghiosoff, K.; Schmidpeter, A. 1,2,3-Diazaphospholo[1,5-a]pyridines. Synthesis 1995, 173–175. [Google Scholar] [CrossRef]
- Bansal, R.K.; Gandhi, N.; Karaghiosoff, K.; Schmidpeter, A. Synthesis of [1,2,4,3]triazaphospholo[1,5-a]pyridines. Z. Naturforsch. 1995, 50b, 558–562. [Google Scholar] [CrossRef]
- Bansal, R.K.; Mahnot, R.; Sharma, D.C.; Karaghiosoff, K.; Schmidpeter, A. 1,3-Azaphospholo[5,1-b]thiazolines and benzothiazoles. Heteroat. Chem. 1992, 3, 351–357. [Google Scholar] [CrossRef]
- Bansal, R.K.; Jain, C.B.; Gupta, N.; Kabra, V.; Karaghiosoff, K.; Schmidpeter, A. Synthesis and properties of a 5,6-dihydro-1,3-azaphospholo[5,1-b]oxazole. Phosphorus Sulfur Silicon 1994, 86, 139–143. [Google Scholar] [CrossRef]
- Bansal, R.K.; Karaghiosoff, K.; Gandhi, N.; Schmidpeter, A. 2-Substituted cycloiminium salts in azaphosphole synthesis. Synthesis 1995, 361–369. [Google Scholar] [CrossRef]
- Bansal, R.K.; Surana, A.; Gupta, N. 2-Phosphaindolizines via 1,5-electrocyclization. Tetrahedron Lett. 1999, 40, 1565–1568. [Google Scholar] [CrossRef]
- Bansal, R.K.; Gupta, N.; Baweja, M.; Hemrajani, L.; Jain, V.K. Pyridinium dichlorophosphinomethylides. Heteroat. Chem. 2001, 12, 602–609. [Google Scholar] [CrossRef]
- Bansal, R.K.; Hemrajani, L.; Gupta, N. 1,3-Azaphospholo[5,1-a]isoquinolines. Heteroat. Chem. 1999, 10, 598–604. [Google Scholar] [CrossRef]
- Singh, D.; Sinha, P.; Gupta, N.; Bansal, R.K. Synthesis of 1,3-azaphospholo[1,5-f]phenanthridines through 1,5-electrocyclization. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 488–492. [Google Scholar] [CrossRef]
- Appel, R.; Knoll, F.; Ruppert, I. Phospha-alkenes and phospha-alkynes, genesis and properties of the (p-p)π-multiple bond. Angew. Chem. Int. Ed. 1981, 20, 731–744. [Google Scholar] [CrossRef]
- Mathey, F.; Mercier, F.; Charrier, C.; Fischer, J.; Mitschler, A. Dicoordinated 2H-phospholes as transient intermediates in the reactions of tervalent phospholes at high temperature. One-step synthesis of 1-phosphabornadienes and phosphorins from phospholes. J. Am. Chem. Soc. 1981, 103, 4595–4597. [Google Scholar] [CrossRef]
- Wannere, C.S.; Bansal, R.K.; Schleyer, P.v.R. Diels-Alder reaction of phosphaethene with 1,3-dienes: An ab-initio study. J. Org. Chem. 2002, 67, 9162–9174. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.K.; Gupta, N.; Kumawat, S.K.; Dixit, G. Diastereo- and regioselective Diels-Alder reactions of 2-phosphaindolizines. Tetrahedron 2008, 64, 6395–6401. [Google Scholar] [CrossRef]
- Bansal, R.K.; Jain, V.K.; Gupta, N.; Gupta, N.; Hemrajani, L.; Baweja, M.; Jones, P.G. Stereo- and regioselectivity in Diels-Alder reactions of 1,3-azaphospholo[5,1-a]isoquinoline and –[5,1-a]pyridine. Tetrahedron 2002, 58, 1573–1579. [Google Scholar] [CrossRef]
- Bansal, R.K.; Karaghiosoff, K.; Gupta, N.; Gandhi, N.; Kumawat, S.K. Diastereo- and regioselectivity in Diels-Alder reaction of [1,4,2]diazaphospholo[4,5-a]pyridines. Tetrahedron 2005, 61, 10521–10528. [Google Scholar] [CrossRef]
- Karaghiosoff, K.; Cleve, C.; Schmidpeter, A. Chloromethyl-dichlorophosphane: A useful reagent for the synthesis of new heterocycles with dicoordinated phosphorus. Phosphorus Sulfur Silicon Relat. Elem. 1986, 28, 289–296. [Google Scholar] [CrossRef]
- Pellegriner, S.C.; Silva, M.A.; Goodman, J.M. Theoretical evaluation of the origin of the regio- and stereoselectivity in the Diels-Alder reactions of dialkylvinylboranes: Studies on the reactions of vinylborane, dimethylvinylborane, and vinyl-9-BBN with trans-piperylene and isoprene. J. Am. Chem. Soc. 2001, 123, 8832–8837. [Google Scholar] [CrossRef]
- Bansal, R.K.; Gupta, N.; Dixit, G.; Kumawat, S.K. Theoretical investigation of an unusual substituent effect on the dienophilicity of >C=P– Functionality in 2-phosphaindolizines. J. Phys. Org. Chem. 2009, 22, 125–129. [Google Scholar] [CrossRef]
- Gupta, N.; Jangid, R.K.; Bansal, R.K.; Hopffgarten, M.V. Catalytic effect of organoaluminium chloride reagents on the dienophilic reactivity of indolizine and 2-phosphaindolizine towards [2+4] cycloaddition: A DFT investigation. Curr. Cat. 2012, 1, 93–99. [Google Scholar] [CrossRef]
- Jangid, R.K.; Gupta, N.; Bansal, R.K.; Hopffgarten, M.V.; Frenking, G. Diels-Alder reaction of 2-phosphaindolizines catalyzed by organoaluminium reagent: Theoretical and experimental results. Tetrahedron Lett. 2011, 52, 1721–1724. [Google Scholar] [CrossRef]
- Alcaraz, J.P.; Mathey, F. Accroissement de la reactivite des phosphorines en tant que dienes et philodienes par complexation du phosphore. Tetrahedron Lett. 1984, 25, 207–210. [Google Scholar] [CrossRef]
- Jangid, R.K.; Sogani, N.; Gupta, N.; Bansal, R.K.; Hopffgarten, M.V.; Frenking, G. Asymmetric Diels-Alder reaction with >C=P– functionality of the 2-phosphaindolizine-η1-P-aluminium(o-menthoxy) dichloride complex: Experimental and theoretical results. Beilstein J. Org. Chem. 2013, 9, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Swinbourne, F.J.; Hunt, J.H.; Klinkert, G. Advances in Indolizine Chemistry in the Series: Advances in Heterocyclic Chemistry; Katritzky, A.R., Boulton, A.J., Eds.; Academic Press: New York, NY, USA, 1979; Volume 23, pp. 103–170. [Google Scholar]
- Bansal, R.K.; Heinicke, J. Anellated heterophospholes and phospholides and analogies with related non-phosphorus systems. Chem. Rev. 2001, 101, 3549–3578. [Google Scholar] [CrossRef] [PubMed]
- Tolmachev, A.A.; Yurchenko, A.A.; Kozlov, E.S.; Shulezhko, V.A.; Pinchuk, A.M. Phosphorylated indolizines. Heteroat. Chem. 1993, 4, 343–360. [Google Scholar] [CrossRef]
- Bansal, R.K.; Gupta, N.; Kabra, V.; Spindler, C.; Karaghiosoff, K.; Schmidpeter, A. Substitution of 2-phosphaindolizines by bromine and by chlorophosphines. Heteroat. Chem. 1992, 3, 359–366. [Google Scholar] [CrossRef]
- Mendez, F.; Gázquez, J.L. Chemical reactivity of enolate ions: The local hard and soft-acids and bases principle viewpoint. J. Am. Chem. Soc. 1994, 116, 9298–9301. [Google Scholar] [CrossRef]
- Kour, M.; Gupta, R.; Bansal, R.K. Experimental and theoretical investigation of the reaction of secondary amines to maleic anhydride. Aust. J. Chem. 2017, 70, 1247–1253. [Google Scholar] [CrossRef]
- Gupta, R.; Paul, B.; Bansal, R.K. Application of Fukui functions for comparing reactivities of indolizine and 2-phosphaindolizine towards electrophilic substitution. 2018; unpublished work. [Google Scholar]
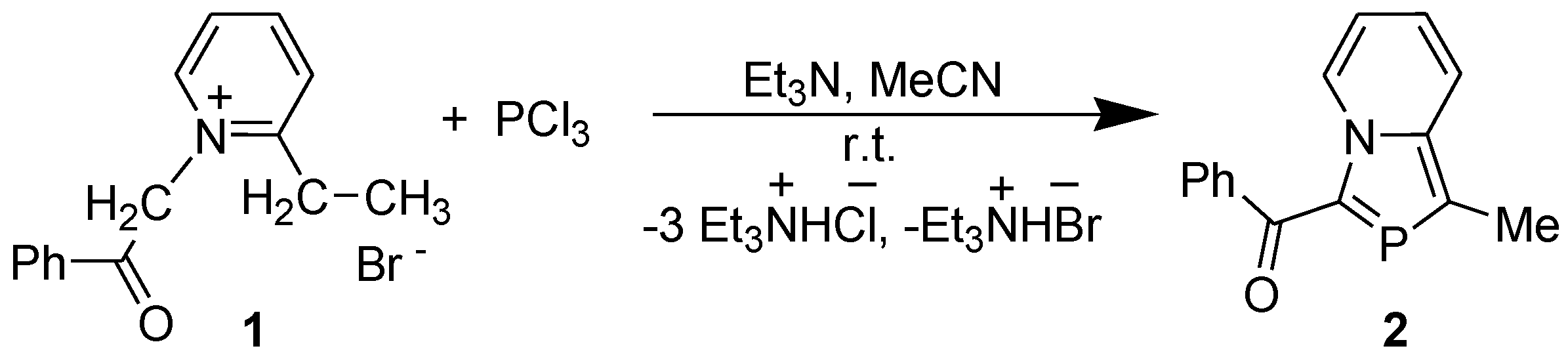
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).