Nanotechnology as a Platform for the Development of Injectable Parenteral Formulations: A Comprehensive Review of the Know-Hows and State of the Art



Abstract
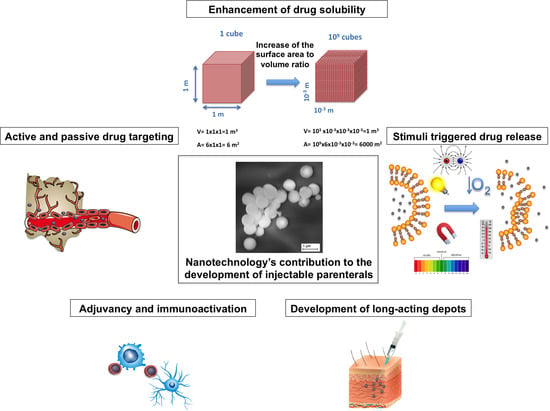
:
1. Introduction
2. Solubility Enhancement
3. Modification of the Drug Release
3.1. Nanoparticles for the Development of Long-Acting Injectables
3.1.1. Locally Injectable Long-Acting Nanoparticle-Based Formulations
3.1.2. Systemically Injectable Long-Acting Nanoparticle-Based Formulations
3.2. Nanoparticles for Tuning the Onset of Drug Release
3.2.1. Temperature-Responsive Nanocarries
3.2.2. Light-Responsive Nanocarriers
3.2.3. Hypoxia-Responsive Nanocarriers
3.2.4. pH-Responsive Nanocarriers
3.2.5. Redox-Responsive Nanocarriers
3.2.6. Enzyme-Responsive Nanocarriers
3.2.7. Electroresponsive Nanocarriers
3.2.8. Magnetically Responsive Nanocarriers
3.2.9. Dual and Multi-Stimuli-Responsive Nanocarriers
4. Targeted Drug Delivery
5. Passive Targeting
5.1. Inflammatory Disorders
5.2. Central Nervous System (CNS)
5.3. Kidneys
5.4. Spleen and Lymphatics
6. Active Targeting
6.1. Active Targeting Based on Affinity Molecules
6.1.1. Affinity Proteins and Peptides
6.1.2. Lectins
6.1.3. Glycans
6.1.4. Aptamers
6.2. Active Targeting Based on Natural Ligand-Receptor Interactions
7. Adjuvancy and Immune Activation
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Gulati, N.; Gupta, H. Parenteral drug delivery: A review. Recent Pat. Drug Deliv. Formul. 2011, 5, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Vega, J.A.; Ochoa, P.S.; Holder, P. Introduction to parenteral preparations. In Concepts in Steril Preparations and Aseptic Techniques; Jones and Bartlett Learning: Burlington, MA, USA, 2015; pp. 1–26. [Google Scholar]
- Packhaeuser, C.B.; Schneider, J.; Oster, C.G.; Kissel, T. In situ forming parenteral drug delivery systems: An overview. Eur. J. Pharm. Biopharm. 2004, 58, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.D.; Müller, R.H. Lipid nanoparticles for parenteral delivery of actives. Eur. J. Pharm. Biopharm. 2009, 71, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Patravale, V.; Prajakta, D.; Jain, R. Nanoparticles as drug carriers. In Nanoparticulate Drug Delivery: Perspectives on the Transition from Laboratory to Market; Woodhead Publishing: New Dehli, India, 2012; pp. 29–86. [Google Scholar]
- Bari, H. A prolonged release parenteral drug delivery system—An overview. Int. J. Pharm. Sci. Rev. Res. 2010, 3, 1–11. [Google Scholar]
- Ogawara, K.; Yoshizawa, Y.; Un, K.; Araki, T.; Kimura, T.; Higaki, K. Nanoparticle-based passive drug targeting to tumors: Considerations and implications for optimization. Biol. Pharm. Bull. 2013, 36, 698–702. [Google Scholar] [CrossRef] [Green Version]
- Weissig, V.; Pettinger, T.K.; Murdock, N. Nanopharmaceuticals (part 1): Products on the market. Int. J. Nanomed. 2014, 9, 4357–4373. [Google Scholar] [CrossRef] [Green Version]
- Ragelle, H.; Danhier, F.; Préat, V.; Langer, R.; Anderson, D.G. Nanoparticle-based drug delivery systems: A commercial and regulatory outlook as the field matures. Expert Opin. Drug Deliv. 2017, 14, 851–864. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in nanomedicine: Approved and investigational nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Narvekar, M.; Xue, H.Y.; Eoh, J.Y.; Wong, H.L. Nanocarrier for poorly water-soluble anticancer drugs—Barriers of translation and solutions. AAPS Pharmscitech 2014, 15, 822–833. [Google Scholar] [CrossRef]
- Tuomela, A.; Hirvonen, J.; Peltonen, L. Stabilizing agents for drug nanocrystals: Effect on bioavailability. Pharmaceutics 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipp, J.E. The role of solid nanoparticle technology in the parenteral delivery of poorly water-soluble drugs. Int. J. Pharm. 2004, 284, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.R.; Weston, N.; Coombes, A.G.A.; Fitzgerald, M.; Perrie, Y. Liposome formulation of poorly water soluble drugs: Optimisation of drug loading and ESEM analysis of stability. Int. J. Pharm. 2004, 285, 23–34. [Google Scholar] [CrossRef]
- Nagarwal, R.C.; Kumar, R.; Dhanawat, M.; Das, N.; Pandit, J.K. Nanocrystal technology in the delivery of poorly soluble drugs: An overview. Curr. Drug Deliv. 2011, 8, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Hörmann, K.; Zimmer, A. Drug delivery and drug targeting with parenteral lipid nanoemulsions—A review. J. Control. Release Off. J. Control. Release Soc. 2016, 223, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, C.; Cheng, W.P. Implementing nanotechnology and novel drug delivery systems to improve dissolution and solubilization. Am. Pharm. Rev. 2012, 15, 7. Available online: http://www.americanpharmaceuticalreview.com/Featured–Articles/126889–Implementing-Nanotechnology–and–Novel–Drug–Delivery–Systems–to–Improve–Dissolution–and–Solubilization/ (accessed on 31 May 2020).
- Trivedi, R.; Kompella, U.B. Nanomicellar formulations for sustained drug delivery: Strategies and underlying principles. Nanomedicine 2010, 5, 485–505. [Google Scholar] [CrossRef] [Green Version]
- Greco, F.; Vicent, M.J. Polymer-drug conjugates: Current status and future trends. Front. Biosci. J. Virtual Libr. 2008, 13, 2744–2756. [Google Scholar] [CrossRef] [Green Version]
- Larson, N.; Ghandehari, H. Polymeric conjugates for drug delivery. Chem. Mater. Publ. Am. Chem. Soc. 2012, 24, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wallace, S. Polymer-drug conjugates: Recent development in clinical oncology. Adv. Drug Deliv. Rev. 2008, 60, 886–898. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Yagui, C.O.; Junior, A.P.; Tavares, L. Micellar solubilization of drugs. J. Pharm. Pharm. Sci. 2005, 8, 147–163. [Google Scholar] [PubMed]
- Gupta, U.; Agashe, H.B.; Jain, N.K. Polypropylene imine dendrimer mediated solubility enhancement: Effect of pH and functional groups of hydrophobes. J. Pharm. Pharm. Sci. 2007, 10, 358–367. [Google Scholar] [PubMed]
- Jain, N.K.; Gupta, U. Application of dendrimer-drug complexation in the enhancement of drug solubility and bioavailability. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Svenson, S.; Chauhan, A.S. Dendrimers for enhanced drug solubilization. Nanomedicine 2008, 3, 679–702. [Google Scholar] [CrossRef] [PubMed]
- Siddalingappa, B.; Nekkanti, V.; Betageri, G. Insoluble drug delivery technologies: Review of health benefits and business potentials. OA Drug Des. Deliv. 2013, 1, 1–6. [Google Scholar]
- Gidwani, B.; Vyas, A. A comprehensive review on cyclodextrin-based carriers for delivery of chemotherapeutic cytotoxic anticancer drugs. BioMed Res. Int. 2015, 2015, 15. [Google Scholar] [CrossRef] [Green Version]
- Lakkakula, J.R.; Maçedo Krause, R.W. A vision for cyclodextrin nanoparticles in drug delivery systems and pharmaceutical applications. Nanomedicine 2014, 9, 877–894. [Google Scholar] [CrossRef]
- Hierrezuelo, J.; Pelaez, L.; Benavente, J.; Lopez-Romero, J.M.; Rico, R.; Armengol, R. Lipid and cyclodextrin nanocarriers loading bioactive agents: Stabilization on polymeric supports. In Bioengineered Nanomaterials; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2014; pp. 225–238. [Google Scholar]
- Nasir, A.; Harikumar, S.; Amanpreet, K. Cyclodextrins: An excipient tool in drug delivery. Int. Res. J. Pharm. 2012, 3, 44–50. [Google Scholar]
- Tejashri, G.; Amrita, B.; Darshana, J. Cyclodextrin based nanosponges for pharmaceutical use: A review. Acta Pharm. Zagreb Croat. 2013, 63, 335–358. [Google Scholar] [CrossRef]
- Olteanu, A.A.; Arama, C.C.; Mihaescu, C.; Moniciu, C.-M. Effect of β-cyclodextrins based nanosponges on the solubility of lipophilic pharmacological active substances (repaglinide). J. Incl. Phenom. Macrocycl. Chem. 2014, 80, 17–24. [Google Scholar] [CrossRef]
- Hu, Q.-D.; Tang, G.-P.; Chu, P.K. Cyclodextrin-based host-guest supramolecular nanoparticles for delivery: From design to applications. Acc. Chem. Res. 2014, 47, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Zerkoune, L.; Angelova, A.; Lesieur, S. Nano-assemblies of modified cyclodextrins and their complexes with guest molecules: Incorporation in nanostructured membranes and amphiphile nanoarchitectonics design. Nanomaterials 2014, 4, 741–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oommen, E.; Dinesh Shenoy, B.; Udupa, N.; Kamath, R.; Uma Devi, P. Antitumour efficiency of cyclodextrin-complexed and niosome-encapsulated plumbagin in mice bearing melanoma. Pahrmacy Pharmacol. Commun. 1999, 5, 281–285. [Google Scholar] [CrossRef]
- Yallapu, M.M.; Ebeling, M.C.; Khan, S.; Sundram, V.; Chauhan, N.; Gupta, B.K.; Puumala, S.E.; Jaggi, M.; Chauhan, S.C. Novel curcumin-loaded magnetic nanoparticles for pancreatic cancer treatment. Mol. Cancer Ther. 2013, 12, 1471–1480. [Google Scholar] [CrossRef] [Green Version]
- Namgung, R.; Mi Lee, Y.; Kim, J.; Jang, Y.; Lee, B.-H.; Kim, I.-S.; Sokkar, P.; Rhee, Y.M.; Hoffman, A.S.; Kim, W.J. Poly-cyclodextrin and poly-paclitaxel nano-assembly for anticancer therapy. Nat. Commun. 2014, 5, 3702. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Liang, M.; Svejkar, D.; Hart-Smith, G.; Lu, H.; Scarano, W.; Stenzel, M.H. Albumin-micelles via a one-pot technology platform for the delivery of drugs. Chem. Commun. 2014, 50, 6394–6397. [Google Scholar] [CrossRef] [Green Version]
- Karimi, M.; Bahrami, S.; Baghaee Ravari, S.; Sahandi, P.; Mirshekari, H.; Bozorgomid, M.; Shahreza, S.; Sori, M.; Hamblin, M.R. Albumin nanostructures as advanced drug delivery systems. Expert Opin. Drug Deliv. 2016, 13, 1609–1623. [Google Scholar] [CrossRef] [Green Version]
- Naveen, R.; Akshata, K.; Pimple, S.; Chaudhari, P. A review on albumin as drug carrier in treating different diseases and disorders. Pharm. Sin 2016, 7, 11–15. [Google Scholar]
- Chen, H.; Khemtong, C.; Yang, X.; Chang, X.; Gao, J. Nanonization strategies for poorly water-soluble drugs. Drug Discov. Today 2011, 16, 354–360. [Google Scholar] [CrossRef]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. 2014, 9, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Möschwitzer, J.P. Drug nanocrystals in the commercial pharmaceutical development process. Int. J. Pharm. 2013, 453, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Yoe, Y. Nanocrystals for the parenteral delivery of poorly water-soluble drugs. Curr. Opin. Solid State Mater. Sci. 2012, 16, 295–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katteboinaa, S.; Chandrasekhar, P.; Balaji, S. Drug nanocrystals: A novel formulation approach for poorly soluble drugs. Int. J. PharmTech Res. 2009, 1, 682–694. [Google Scholar]
- Wang, Y.; Ma, Y.; Ma, Y.; Du, Y.; Liu, Z.; Zhang, D.; Zhang, Q. Formulation and pharmacokinetics evaluation of puerarin nanocrystals for intravenous delivery. J. Nanosci. Nanotechnol. 2012, 12, 6176–6184. [Google Scholar] [CrossRef]
- Gu, L.; Fang, R.H.; Sailor, M.J.; Park, J.-H. In Vivo clearance and toxicity of monodisperse iron oxide nanocrystals. ACS Nano 2012, 6, 4947–4954. [Google Scholar] [CrossRef] [Green Version]
- Mouton, J.; van Peer, A.; de Beule, K.; Van Vliet, A.; Donnelly, J.; Soons, P. Pharmacokinetics of itraconazole and OH-itraconazole as a nanocrystal formulation in healthy volunteers after single and multiple doses|Aspergillus & Aspergillosis Website. Antimicrob. Agents Chemother. 2006, 50, 4096–4102. [Google Scholar]
- Wei, L.; Ji, Y.; Gong, W.; Kang, Z.; Meng, M.; Zheng, A. Preparation, physical characterization and pharmacokinetic study of paclitaxel nanocrystals. Drug Dev. Ind. Pharm. 2015, 41, 1343–1352. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, Z.; Li, T.; McNally, H.; Park, K.; Sturek, M. Development and evaluation of transferrin-stabilized paclitaxel nanocrystal formulation. J. Control. Release 2015, 176, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Hollis, C.P.; Weiss, H.L.; Leggas, M.; Evers, B.M.; Gemeinhart, R.A.; Li, T. Biodistribution and bioimaging studies of hybrid paclitaxel nanocrystals: Lessons learned of the EPR effect and image-guided drug delivery. J. Control. Release Off. J. Control. Release Soc. 2013, 172, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, Y.; Zhang, J.; Hao, L.; Guo, H.; Lou, H.; Zhang, D. Bexarotene nanocrystal—Oral and parenteral formulation development, characterization and pharmacokinetic evaluation. Eur. J. Pharm. Biopharm. 2014, 87, 160–169. [Google Scholar] [CrossRef]
- Chiang, P.-C.; Ran, Y.; Chou, K.-J.; Cui, Y.; Wong, H. Investigation of utilization of nanosuspension formulation to enhance exposure of 1,3-dicyclohexylurea in rats: Preparation for PK/PD study via subcutaneous route of nanosuspension drug delivery. Nanoscale Res. Lett. 2011, 6, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, R.; Lu, W.; Li, J.; Wang, P.; Xu, R.; Chen, T. Preparation and characterization of intravenously injectable nimodipine nanosuspension. Int. J. Pharm. 2008, 350, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Junghanns, J.-U.A.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar]
- Yun, Y.H.; Lee, B.K.; Park, K. Controlled drug delivery: Historical perspective for the next generation. J. Control. Release 2015, 219, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Schwendeman, S.P.; Shah, R.B.; Bailey, B.A.; Schwendeman, A.S. Injectable controlled release depots for large molecules. J. Control. Release Off. J. Control. Release Soc. 2014, 190, 240–253. [Google Scholar] [CrossRef] [Green Version]
- Karode, N.P.; Prajapati, V.D.; Solanki, H.K.; Jani, G.K. Sustained release injectable formulations: Its rationale, recent progress and advancement. World J. Pharm. Pharm. Sci. 2015, 4, 702–722. [Google Scholar]
- Yang, W.-W.; Pierstorff, E. Reservoir-based polymer drug delivery systems. J. Lab. Autom. 2012, 17, 50–58. [Google Scholar] [CrossRef]
- Kalyani, M.; Surendra, P.; Sirisha, V. Parenteral controlled drug delivery system. Int. J. Res. Pharm. Nano Sci. 2013, 2, 572–580. [Google Scholar]
- Larsen, C.; Weng Larsen, S.; Jensen, H.; Jaghmur, A.; Ostergaard, J. Role of in vitro release models in formulation development and quality control of parenteral depots. Expert Opin. Drug Deliv. 2009, 6, 1283–1295. [Google Scholar] [CrossRef]
- Morais, J.M.; Burgess, D.J. Micro- and nanoemulsions (controlled release parenteral drug delivery systems). In Long Acting Injectables and Implants; Controlled Release Society, Springer: New York, NY, USA, 2012; pp. 221–238. [Google Scholar]
- McLennan, D.N.; Porter, C.J.; Charman, S.A. Subcotaneous drug delivery and the role of the lymphatics. Drug Discov. Today 2005, 2, 89–96. [Google Scholar] [CrossRef]
- Wei, X.-L.; Han, Y.-R.; Quan, L.-H.; Liu, C.-Y.; Liao, Y.-H. Oily nanosuspension for long-acting intramuscular delivery of curcumin didecanoate prodrug: Preparation, characterization and In Vivo evaluation. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2013, 49, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.W.; Østergaard, J.; Friberg-Johansen, H.; Jessen, M.N.B.; Larsen, C. In Vitro assessment of drug release rates from oil depot formulations intended for intra-articular administration. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2006, 29, 348–354. [Google Scholar] [CrossRef]
- Sartorius, G.; Fennell, C.; Spasevska, S.; Turner, L.; Conway, A.J.; Handelsman, D.J. Factors influencing time course of pain after depot oil intramuscular injection of testosterone undecanoate. Asian J. Androl. 2010, 12, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, Y.-S.; Park, C.-W.; DeLuca, P.; Mansour, H.M. Sustained-release injectable drug delivery. Pharm. Technol. 2010, 2010, 6. [Google Scholar]
- Jogala, S.; Rachamalla, S.S.; Aukunuru, J. Development of subcotaneous sustained release nanoparticles encapsulating low molecular weight heparin. J. Adv. Pharm. Technol. Res. 2015, 6, 58–64. [Google Scholar] [PubMed]
- Hassan, A.H.; Hosny, K.M.; Alhadlaq, A.; Alyamani, A.; Naguib, G. Depot injectable biodegradable nanoparticles loaded with recombinant human bone morphogenetic protein-2: Preparation, characterization, and In Vivo evaluation. Drug Des. Devel. Ther. 2015, 2015, 3599–3606. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wang, Y.; Chen, R.; Feng, C.; Yao, F.; Tong, S.; Wang, L.; Yamashita, F.; Yu, J. Formulation and pharmacokinetic evaluation of tetracycline-loaded solid lipid nanoparticles for subcotaneous injection in mice. Chem. Pharm. Bull. 2011, 59, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Suitthimeathegorn, O.; Turton, J.A.; Mizuuchi, H.; Florence, A.T. Intramuscular absorption and biodistribution of dexamethasone from non-aqueous emulsions in the rat. Int. J. Pharm. 2007, 331, 204–210. [Google Scholar] [CrossRef]
- Allen, T.M.; Hansen, C.B.; Peliowski, A. Subcutaneous administration of sterically stabilized (stealth) liposomes is an effective sustained release system for 1-β-d-arabinofuranosylcytosine. Drug Deliv. 1993, 1, 55–60. [Google Scholar] [CrossRef]
- Corvo, M.L.; Boerman, O.C.; Oyen, W.J.; Jorge, J.C.; Cruz, M.E.; Crommelin, D.J.; Storm, G. Subcutaneous administration of superoxide dismutase entrapped in long circulating liposomes: In Vivo fate and therapeutic activity in an inflammation model. Pharm. Res. 2000, 17, 600–606. [Google Scholar] [CrossRef]
- Freeling, J.P.; Koehn, J.; Shu, C.; Sun, J.; Ho, R.J. Long-acting three-drug combination anti-HIV nanoparticles enhance drug exposure in primate plasma and cells within lymph nodes and blood. AIDS 2014, 13, 2625–2627. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef]
- Liu, R.; Priestley, R.D. Rational design and fabrication of core–shell nanoparticles through a one-step/pot strategy. J. Mater. Chem. A 2016, 4, 6680–6692. [Google Scholar] [CrossRef]
- Mandal, B.; Bhattacharjee, H.; Mittal, N.; Sah, H.; Balabathula, P.; Thoma, L.A.; Wood, G.C. Core-shell-type lipid-polymer hybrid nanoparticles as a drug delivery platform. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 474–491. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wrezesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Limon, P.L.; et al. Combination delivery of TGF-b inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumor immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef]
- Chan, J.M.; Zhang, L.; Yuet, K.P.; Liao, G.; Rhee, J.-W.; Langer, R.; Farokhzad, O.C. PLGA-Lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials 2009, 30, 1627–1634. [Google Scholar] [CrossRef]
- Haidar, Z.S.; Hamdy, R.C.; Tabrizian, M. Protein release kinetics for cor-shell hybrid nanoparticles based on the layer-by-layer assembly of alginate and chitosan on liposomes. Biomaterials 2008, 29, 1207–1215. [Google Scholar] [CrossRef]
- Mittapelly, N.; Rachumallu, R.; Pandey, G.; Sharma, S.; Arya, A.; Bhatta, R.S.; Mishra, P.R. Investigation of the salt formation between memantine and pamoic acid: Its exploitation in nanocrystalline form as long-acting injection. Eur. J. Pharm. Biopharm. 2016, 101, 62–71. [Google Scholar] [CrossRef]
- Hu, L.; Yang, C.; Kong, D.; Gao, N.; Zhai, F. Development of a long-acting intramuscularly injectable formulation with nanosuspension of andrographolide. J. Drug Deliv. Sci. Technol. 2016, 35, 327–332. [Google Scholar] [CrossRef]
- Baert, L.; van Klooster, G.; Dries, W.; Francois, M.; Wouters, A.; Basstanie, E.; Iterbeke, K.; Stappers, F.; Stevens, P.; Schueller, L.; et al. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. Eur. J. Pharm. Biopharm. 2009, 72, 502–508. [Google Scholar] [CrossRef]
- Hollis, C.P. Nanocrystals of Chemotherapeutic Agents for Cancer Theranostics: Development and In Vitro and In Vivo Evaluation; University of Kentucky: Lexington, Kentucky, 2012. [Google Scholar]
- Kempe, S.; Mäder, K. In Situ forming implants—An attractive formulation principle for parenteral depot formulations. J. Control. Release Off. J. Control. Release Soc. 2012, 161, 668–679. [Google Scholar] [CrossRef]
- Hatefi, A.; Amsden, B. Biodegradable injectable in situ forming drug delivery systems. J. Control. Release Off. J. Control. Release Soc. 2002, 80, 9–28. [Google Scholar] [CrossRef]
- Thoniyot, P.; Tan, M.J.; Karim, A.A.; Young, D.J.; Loh, X.J. Nanoparticle–hydrogel composites: Concept, design, and applications of these promising, multi-functional materials. Adv. Sci. 2015, 2, 1400010. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, L.C. Current advances in sustained-release systems for parenteral drug delivery. Expert Opin. Drug Deliv. 2005, 2, 1039–1058. [Google Scholar] [CrossRef]
- Packhaeuser, C.B.; Kissel, T. On the design of in situ forming biodegradable parenteral depot systems based on insulin loaded dialkylaminoalkyl-amine-poly(vinyl alcohol)-g-poly(lactide-co-glycolide) nanoparticles. J. Control. Release Off. J. Control. Release Soc. 2007, 123, 131–140. [Google Scholar] [CrossRef]
- Yu, L.; Ding, J. Injectable hydrogels as unique biomedical materials. Chem. Soc. Rev. 2008, 37, 1473–1481. [Google Scholar] [CrossRef]
- Amin, S.; Rajabnezhad, S.; Kohli, K. Hydrogels as potential drug dleivery systems. Sci. Res. Essay 2009, 3, 1175–1183. [Google Scholar]
- Lin, C.-C.; Metters, A.T. Hydrogels in controlled release formulations: Network design and mathematical modeling. Adv. Drug Deliv. Rev. 2006, 58, 1379–1408. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Cholkar, K.; Mitra, A.K. Recent developments in protein and peptide parenteral delivery approaches. Ther. Deliv. 2014, 5, 337–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grijalvo, S.; Mayr, J.; Eritja, R.; Diaz Diaz, D. Biodegradable liposome-encapsulated hydrogels for biomedical applications: A marriage of convenience. Biomater. Sci. 2016, 4, 555–574. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Yao, D.; Ruiwei, G.; Deng, L.; Dong, A.; Zhang, J. Composites of polymer hydrogels and nanoparticulate systems for biomedical and pharmaceutical applications. Nanomaterials 2015, 5, 2054–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, G.P. Development and Evaluation of Novel In Situ Depot-Forming Controlled Release Formulations. Ph.D. Thesis, University of Missouri-Kensas City, Kansas City, MO, USA, 2006. [Google Scholar]
- Appel, E.A.; Tibbitt, M.W.; Webber, M.J.; Mattix, B.A.; Veiseh, O.; Langer, R. Self-assembled hydrogels utilizing polymer–nanoparticle interactions. Nat. Commun. 2015, 6, 6295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satarkar, N.S.; Biswal, D.; Hilt, J.Z. Hydrogel nanocomposites: A review of applications as remote controlled biomaterials. Soft Matter 2010, 6, 2364–2371. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, S.M.; Chen, P.P.; Cheng, L.; Zhou, W.; Tang, W.X.; Chen, Z.W.; Ke, C.M. Controlled release of insulin from PLGA nanoparticles embedded within PVA hydrogels. J. Mater. Sci. Mater. Med. 2007, 18, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Sun, X.; Gong, T.; Wu, C.-Y.; Zhang, T.; Tan, J.; Zhang, Z.-R. Injectable and biodegradable thermosensitive hydrogels loaded with PHBHHx nanoparticles for the sustained and controlled release of insulin. Acta Biomater. 2013, 9, 5063–5069. [Google Scholar] [CrossRef]
- Ciobanu, B.C.; Cadinoiu, A.N.; Popa, M.; Desbrieres, J.; Peptu, C.A. Modulated release from liposomes entapped in chitosan/gelatin hydrogels. Mater. Sci. Eng. C 2014, 43, 383–391. [Google Scholar] [CrossRef]
- Chung, Y.-I.; Ahn, K.-M.; Jeon, S.-Y.; Lee, J.-H.; Tae, G. Enhanced bone generation with BMP-2 loaded functional nanoparticle-hydrogel complex. J. Control. Release 2007, 121, 91–99. [Google Scholar] [CrossRef]
- Alinaghi, A.; Rouini, M.; Johari Daha, F.; Moghimi, H. Hydrogel-embeded vesicles as a novel approach for prolonged release and delivery of liposomes In Vitro and In Vivo. J. Liposome Res. 2013, 23, 235–243. [Google Scholar] [CrossRef]
- Dorraj, G.; Moghimi, H.R. Preparation of SLN-containing thermoresponsive in-situ Forming gel as a controlled nanoparticle delivery system and investigating its rheological, thermal and erosion behavior. Iran. J. Pharm. Res. Ijpr 2015, 14, 347–358. [Google Scholar]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef]
- Goncalves, C.; Pereira, P.; Gama, M. Self-assembled hydrogel nanoparticles for drug delivery. Materials 2010, 2, 1430–1460. [Google Scholar] [CrossRef] [Green Version]
- Arnfast, L.; Madsen, C.G.; Jorgensen, L.; Baldursdottir, S. Design and processing of nanogels as delivery systems for peptides and proteins. Ther. Deliv. 2014, 5, 691–708. [Google Scholar] [CrossRef]
- Akiyoshi, K.; Kobayashi, S.; Shichibe, S.; Mix, D.; Baudys, M.; Kim, S.W.; Sunamoto, J. Self-assembeled hydrogel nanoparticle of cholesterol-bearing pllulan as a carrier of protein drugs: Complexation and stabilization of insulin. J. Control. Release 1998, 54, 313–320. [Google Scholar] [CrossRef]
- Shimizu, T.; Kishida, T.; Hasegawa, U.; Ueda, Y.; Imanishi, J.; Yamagishi, H.; Akiyoshi, K.; Otsuji, E.; Mazda, O. Nanogel DDS enables sustained release of IL-12 for tumor immunotherapy. Biochem. Biophys. Res. Commun. 2008, 367, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Kitano, S.; Kageyama, S.; Nagata, Y.; Miyahara, Y.; Hiasa, A.; Naota, H.; Okumura, S.; Imai, H.; Shiraishi, T.; Masuya, M.; et al. HER2-specific T-cell immune responses in patients vaccinated with truncated HER2 protein complexed with nanogels of cholesteryl pullulan. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 7397–7405. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, S.; Kitano, S.; Hirayama, M.; Nagata, Y.; Imai, H.; Shiraishi, T.; Akiyoshi, K.; Scott, A.M.; Murphy, R.; Hoffman, E.W.; et al. Humoral immune responses in patients vaccinated with 1-146 HER2 protein complexed with cholesteryl pullulan nanogel. Cancer Sci. 2008, 99, 601–607. [Google Scholar] [CrossRef]
- Shimoda, A.; Yamamoto, Y.; Sawada, S.; Akiyoshi, K. Biodegradable nanogel-integrated hydrogels for sustained protein delivery. Macromol. Res. 2012, 20, 266–270. [Google Scholar] [CrossRef]
- Hoare, T.; Young, S.; Lawlor, M.W.; Kohane, D.S. Thermoresponsive nanogels for prolonged duration local anesthesia. Acta Biomater. 2012, 8, 3596–3605. [Google Scholar] [CrossRef] [Green Version]
- Blanco, M.D.; Guerrero, S.; Benito, M.; Jaghmur, A.; Teijon, C.; Olmo, R.; Katime, I.; Teijon, J.M. In Vitro and In Vivo evaluation of a folate-targeted copolymeric submicrohydrogel based on N-isopropylacrylamide as 5-fluorouracil delivery system. Polymers 2011, 3, 1107–1125. [Google Scholar] [CrossRef] [Green Version]
- Hirlekar, R.; Jain, S.; Patel, M.; Garse, H.; Kadam, V. Hexosomes: A novel drug delivery system. Curr. Drug Deliv. 2010, 7, 28–35. [Google Scholar] [CrossRef]
- Boyd, B.J.; Whittaker, D.V.; Khoo, S.-M.; Davey, G. Hexosomes formed from glycerate surfactants—Formulation as a colloidal carrier for irinotecan. Int. J. Pahrmaceutics 2006, 318, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Omray, L. Liquid crystals as novel vesicular delivery system: A review. Curr. Trends Technol. Sci. 2013, 2, 347–353. [Google Scholar]
- Boyd, B.J.; Whittaker, D.V.; Khoo, S.-M.; Davey, G. Lyotropic liquid crystalline phases formed from glycerate surfactants as sustained release drug delivery systems. Int. J. Pharm. 2006, 309, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Phan, S.; Fong, W.-K.; Kirby, N.; Hanley, T.; Boyd, B.J. Evaluating the link between self-assembled mesophase structure and drug release. Int. J. Pharm. 2011, 421, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Couffin-Hoarau, A.-C.; Motulsky, A.; Delmas, P.; Leroux, J.-C. In Situ-forming pharmaceutical organogels based on the self-assembly of L-alanine derivatives. Pharm. Res. 2004, 21, 454–457. [Google Scholar] [CrossRef]
- Angelova, A.; Angelov, B.; Mutafchieva, R.; Lesieur, S.; Couvreur, P. Self-assembled multicompartment liquid crystalline lipid carriers for protein, peptide, and nucleic acid drug delivery. Acc. Chem. Res. 2011, 44, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ma, P.; Gui, S. Cubic and hexagonal liquid crystals as drug delivery systems. BioMed Res. Int. 2014, 2014, 815981. [Google Scholar] [CrossRef]
- Ki, M.-H.; Lim, J.-L.; Ko, J.-Y.; Park, S.-H.; Kim, J.-E.; Cho, H.-J.; Park, E.-S.; Kim, D.-D. A new injectable liquid crystal system for one monthe delivery of leuprolide. J. Control. Release 2014, 185, 62–70. [Google Scholar] [CrossRef]
- Rizwan, S.; McBurney, W.; Young, K.; Hanley, T.; Boyd, B.J.; Rades, T.; Hook, S. Cubosomes containing adjuvants imiquimod and monophosphoryl lipid A stimulate robust cellular and humoral immune responses. J. Control. Release 2013, 165, 16–21. [Google Scholar] [CrossRef]
- Nasr, M.; Ghorab, M.K.; Abdelazem, A. In Vitro and In Vivo evaluation of cubosomes containing 5-fluorouracil for liver targetting. Acta Pharm. Sin. B 2015, 5, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Luo, L.; Zheng, S.; Niu, Y.; Bo, R.; Huang, Y.; Xing, J.; Li, Z.; Wang, D. Cubosome nanoparticles potentiate immune properties of immunostimulants. Int. J. Nanomed. 2016, 11, 3571–3583. [Google Scholar]
- Jogala, S.; Rachamalla, S.S.; Aukunuru, J. Development of PEG-PLGA based intravenous low molecular wight heparin (LMWH) nanoparticles intended to treat venous thrombosis. Curr. Drug Deliv. 2016, 13, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pei, Y.; Zhang, X.; Gu, Z.; Zhou, Z.; Yuan, W.; Zhou, J.; Zhu, J.; Gao, X. PEGylated PLGA nanoparticles as protein carriers: Synthesis, preparation and biodistribution in rats. J. Control. Release Off. J. Control. Release Soc. 2001, 71, 203–211. [Google Scholar] [CrossRef]
- Mei, B.; Jiang, H.; Tjandra, H.; Strauss, J.; Chen, Y.; Liu, T.; Zhang, X.; Severs, J.; Newgern, J.; Chen, J.; et al. Rational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatment. Blood 2010, 116, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Yao, C.; Wang, L.; Min, W.; Xu, J.; Xiao, J.; Huang, M.; Chen, B.; Liu, B.; Li, X.; et al. An albumin-conjugated peptide exhibits potent anti-HIV activity and long in vivo half-life. Antimicrob. Agents Chemother. 2010, 54, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Liechty, W.B.; Peppas, N.A. Expert opinion: Responsive polymer nanoparticles in cancer therapy. Eur. J. Pharm. Biopharm. 2012, 80, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release Off. J. Control. Release Soc. 2008, 126, 187–204. [Google Scholar] [CrossRef]
- MacEwan, S.R.; Callahan, D.J.; Chilkoti, A. Stimulus-responsive macromolecules and nanoparticles for cancer drug delivery. Nanomedicine 2010, 5, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef]
- Bennet, D.; Kim, S. Polymer nanoparticles for smart drug delivery. In Application of Nanotechnology in Drug Delivery; InTech: London, UK, 2014. [Google Scholar]
- Shao, P.; Wang, B.; Yazhou, W.; Li, J.; Zhnag, Y. The application of thermosensitive nanocarriers in controlled drug delivery. J. Nanomater. 2011, 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-C.; Kim, M.-S.; Kim, J.-D. Temperature-sensitive releases from liposomes containing hydrophobically modified poly(N-isopropylacrylamide). Korean J. Chem. Eng. 1999, 16, 28–33. [Google Scholar] [CrossRef]
- Kono, K.; Ozawa, T.; Yoshida, T.; Ozaki, F.; Ishizaka, Y.; Maruyama, K.; Kojima, C.; Harada, A.; Aoshima, S. Highly temperature-sensitive liposomes based on a thermosensitive block copolymer for tumor-specific chemotherapy. Biomaterials 2010, 31, 7096–7105. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Sahandi Zangabad, P.; Ghasemi, A.; Amiri, M.; Bahrami, M.; Malekzad, H.; Ghahramanzadeh Asl, H.; Mahdieh, Z.; Bozorgomid, M.; Ghasemi, A.; et al. Temperature-responsive smart nanocarriers for the delivery of therapeutic agents: Applications and recent advances. ASC Appl. Mater. Interfaces 2016, 8, 21107–21133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokardekar, R.R.; Shah, V.; Mody, H.R. PNIPAM poly (N-isopropylacrylamide): A thermosensitive “smart” polymer in novel drug delivery systems. Internet J. Med. Update 2012, 7, 60–63. [Google Scholar]
- Pang, C.-L.; Tsai, H.-M.; Yang, S.-J.; Luo, T.-Y.; Lin, C.-F.; Lin, W.-J.; Shieh, M.-J. Development of thermosensitive poly(n-isopropylacrylamide-co-((2-dimethylamino)ethyl methacrylate))-based nanoparticles for controlled drug release. Nanotechnology 2011, 22, 11. [Google Scholar] [CrossRef]
- Luo, Y.-L.; Yu, W.; Xu, F.; Zhang, L.-L. Novel thermo-responsive self-assembly micelles from a double brush-shaped PNIPAM-g-(PA-b-PEG-b-PA)-g-PNIPAM block copolymer with PNIPAM polymers as side chains. J. Polym. Sci. Part Polym. Chem. 2012, 50, 2053–2067. [Google Scholar] [CrossRef]
- Hamner, K.L.; Maye, M.M. Thermal aggregation properties of nanoparticles modified with temperature sensitive copolymers. Langmuir 2013, 29, 15217–15223. [Google Scholar] [CrossRef]
- Tamarov, K.; Xu, W.; Osminkina, L.; Zinovyev, S.; Soininen, P.; Kurdyavtsev, A.; Gaydarova, M.; Närvänen, A.; Timoshenko, V.; Lehto, V.-P. Temperature responsive porous silicon nanoparticles for cancer therapy-spatiotemporal triggering through infrared and radiofrequency electromagnetic heating. J. Control. Release 2016, 241, 220–228. [Google Scholar] [CrossRef]
- Han, H.D.; Shin, B.C.; Choi, H.S. Doxorubicin-encapsulated thermosensitive liposomes modified with poly(N-isopropylacrylamide-co-acrylamide): Drug release behavior and stability in the presence of serum. Eur. J. Pharm. Biopharm. 2006, 62, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Shirakura, T.; Kelson, T.J.; Ray, A.; Malyarenko, A.E.; Kopelman, R. Hydrogel nanoparticles with thermally controlled drug release. ACS Macro Lett. 2014, 3, 602–606. [Google Scholar] [CrossRef] [Green Version]
- Kneidl, B.; Peller, M.; Winter, G.; Lindner, L.H.; Hossann, M. Thermosensitive liposomal drug delivery systems: State of the art review. Int. J. Nanomed. 2014, 9, 4387–4398. [Google Scholar]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Ta, T.; Porter, T.M. Thermosensetive liposomes for localized delivery and triggered release of chemotherapy. J. Control. Release 2013, 169, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-Y.; Zhang, H.; Yang, Y.; Xie, X.-Y.; Yang, Y.-F.; Li, Z.; Li, Y.; Gong, W.; Yu, F.-L.; Yang, Z.; et al. Preparation, characterization, and efficacy of thermosensitive liposomes containing paclitaxel. Drug Deliv. 2016, 23, 1222–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, J.P.; Ernsting, M.J.; Undzys, E.; Li, S.-D. Thermosensitive liposomes for the delivery of gemcitabine and oxaliplatin to tumors. Mol. Pharm. 2013, 10, 4499–4508. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Yu, F.; Yang, Y.; Cheng, X.; Liu, Y.; Zhang, H.; Zhao, S.; Yang, Z.; Li, M.; Li, Z.; et al. Preparation and evaluation of oxaliplatin thermosensitive liposomes with rapid release and high stability. PLoS ONE 2016, 11, e0158517. [Google Scholar] [CrossRef]
- Stover, T.C.; Kim, Y.S.; Lowe, T.L.; Kester, M. Thermoresponsive and biodegradable linear-dendritic nanoparticles for targeted and sustained release of a pro-apoptotic drug. Biomaterials 2008, 29, 359–369. [Google Scholar] [CrossRef]
- Yan, B.; Boyer, J.-C.; Habault, D.; Branda, N.R.; Zhao, Y. Near infrared light triggered release of biomacromolecules from hydrogels loaded with upconversion nanoparticles. J. Am. Chem. Soc. 2012, 134, 16558–16561. [Google Scholar] [CrossRef]
- Fomina, N.; Sankaranarayanan, J.; Almutairi, A. Photochemical mechanisms of light-triggered release from nanocarriers. Adv. Drug Deliv. Rev. 2012, 64, 1005–1020. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Lorenzo, C.; Bromberg, L.; Concheiro, A. Light-sensitive intelligent drug delivery systems. Photochem. Photobiol. 2009, 85, 848–860. [Google Scholar] [CrossRef]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.; Shin, E.; Kim, B.-S. Light-responsive micelles of spiropyran initiated hyperbranched polyglycerol for smart drug delivery. Biomacromolecules 2014, 15, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, N.; Iriyama, A.; Jang, W.-D.; Miyata, K.; Itaka, K.; Inoue, Y.; Takahashi, H.; Yanagi, Y.; Tamaki, Y.; Koyama, H.; et al. Light-induced gene transfer from packaged DNA enveloped in a dendrimeric photosensitizer. Nat. Mater. 2005, 4, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Rijcken, C.J.F.; Soga, O.; Hennink, W.E.; van Nostrum, C.F. Triggered destabilisation of polymeric micelles and vesicles by changing polymers polarity: An attractive tool for drug delivery. J. Control. Release Off. J. Control. Release Soc. 2007, 120, 131–148. [Google Scholar] [CrossRef]
- He, J.; Tong, X.; Zhao, Y. Photoresponsive nanogels based on photocontrollable cross-links. Macromolecules 2009, 42, 4845–4852. [Google Scholar] [CrossRef]
- Paasonen, L.; Laaksonen, T.; Johans, C.; Yliperttula, M.; Kontturi, K.; Urtti, A. Gold nanoparticles enable selective light-induced contents release from liposomes. J. Control. Release Off. J. Control. Release Soc. 2007, 122, 86–93. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Jiang, C. Charge-reversal nanoparticles: Novel targeted drug delivery carriers. Acta Pharm. Sin. B 2016, 4, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Yao, R.; Ding, B.; Shen, Y.; Shui, S.; Wang, L.; Li, Y.; Yang, X.; Tao, W. Fabrication of upconverting hybrid nanoparticles for near-infrared light triggered drug release. Adv. Mater. Sci. Eng. 2014, 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Volodkin, D.V.; Skirtach, A.G.; Möhwald, H. Near-IR remote release from assemblies of liposomes and nanoparticles. Angew. Chem. Int. Ed. 2009, 48, 1807–1809. [Google Scholar] [CrossRef]
- Jayakumar, M.K.G.; Idris, N.M.; Zhang, Y. Remote activation of biomolecules in deep tissues using near-infrared-to-UV upconversion nanotransducers. Proc. Natl. Acad. Sci. USA 2012, 109, 8483–8488. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, L.; Wang, J.; Jiang, X.; Li, X.; Hu, Z.; Ji, Y.; Wu, X.; Ceh, C. Mesoporous silica-coated gold nanorods as a light-mediated multifunctional theranostic platform for cancer treatment. Adv. Mater. 2012, 24, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Thambi, T.; Deepagan, V.G.; Yoon, H.Y.; Han, H.S.; Kim, S.-H.; Son, S.; Jo, D.-G.; Ahn, C.-H.; Suh, Y.D.; Kim, K.; et al. Hypoxia-responsive polymeric nanoparticles for tumor-targeted drug delivery. Biomaterials 2014, 35, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Haldar, M.K.; You, S.; Choi, Y.; Mallik, S. Hypoxia-responsive polymersomes for drug delivery to hypoxic pancreatic cancer cells. Biomacromolecules 2016, 17, 2507–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lue, Y.; Aimmetti, A.A.; Langer, R.; Gu, Z. Bioresponsive materials. Nat. Rev. Mater. 2016, 1, 17. [Google Scholar] [CrossRef]
- Zhang, R.; Li, Y.; Zhang, M.; Tang, Q.; Zhang, X. Hypoxia-responsive drug-drug conjugated nanoparticles for breast cancer synergistic therapy. Recent Adv. 2016, 6, 30268–30276. [Google Scholar] [CrossRef]
- Kulkarni, P.; Haldar, M.K.; Katti, P.; Dawes, C.; You, S.; Choi, Y.; Mallik, S. Hypoxia responsive, tumor penetrating lipid nanopyrticles for delivery of chemotherapeutics to pancreatic cancer cell spheroids. Bioconjug. Chem. 2016, 27, 1830–1838. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, R.; Niu, Y.; Qiao, C.; Weng, J.; Wang, X.; Xiao, Z.; Zhang, X. Development of hypoxia triggered prodrug micelles as doxorubicin carriers for tumor therapy. RSC Adv. 2015, 5, 20848–20857. [Google Scholar] [CrossRef]
- Aldea, M.; Florian, I.A.; Kacso, G.; Craciun, L.; Boca, S.; Soritau, O.; Florian, I.S. Nanoparticles for targeting intratumoral hypoxia: Exploiting a potential weakness of glioblasoma. Pharm. Res. 2016, 33, 2059–2077. [Google Scholar] [CrossRef]
- Qian, C.; Yu, J.; Chen, Y.; Hu, Q.; Xiao, X.; Sun, W.; Wan, C.; Feng, P.; Shen, Q.-D.; Gu, Z. Light-activated hypoxia-responsive nanocarriers for enhanced anticancer therapy. Adv. Mater. 2016, 28, 3313–3320. [Google Scholar] [CrossRef]
- Gao, W.; Chan, J.M.; Farokhzad, O.C. pH-Responsive nanoparticles for drug delivery. Mol. Pharm. 2010, 7, 1913–1920. [Google Scholar] [CrossRef]
- Shen, Y.; Tang, H.; Radosz, M.; Van Kirk, E.; Murdoch, W.J. pH-Responsive nanoparticles for cancer drug delivery. In Drug Delievery Systems; Humana Press: Totowa, NJ, USA, 2008; pp. 183–216. [Google Scholar]
- Chang, G.; Li, C.; Lu, W.; Ding, J. N-Boc-histidine-capped PLGA-PEG-PLGA as a smart polymer for drug delivery sensitive to tumor extracellular pH. Macromol. Biosci. 2010, 10, 1248–1256. [Google Scholar] [CrossRef]
- Ko, J.; Park, K.; Kim, Y.-S.; Kim, M.S.; Han, J.K.; Kim, K.; Park, R.-W.; Kim, I.-S.; Song, H.K.; Lee, D.S.; et al. Tumoral acidic extracellular pH targeting of pH-responsive MPEG-poly(beta-amino ester) block copolymer micelles for cancer therapy. J. Control. Release Off. J. Control. Release Soc. 2007, 123, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Poon, Z.; Chang, D.; Zhao, X.; Hammond, P.T. Layer-by-layer nanoparticles with a pH-sheddable layer for in vivo targeting of tumor hypoxia. ACS Nano 2011, 5, 4284–4292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.H.; Ramasamy, T.; Choi, J.Y.; Nguyen, H.T.; Pham, T.T.; Jeong, J.-H.; Ku, S.K.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Tumor-targeting, pH-sensitive nanoparticles for docetaxel delivery to drug-resistant cancer cells. Int. J. Nanomed. 2015, 10, 5249–5262. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Zhong, Y.; Cheng, R.; Deng, C.; Zhong, Z. pH-sensitive polymeric nanoparticles for tumor-targeting doxorubicin delivery: Concept and recent advances. Nanomedicine 2014, 9, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, M.J.; Murthy, N. Polyketal nanoparticles: A new pH-sensitive biodegradable drug delivery vehicle. Bioconjug. Chem. 2005, 16, 1340–1342. [Google Scholar] [CrossRef]
- Qiu, L.; Hu, Q.; Cheng, L.; Li, L.; Tian, C.; Chen, W.; Chen, Q.; Hu, W.; Xu, L.; Yang, J.; et al. cRGDyK modified pH responsive nanoparticles for specific intracellular delivery of doxorubicin. Acta Biomater. 2016, 30, 285–298. [Google Scholar] [CrossRef]
- Kanamala, M.; Wilson, W.R.; Yang, M.; Palmer, B.D.; Wu, Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review. Biomaterials 2016, 85, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-Z.; Du, X.-J.; Mao, C.-Q.; Wang, J. Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery. J. Am. Chem. Soc. 2011, 133, 17560–17563. [Google Scholar] [CrossRef]
- She, W.; Luo, K.; Zhang, C.; Wang, G.; Geng, Y.; Li, L.; He, B.; Gu, Z. The potential of self-assembled, pH-responsive nanoparticles of mPEGylated peptide dendron-doxorubicin conjugates for cancer therapy. Biomaterials 2013, 34, 1613–1623. [Google Scholar] [CrossRef]
- He, H.; Chen, S.; Zhou, J.; Dou, Y.; Song, L.; Che, L.; Zhou, X.; Chen, X.; Jia, Y.; Zhang, J.; et al. Cyclodextrin-derived pH-responsive nanoparticles for delivery of paclitaxel. Biomaterials 2013, 34, 5344–5358. [Google Scholar] [CrossRef] [PubMed]
- Aryal, S.; Hu, C.-M.J.; Zhang, L. Polymer—Cisplatin conjugate nanoparticles for acid-responsive drug delivery. ACS Nano 2010, 4, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-H.; Cheng, S.-H.; Huang, I.-P.; Souris, J.S.; Yang, C.-S.; Mou, C.-Y.; Lo, L.-W. Intracellular pH-responsive mesoporous silica nanoparticles for the controlled release of anticancer chemotherapeutics. Angew. Chem. Int. Ed Engl. 2010, 122, 8390–8395. [Google Scholar] [CrossRef]
- Zhao, H.; Duong, H.H.P.; Yung, L.Y.L. Folate-conjugated polymer micelles with pH-triggered drug release properties. Macromol. Rapid Commun. 2010, 31, 1163–1169. [Google Scholar] [CrossRef]
- Duan, C.; Gao, J.; Zhang, D.; Jia, L.; Liu, Y.; Zheng, D.; Liu, G.; Tian, X.; Wang, F.; Zhang, Q. Galactose-decorated pH-responsive nanogels for hepatoma-targeted delivery of oridonin. Biomacromolecules 2011, 12, 4335–4343. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Wu, H.; Zhang, H.; Li, F.; Yang, T.; Gu, C.; Yang, Q. Novel super pH-sensitive nanoparticles responsive to tumor extracellular pH. Carbohydr. Polym. 2008, 73, 390–400. [Google Scholar] [CrossRef]
- Wu, X.L.; Kim, J.H.; Koo, H.; Bae, S.M.; Shin, H.; Kim, M.S.; Lee, B.-H.; Park, R.-W.; Kim, I.-S.; Choi, K.; et al. Tumor-targeting peptide conjugated pH-responsive micelles as a potential drug carrier for cancer therapy. Bioconjug. Chem. 2010, 21, 208–213. [Google Scholar] [CrossRef]
- Oh, K.; Oh, Y.; Oh, N.; Kim, K.; Lee, E. A smart flower-like polymeric micelle for pH-triggered anticancer drug release. Int. J. Pahrmaceutics 2009, 375, 163–169. [Google Scholar] [CrossRef]
- Filippov, S.; Hrubý, M.; Koňák, Č.; Macková, H.; Špírková, M.; Štěpánek, P. Novel pH-responsive nanoparticles. Langmuir 2008, 24, 9295–9301. [Google Scholar] [CrossRef]
- Andrieu, J. Specific Enzymatic Cleavage and Payload Release from Peptide-Based Hybrid Nanocapsules. Ph.D. Thesis, Johannes Gutenberg-Universität, Mainz, Germany, 2011. [Google Scholar]
- Park, H.-K.; Lee, S.J.; Oh, J.-S.; Lee, S.-G.; Jeong, Y.-I.; Lee, H.C. Smart nanoparticles based on hyaluronic acid for redox-responsive and CD44 receptor-mediated targeting of tumor. Nanoscale Res. Lett. 2015, 10, 981. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Biswas, A.; Hu, B.; Joo, K.-I.; Wang, P.; Gu, Z.; Tang, Y. Redox-responsive nanocapsules for intracellular protein delivery. Biomaterials 2011, 32, 5223–5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuan, X.; Song, Q.; Lin, J.; Chen, X.; Zhang, H.; Dai, W.; He, B.; Wang, X.; Zhang, Q. Novel free-paclitaxel-loaded redox-responsive nanoparticles based on a disulfide-linked poly(ethylene glycol)−drug conjugate for intracellular drug delivery: Synthesis, characterization, and antitumor activity In Vitro and In Vivo. Mol. Pharm. 2014, 11, 2656–3670. [Google Scholar] [CrossRef]
- Gong, H.; Xie, Z.; Liu, M.; Sun, H.; Zhu, H.; Guo, H. Research on redox-responsive mesoporous silica nanoparticles functionalized with PEG via a disulfide bond linker as drug carrier materials. Colloid Polym. Sci. 2015, 293, 2121–2128. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, J.; Sun, Q.; Murdoch, W.J.; Shen, Y. A multifunctional PEG–PLL drug conjugate forming redox-responsive nanoparticles for intracellular drug delivery. J. Mater. Chem. B 2015, 3, 7594–7603. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Cai, K.; Hu, Y.; Li, J.; Ding, X.; Zhang, B.; Xu, D.; Yang, W.; Liu, P. Redox-responsive molecular nanoreservoirs for controlled intracellular anticancer drug delivery based on magnetic nanoparticles. Adv. Mater. 2012, 24, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Ariza, R.; Kronig, G.A.; Huskens, J. Size-controlled and redox-responsive supramolecular nanoparticles. Beilstein J. Org. Chem. 2015, 11, 2388–2399. [Google Scholar] [CrossRef]
- Hu, X.-Y.; Chen, Y.; Liu, Y. Redox-responsive supramolecular nanoparticles based on amphiphilic sulfonatocalixarene and selenocystamine dihydrochloride. Chin. Chem. Lett. 2015, 26, 862–866. [Google Scholar] [CrossRef]
- Wen, H.; Li, Y. Redox sensitive nanoparticles with disulfide bond linked sheddable shell for intracellular drug delivery. Med. Chem. 2014, 4, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Liu, W.; Tu, Q.; Liu, R.; Zhang, Y.; Wang, J. Preparation and in vitro properties of redox-responsive polymeric nanoparticles for paclitaxel delivery. Colloids Surf. B Biointerfaces 2011, 87, 454–463. [Google Scholar] [CrossRef]
- Xu, J.; Singh, A.; Amiji, M.M. Redox-responsive targeted gelatin nanoparticles for delivery of combination wt-p53 expressing plasmid DNA and gemcitabine in the treatment of pancreatic cancer. BMC Cancer 2014, 14, 75. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Berríos, M.P.; Vivero-Escoto, J.L. In Vitro evaluation of folic acid-conjugated redox-responsive mesoporous silica nanoparticles for the delivery of cisplatin. Int. J. Nanomed. 2016, 11, 6251–6265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Rica, R.; Aili, D.; Stevens, M.M. Enzyme-responsive nanoparticles for drug release and diagnostics. Adv. Drug Deliv. Rev. 2012, 64, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Callmann, C.E. Therapeutic enzyme-responsive nanoparticles for targeted delivery and accumulation in tumors. Adv. Mater. 2015, 27, 4611–4615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.M.; Carlini, A.S.; Chien, M.-P.; Sonnenberg, S.; Luo, C.; Braden, R.L.; Osborn, K.G.; Li, Y.; Gianneschi, N.C.; Christman, K.L. Enzyme-responsive nanoparticles for targeted accumulation and prolonged retention in heart tissue after myocardial infarction. Adv. Mater. 2015, 27, 5547–5552. [Google Scholar] [CrossRef] [PubMed]
- Insua, I.; Liamas, E.; Zhang, Z.; Peacock, A.F.; Krachler, A.M.; Fernandez-Trillo, F. Enzyme-responsive polyion complex (PIC) nanoparticles for the targeted delivery of antimicrobial polymers. Polym. Chem. 2016, 7, 2684–2690. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Katti, P.; Gu, Z. Enzyme-responsive destabilization of stabilized plasmid-lipid nanoparticles as an efficient gene delivery. Nanoscale 2014, 6, 12273–12286. [Google Scholar] [CrossRef]
- Choi, K.Y.; Swierczewska, M.; Lee, S.; Chen, X. Protease-activated drug development. Theranostics 2012, 2, 156–178. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Jeong, Y.-I.; Park, H.-K.; Kang, D.H.; Oh, J.-S.; Lee, S.-G.; Lee, H.C. Enzyme-responsive doxorubicin release from dendrimer nanoparticles for anticancer drug delivery. Int. J. Nanomed. 2015, 10, 5489–5503. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Aw, J.; Chen, K.; Liu, F.; Padmanabhan, P.; Hou, Y.; Cheng, Z.; Xing, B. Enzyme-responsive multifunctional magnetic nanoparticles for tumor intracellular drug delivery and imaging. Chem. Asian J. 2011, 6, 1381–1389. [Google Scholar] [CrossRef]
- Hou, X.-F.; Chen, Y.; Liu, Y. Enzyme-responsive protein/polysaccharide supramolecular nanoparticles. Soft Matter 2015, 11, 2488–2493. [Google Scholar] [CrossRef]
- Bernardos, A.; Mondragon, L.; Aznar, E.; Marcos, M.D.; Martinez-Mañez, R.; Sancenon, F.; Soto, J.; Barat, J.M.; Perez-Paya, E.; Guillem, C.; et al. Enzyme-responsive intracellular controlled release using nanometric silica mesoporous supports capped with “saccharides”. ACS Nano 2010, 4, 6353–6368. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Angelos, S.; Dichtel, W.R.; Coskun, A.; Yang, Y.-W.; Zink, J.I.; Stoddart, J.F. Enzyme-responsive snap-top covered silica nanocontainers. J. Am. Chem. Soc. 2008, 130, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basel, M.T.; Shrestha, T.B.; Troyer, D.L.; Bossmann, S.H. Protease-sensitive, polymer-caged liposomes: A method for making highly targeted liposomes using triggered release. ACS Nano 2011, 5, 2162–2175. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Hosseini-Nassab, N.; Zare, R.N. Electroresponsive nanoparticles for drug delivery on demand. Nanoscale 2016, 8, 9310–9317. [Google Scholar] [CrossRef]
- Ge, J.; Neofytou, E.; Cahill, T.J.; Beygui, R.E.; Zare, R.N. Drug release from electric-field-responsive nanoparticles. ACS Nano 2012, 6, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhu, Y.; Wang, Y. Dual-responsive drug delivery system with real time tunable release behavior. Microporous Mesoporous Mater. 2014, 200, 46–51. [Google Scholar] [CrossRef]
- Yan, Q.; Yuan, J.; Cai, Z.; Xin, Y.; Kang, Y.; Yin, Y. Voltage-responsive vesicles based on orthogonal assembly of two homopolymers. J. Am. Chem. Soc. 2010, 132, 9268–9270. [Google Scholar] [CrossRef]
- Ying, X.; Wang, Y.; Liang, J.; Yue, J.; Xu, C.; Lu, L.; Xu, Z.; Gao, J.; Du, Y.; Chen, Z. Angiopep-conjugated electro-responsive hydrogel nanoparticles: Therapeutic potential for epilepsy. Angew. Chem. 2014, 53, 12436–12440. [Google Scholar] [CrossRef]
- Wang, Y.; Ying, X.; Chen, L.; Liu, Y.; Wang, Y.; Liang, J.; Xu, C.; Guo, Y.; Wang, S.; Hu, W.; et al. Electroresponsive nanoparticles improve antiseizure effect of phenytoin in generalized tonic-clonic seizures. Neurotherapeutics 2016, 13, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Pankhurst, Q.A.; Connolly, J.; Jones, S.K.; Dobson, J. Applications of magnetic nanoparticles in biomedicine. J. Phys. D Appl. Phys. 2003, 36, R167. [Google Scholar] [CrossRef] [Green Version]
- Bucak, S.; Yavuztürk, B.; Sezer, A.D. Magnetic nanoparticles: Synthesis, surface modifications and application in drug delivery. In Recent Advances in Nivel Drug Carrier System; InTech: London, UK, 2012; pp. 165–200. [Google Scholar]
- McBain, S.C.; Yiu, H.H.; Dobson, J. Magnetic nanoparticles for gene and drug delivery. Int. J. Nanomed. 2008, 3, 169–180. [Google Scholar]
- Estelrich, J.; Escribano, E.; Queralt, J.; Busquets, M.A. Iron oxide nanoparticles for magnetically-guided and magnetically-responsive drug delivery. Int. J. Mol. Sci. 2015, 16, 8070–8101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Asempah, I.; Laurent, S.; Fornara, A.; Muller, R.N.; Muhammed, M. Injectable superparamagnetic ferrogels for controlled release of hydrophobic drugs. Adv. Mater. 2009, 21, 1354–1357. [Google Scholar] [CrossRef]
- Kumar, C.S.S.R.; Mohammad, F. Magnetic nanomaterials for hyperthermia-based therapy and controlled drug delivery. Adv. Drug Deliv. Rev. 2011, 63, 789–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monnier, C.A.; Burnand, D.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Magnetoliposomes: Opportunities and challenges. Eur. J. Nanomed. 2014, 6, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.D.; Zhang, W.; Lee, J.H.; Brammer, K.; Lal, R.; Karin, M.; Jin, S. Magnetically vectored nanocapsules for tumor penetration and remotely switchable on-demand drug release. Nano Lett. 2010, 10, 5088–5092. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.-Y.; Hu, S.-H.; Liu, K.-H.; Shaiu, R.-S.; Liu, D.-M.; Chen, S.-Y. Instantaneous drug delivery of magnetic/thermally sensitive nanospheres by a high-frequency magnetic field. Langmuir ACS J. Surf. Colloids 2008, 24, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Giri, S.; Trewyn, B.G.; Stellmaker, M.P.; Lin, V.S.-Y. Stimuli-responsive controlled-release delivery system based on mesoporous silica nanorods capped with magnetic nanoparticles. Angew. Chem. Int. Ed Engl. 2005, 44, 5038–5044. [Google Scholar] [CrossRef]
- Ruiz-Hernández, E.; Baeza, A.; Vallet-Regí, M. Smart drug delivery through DNA/magnetic nanoparticle gates. ACS Nano 2011, 5, 1259–1266. [Google Scholar] [CrossRef]
- Hayashi, K.; Ono, K.; Suzuki, H.; Sawada, M.; Moriya, M.; Sakamoto, W.; Yogo, T. High-frequency, magnetic-field-responsive drug release from magnetic nanoparticle/organic hybrid based on hyperthermic effect. ACS Appl. Mater. Interfaces 2010, 2, 1903–1911. [Google Scholar] [CrossRef]
- Tan, S.; Wang, G. Redox-responsive and pH-sensitive nanoparticles enhanced stability and anticancer ability of erlotinib to treat lung cancer In Vivo. Drug Des. Devel. Ther. 2017, 11, 3519–3529. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, S.; Liu, J.; Liu, Z.; Su, L.; Wang, H.; Chang, J. pH- and reduction-responsive polymeric lipid vesicles for enhanced tumor cellular internalization and triggered drug release. ACS Appl. Mater. Interfaces 2014, 6, 10706–10713. [Google Scholar] [CrossRef] [PubMed]
- Unsoy, G.; Khodadust, R.; Yalcin, S.; Mutlu, P.; Gunduz, U. Synthesis of Doxorubicin loaded magnetic chitosan nanoparticles for pH responsive targeted drug delivery. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2014, 62, 243–250. [Google Scholar] [CrossRef]
- Klaikherd, A.; Nagamani, C.; Thayumanavan, S. Multi-stimuli sensitive amphiphilic block copolymer assemblies. J. Am. Chem. Soc. 2009, 131, 4830–4838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Wu, H.; Dong, J.; Wang, G. Quadruple-stimuli-sensitive polymeric nanocarriers for controlled release under combined stimulation. Macromolecules 2014, 47, 8777–8783. [Google Scholar] [CrossRef]
- Wen, J.; Sun, S. Recent advances of multi-stimuli-responsive drug delivery systems for cancer therapy. Curr. Trends Biomed. Eng. Biosci. 2017, 3, 5555607. [Google Scholar]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Weissig, V.; Guzman-Villanueva, D. Nanopharmaceuticals (part 2): Products in the pipeline. Int. J. Nanomed. 2015, 10, 1245–1257. [Google Scholar] [CrossRef] [Green Version]
- Lammers, T.; Kiessling, F.; Ashford, M.; Hennink, W.; Crommelin, D.; Storm, G. Cancer nanomedicine: Is targeting our target? Nat. Rev. Mater. 2016, 1, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Ojha, T.; Kiessling, F.; Lammers, T.; Shi, Y. Enhancing tumor penetration of nanomedicines. Biomacromolecules 2017, 18, 1449–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, A.; Liu, M.; Ojha, T.; Storm, G.; Kiessling, F.; Lammers, T. Ultrasound-mediated drug delivery to the brain: Principles, progress and prospects. Drug Discov. Today Technol. 2016, 20, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, S. Understanding and overcoming major barriers in cancer nanomedicine. Nanomedicine 2010, 5, 523–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammam, S.; Azzazy, H.M.; Lamprecht, A. Biodegradable particulate carrier formulation and tuning for targeted drug delivery. J. Biomed. Nanotechnol. 2015, 11, 555–577. [Google Scholar] [CrossRef]
- Li, S.-D.; Huang, L. Nanoparticles evading the reticuloendotherlial system: Role of the supported bilayer. Biochim. Biophys. Acta BBA Biomembr. 2009, 1788, 2259–2266. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.M.; Juettner, V.V.; Hong, S. Biomolecular corona on nanoparticles: A survey of recent literature and its implications in targeted drug delivery. Front. Chem. 2014, 2, 108. [Google Scholar] [CrossRef]
- Guo, S.; Huang, L. Nanoparticles escaping RES and endosome: Challenges for siRNA delivery for cancer therapy. J. Nanomater. 2011, 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, M.; Hata, K.; Higashisaka, K.; Nagano, K.; Yoshioka, Y.; Tsutsumi, Y. Clusterin in the protein corona plays a key role in the stealth effect of nanoparticles against phagocytes. Biochem. Biophys. Res. Commun. 2016, 480, 690–695. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Rao, L.; Xu, J.-H.; Cai, B.; Liu, H.; Li, M.; Jia, Y.; Xiao, L.; Guo, S.-S.; Liu, W.; Zhao, X.-Z. Synthetic nanoparticles camouflaged with biomimetic erythrocyte membranes for reduced reticuloendothelial system uptake. Nanotechnology 2016, 27, 085106. [Google Scholar] [CrossRef]
- Magaña, I.B.; Yendluri, R.B.; Adhikari, P.; Goodrich, G.P.; Schwartz, J.A.; Sherer, E.A.; O’Neal, D.P. Suppression of the reticuloendothelial system using λ-carrageenan to prolong the circulation of gold nanoparticles. Ther. Deliv. 2015, 6, 777–783. [Google Scholar] [CrossRef]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release Off. J. Control. Release Soc. 2012, 157, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.I.; Minzuta, Y.; Takasu, A.; Hahn, Y.S.; King, Y.H.; Kwon, I. Site-specific fatty acid-conjugation to prolong protein half-life In Vivo. J. Control. Release 2013, 170, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazak, R.; Houri, M.; Achy, S.E.; Hussein, W.; Refaat, T. Passive targeting of nanoparticles to cancer: A comprehensive review of the literature. Mol. Clin. Oncol. 2014, 2, 904–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talekar, M.; Tran, T.-H.; Amiji, M. Translational nano-medicines: Targeted therapeutic delivery for cancer and inflammatory diseases. AAPS J. 2015, 17, 813–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbrich, W.; Lamprecht, A. Targeted drug-delivery approaches by nanoparticulate carriers in the therapy of inflammatory diseases. J. R. Soc. Interface 2010, 7, S55–S66. [Google Scholar] [CrossRef] [Green Version]
- Look, M.; Stern, E.; Wang, Q.A.; DiPlacido, L.D.; Kashgarian, M.; Craft, J.; Fahmy, T.M. Nanogel-based delivery of mycophenolic acid ameliorates systemic lupus erythematosus in mice. J. Clin. Investig. 2013, 123, 1741–1749. [Google Scholar] [CrossRef] [Green Version]
- Lundy, D.J.; Chen, K.-H.; Toh, E.K.-W.; Hsieh, P.C.-H. Distribution of systemically administered nanoparticles reveals a size-dependent effect immediately following cardiac ischaemia-reperfusion injury. Sci. Rep. 2016, 6, 25613. [Google Scholar] [CrossRef] [Green Version]
- Goyal, K.; Koul, V.; Singh, Y.; Anand, A. Targeted drug delivery to central nervous system (CNS) for the treatment of neurodegenerative disorders: Trends and advances. Cent. Nerv. Syst. Agents Med. Chem. 2014, 14, 43–59. [Google Scholar] [CrossRef]
- Olivier, J.C.; Fenart, L.; Chauvet, R.; Pariat, C.; Cecchelli, R.; Couet, W. Indirect evidence that drug brain targeting using polysorbate 80-coated polybutylcyanoacrylate nanoparticles is related to toxicity. Pharm. Res. 1999, 16, 1836–1842. [Google Scholar] [CrossRef]
- Xu, L.; Dan, M.; Shao, A.; Cheng, X.; Zhang, C.; Yokel, R.A.; Takemura, T.; Hanagata, N.; Niwa, M.; Watanabe, D. Silver nanoparticles induce tight junction disruption and astrocyte neurotoxicity in a rat blood–brain barrier primary triple coculture model. Int. J. Nanomed. 2015, 10, 6105–6119. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, J.V.; Kalicharan, D.; Couraud, P.-O.; Romero, I.A.; Weksler, B.; Hoekstra, D.; Zuhorn, I.S. Surface characteristics of nanoparticles determine their intracellular fate in and processing by human blood–brain barrier endothelial cells In Vitro. Mol. Ther. 2011, 19, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Release Off. J. Control. Release Soc. 2016, 235, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.S.; Tammam, S.N.; Shetab Boushehri, M.A.; Lamprecht, A. MDR in cancer: Addressing the underlying cellular alterations with the use of nanocarriers. Pharmacol. Res. 2017, 126, 2–30. [Google Scholar] [CrossRef] [PubMed]
- Nieto Montesinos, R.; Béduneau, A.; Pellequer, Y.; Lamprecht, A. Delivery of P-glycoprotein substrates using chemosensitizers and nanotechnology for selective and efficient therapeutic outcomes. J. Control. Release 2012, 161, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.M.; Jaimes, E.A.; Heller, D.A. Nanomedicines for kidney diseases. Kidney Int. 2016, 90, 740–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.M.; Shah, J.; Ng, B.D.; Minton, D.R.; Gudas, L.J.; Park, C.Y.; Heller, D.A. Mesoscale nanoparticles selectively target the renal proximal tubule epithelium. Nano Lett. 2015, 15, 2358–2364. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.M.; Shah, J.; Tian, H.S.; Chen, X.; Geissmann, F.; Jaimes, E.A.; Heller, D.A. Selective nanoparticle targeting of the renal tubules. Hypertens. Dallas Tex 1979 2018, 71, 87–94. [Google Scholar] [CrossRef]
- Nair, A.V.; Keliher, E.J.; Core, A.B.; Brown, D.; Weissleder, R. Characterizing the interactions of organic nanoparticles with renal epithelial cells in vivo. ACS Nano 2015, 9, 3641–3653. [Google Scholar] [CrossRef] [Green Version]
- Bennett, K.M.; Zhou, H.; Sumner, J.P.; Dodd, S.J.; Bouraoud, N.; Doi, K.; Star, R.A.; Koretsky, A.P. MRI of the basement membrane using charged nanoparticles as contrast agents. Magn. Reson. Med. 2008, 60, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Torres Andón, F.; Alonso, M.J. Nanomedicine and cancer immunotherapy—Targeting immunosuppressive cells. J. Drug Target. 2015, 23, 656–671. [Google Scholar] [CrossRef]
- Mishra, H.; Mishra, D.; Mishra, P.K.; Nahar, M.; Dubey, V.; Jain, N.K. Evaluation of solid lipid nanoparticles as carriers for delivery of hepatitis B surface antigen for vaccination using subcutaneous route. J. Pharm. Pharm. Sci. 2010, 13, 495–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, Y.; Srinivas, A.; Gangwar, M.; Meher, J.G.; Misra-Bhattacharya, S.; Chourasia, M.K. Subcutaneously administered ultrafine PLGA nanoparticles containing doxycycline hydrochloride target lymphatic filarial parasites. Mol. Pharm. 2016, 13, 2084–2094. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Trase, I.; Ren, M.; Duval, K.; Guo, X.; Chen, Z. Design of nanoparticle-based carriers for targeted drug delivery. J. Nanomater. 2016, 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Zhang, A.X.; Li, S.-D. Limitations and niches of the active targeting approach for nanoparticle drug delivery. Eur. J. Nanomed. 2012, 4, 89–93. [Google Scholar] [CrossRef]
- Arruebo, M.; Valladares, M.; González-Fernández, Á. Antibody-conjugated nanoparticles for biomedical applications. J. Nanomater. 2009, 2009, 439389. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.A.; Maruani, A.; Chudasama, V. Antibody fragments as nanoparticle targeting ligands: A step in the right direction. Chem. Sci. 2016, 8, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.J.; Perreira, I.; Ferreira, D.; Nestor, M.; Oliveira, C.; Granja, P.L.; Sarmento, B. Impact of surfactants on the target recognition of Fab-conjugated PLGA nanoparticles. Eur. J. Pharm. Biopharm. 2018, 127, 366–370. [Google Scholar] [CrossRef]
- Rezaei, G.; Habibi-Anbouhi, M.; Mahmoudi, M.; Azadmanesh, K.; Moradi-Kalbolandi, S.; Behdani, M.; Ghazizadeh, L.; Abolhassani, M.; Shokrgozar, M.A. Development of anti-CD47 single-chain variable fragment targeted magnetic nanoparticles for treatment of human bladder cancer. Nanomedicine 2017, 12, 597–613. [Google Scholar] [CrossRef]
- Hu, C.-M.J.; Kaushal, S.; Tran Cao, H.S.; Aryal, S.; Sartor, M.; Esener, S.; Bouvet, M.; Zhang, L. Half-antibody functionalized lipid-polymer hybrid nanoparticles for targeted drug delivery to carcinoembryonic antigen presenting pancreatic cancer cells. Mol. Pharm. 2010, 7, 914–920. [Google Scholar] [CrossRef]
- Girgis, M.D.; Federman, N.; Rochefort, M.M.; McCabe, K.E.; Wu, A.M.; Nagy, J.O.; Denny, C.; Tomlinson, J.S. An engineered anti-CA19-9 cys-diabody for positron emission tomography imaging of pancreatic cancer and targeting of polymerized liposomal nanoparticles. J. Surg. Res. 2013, 185, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontermann, R. Dual targeting strategies with bispecific antibodies. mAbs 2012, 4, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Pietersz, G.A.; Wang, X.; Yap, M.L.; Lim, B.; Peter, K. Therapeutic targeting in nanomedicine: The future lies in recombinant antibodies. Nanomedicine 2017, 12, 1873–1889. [Google Scholar] [CrossRef] [Green Version]
- Nygren, P.-A.; Skerra, A. Binding proteins from alternative scaffolds. J. Immunol. Methods 2004, 290, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Löfblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Ståhl, S.; Frejd, F.Y. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010, 584, 2670–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ståhl, S.; Gräslund, T.; Eriksson Karlström, A.; Frejd, F.Y.; Nygren, P.-Å.; Löfblom, J. Affibody molecules in biotechnological and medical applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef]
- Satpathy, M.; Wang, L.; Zielinski, R.; Qian, W.; Lipowska, M.; Capala, J.; Lee, G.Y.; Xu, H.; Wang, Y.A.; Mao, H.; et al. Active targeting using HER-2-affibody-conjugated nanoparticles enabled sensitive and specific imaging of orthotopic HER-2 positive ovarian tumors. Small Weinh. Bergstr. Ger. 2014, 10, 544–555. [Google Scholar] [CrossRef] [Green Version]
- Alexis, F.; Basto, P.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Zhang, L.; Pridgen, E.; Wang, A.Z.; Marein, S.L.; Westerhof, K.; Molnar, L.K.; et al. HER-2-targeted nanoparticle-affibody bioconjugates for cancer therapy. ChemMedChem 2008, 3, 1839–1843. [Google Scholar] [CrossRef] [Green Version]
- Beuttler, J.; Rothdiener, M.; Müller, D.; Frejd, F.Y.; Kontermann, R.E. Targeting of epidermal growth factor receptor (EGFR)-expressing tumor cells with sterically stabilized affibody liposomes (SAL). Bioconjug. Chem. 2009, 20, 1201–1208. [Google Scholar] [CrossRef]
- Tiede, C.; Bedford, R.; Heseltine, S.J.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer proteins are versatile and renewable affinity reagents. eLife 2017, 6, e24903. [Google Scholar] [CrossRef]
- Khaled, Y.S.; Shamsuddin, S.; Tiernan, J.; McPherson, M.; Hughes, T.; Millner, P.; Jayne, D.G. Theranostic CEA-Affimer functionalised silica nanoparticles allow specific in vitro fluorescent imaging of colorectal cancer cells. Eur. J. Surg. Oncol. 2018, 44, S1. [Google Scholar] [CrossRef]
- Rawlings, A.E.; Bramble, J.P.; Tang, A.A.S.; Somner, L.A.; Monnington, A.E.; Cooke, D.J.; McPherson, M.J.; Tomlinson, D.C.; Staniland, S.S. Phage display selected magnetite interacting Adhirons for shape controlled nanoparticle synthesis. Chem. Sci. 2015, 6, 5586–5594. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.-Y.; Deng, W.; Engelke, H.; Helma, J.; Leonhardt, H.; Bein, T. Intracellular chromobody delivery by mesoporous silica nanoparticles for antigen targeting and visualization in real time. Sci. Rep. 2016, 6, 25019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelstein, J. Fabrication of Protein Scaffold-Nanoparticle Conjugates for Targeted Cancer Therapeutics; Princeton University: Princeton, NJ, USA, 2014. [Google Scholar]
- Deyev, S.; Proshkina, G.; Ryabova, A.; Tavanti, F.; Menziani, M.C.; Eidelshtein, G.; Avishai, G.; Kotlyar, A. Synthesis, characterization, and selective delivery of DARPin-gold nanoparticle conjugates to cancer cells. Bioconjug. Chem. 2017, 28, 2569–2574. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-J.; Kang, J.A.; Ryu, Y.; Han, S.-S.; Nam, Y.R.; Rho, J.K.; Choi, D.S.; Kang, S.-W.; Lee, D.-E.; Kim, H.-S. Genetically engineered and self-assembled oncolytic protein nanoparticles for targeted cancer therapy. Biomaterials 2017, 120, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Z.; Gu, F.; Zhang, L.; Chan, J.M.; Radovic-Moreno, A.; Shaikh, M.R.; Farokhzad, O.C. Biofunctionalized targeted nanoparticles for therapeutic applications. Expert Opin. Biol. Ther. 2008, 8, 1063–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasongkla, N.; Shuai, X.; Ai, H.; Weinberg, B.D.; Pink, J.; Boothman, D.A.; Gao, J. cRGD-functionalized polymer micelles for targeted doxorubicin delivery. Angew. Chem. Int. Ed Engl. 2004, 43, 6323–6327. [Google Scholar] [CrossRef]
- Yu, X.; Song, Y.; Di, Y.; He, H.; Fu, D.; Jin, C. Enhanced tumor targeting of cRGD peptide-conjugated albumin nanoparticles in the BxPC-3 cell line. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Danhier, F.; Pourcelle, V.; Marchand-Brynaert, J.; Jérôme, C.; Feron, O.; Préat, V. Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles. Methods Enzymol. 2012, 508, 157–175. [Google Scholar] [CrossRef]
- Endo-Takahashi, Y.; Ooaku, K.; Ishida, K.; Suzuki, R.; Maruyama, K.; Negishi, Y. Preparation of Angiopep-2 peptide-modified bubble liposomes for delivery to the brain. Biol. Pharm. Bull. 2016, 39, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-J.; Su, Y.-Z.; Hsu, C.; Lo, Y.-L.; Huang, S.-J.; Ke, J.-H.; Kuo, Y.-C.; Wang, L.-F. Angiopep-pluronic F127-conjugated superparamagnetic iron oxide nanoparticles as nanotheranostic agents for BBB targeting. J. Mater. Chem. B 2014, 2, 5666–5675. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, G.L.; Mahdi, F.; Shao, Q.; Logue, O.C.; Waller, J.P.; Reese, C.; Chade, A.R. A kidney-selective biopolymer for targeted drug delivery. Am. J. Physiol. Ren. Physiol. 2017, 312, F54–F64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Q.; Sun, X.; Gong, T.; Zhang, Z.-R. Peptide-drug conjugate linked via a disulfide bond for kidney targeted drug delivery. Bioconjug. Chem. 2012, 23, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Tammam, S.N.; Azzazy, H.M.E.; Breitinger, H.G.; Lamprecht, A. Chitosan nanoparticles for nuclear targeting: The effect of nanoparticle size and nuclear localization sequence density. Mol. Pharm. 2015, 12, 4277–4289. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Meng, F.; Deng, C.; Zhong, Z. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules 2014, 15, 1955–1969. [Google Scholar] [CrossRef]
- Bruschi, M.L. Lectins and nanostructured drug delivery systems. Curr. Drug Deliv. 2019, 16, 268–269. [Google Scholar] [CrossRef]
- Iskratsch, T.; Braun, A.; Paschinger, K.; Wilson, I.B.H. Specificity analysis of lectins and antibodies using remodeled glycoproteins. Anal. Biochem. 2009, 386, 133–146. [Google Scholar] [CrossRef]
- Hirabayashi, J.; Arai, R. Lectin engineering: The possible and the actual. Interface Focus 2019, 9, 20180068. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wu, H.; Hong, J.; Xu, X.; Yang, H.; Wu, B.; Wang, Y.; Zhu, J.; Lai, R.; Jiang, X.; et al. Odorranalectin is a small peptide lectin with potential for drug delivery and targeting. PLoS ONE 2008, 3, e2381. [Google Scholar] [CrossRef] [Green Version]
- Vineethkumar, T.V.; Shyla, G.; George, S. Smallest lectin-like peptide identified from the skin secretion of an endemic frog, Hydrophylax bahuvistara. Acta Biol. Hung. 2018, 69, 110–113. [Google Scholar] [CrossRef]
- Smart, J.D. Lectin-mediated drug delivery in the oral cavity. Adv. Drug Deliv. Rev. 2004, 56, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Moulari, B.; Béduneau, A.; Pellequer, Y.; Lamprecht, A. Lectin-decorated nanoparticles enhance binding to the inflamed tissue in experimental colitis. J. Control. Release Off. J. Control. Release Soc. 2014, 188, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Surti, N.; Misra, A. Wheat germ agglutinin-conjugated nanoparticles for sustained cellular and lung delivery of budesonide. Drug Deliv. 2008, 15, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, T.J.; Green, K.L.; Rogers, D.J.; Cook, J.D.; Wolowacz, S.; Smart, J.D. Lectins in ocular drug delivery: An investigation of lectin binding sites on the corneal and conjunctival surfaces. Int. J. Pharm. 1996, 138, 175–183. [Google Scholar] [CrossRef]
- Gao, X.; Tao, W.; Lu, W.; Zhang, Q.; Zhang, Y.; Jiang, X.; Fu, S. Lectin-conjugated PEG–PLA nanoparticles: Preparation and brain delivery after intranasal administration. Biomaterials 2006, 27, 3482–3490. [Google Scholar] [CrossRef] [PubMed]
- Bies, C.; Lehr, C.-M.; Woodley, J.F. Lectin-mediated drug targeting: History and applications. Adv. Drug Deliv. Rev. 2004, 56, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Poiroux, G.; Barre, A.; van Damme, E.J.M.; Benoist, H.; Rougé, P. Plant lectins targeting O-glycans at the cell surface as tools for cancer diagnosis, prognosis and therapy. Int. J. Mol. Sci. 2017, 18, 1232. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Carmona, M.; Lozano, D.; Colilla, M.; Vallet-Regí, M. Lectin-conjugated pH-responsive mesoporous silica nanoparticles for targeted bone cancer treatment. Acta Biomater. 2018, 65, 393–404. [Google Scholar] [CrossRef]
- Singh, A.; Dilnawaz, F.; Sahoo, S.K. Long circulating lectin conjugated paclitaxel loaded magnetic nanoparticles: A new theranostic avenue for leukemia therapy. PLoS ONE 2011, 6, e26803. [Google Scholar] [CrossRef] [Green Version]
- Obaid, G.; Chambrier, I.; Cook, M.J.; Russell, D.A. Targeting the oncofetal Thomsen-Friedenreich disaccharide using jacalin-PEG phthalocyanine gold nanoparticles for photodynamic cancer therapy. Angew. Chem. Int. Ed Engl. 2012, 51, 6158–6162. [Google Scholar] [CrossRef]
- Obaid, G.; Chambrier, I.; Cook, M.J.; Russell, D.A. Cancer targeting with biomolecules: A comparative study of photodynamic therapy efficacy using antibody or lectin conjugated phthalocyanine-PEG gold nanoparticles. Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2015, 14, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulibaly, F.S.; Youan, B.-B.C. Current status of lectin-based cancer diagnosis and therapy. AIMS Mol. Sci. 2017, 4, 1–27. [Google Scholar] [CrossRef]
- Lord, J.M. The use of cytotoxic plant lectins in cancer therapy. Plant Physiol. 1987, 85, 1–3. [Google Scholar] [CrossRef]
- Kröger, A.P.P.; Komil, M.I.; Hamelmann, N.M.; Juan, A.; Stenzel, M.H.; Paulusse, J.M.J. Glucose single-chain polymer nanoparticles for cellular targeting. ACS Macro Lett. 2019, 8, 95–101. [Google Scholar] [CrossRef]
- Carrillo-Conde, B.; Song, E.H.; Chavez-Santoscoy, A.; Phanse, Y.; Ramer-Tait, A.E.; Pohl, N.L.B.; Wannemuehler, M.J.; Bellaire, B.H.; Narasimhan, B. Mannose-functionalized “pathogen-like” polyanhydride nanoparticles target C-type lectin receptors on dendritic cells. Mol. Pharm. 2011, 8, 1877–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Zhong, J.; Zhao, M.; Tang, Y.; Han, N.; Hua, L.; Xu, T.; Wang, C.; Zhu, B. Galactose-modified selenium nanoparticles for targeted delivery of doxorubicin to hepatocellular carcinoma. Drug Deliv. 2019, 26, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chan, J.W.; Moretti, A.; Uhrich, K.E. Designing polymers with sugar-based advantages for bioactive delivery applications. J. Control. Release Off. J. Control. Release Soc. 2015, 219, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Kikkeri, R.; Lepenies, B.; Adibekian, A.; Laurino, P.; Seeberger, P.H. In Vitro imaging and In Vivo liver targeting with carbohydrate capped quantum dots. J. Am. Chem. Soc. 2009, 131, 2110–2112. [Google Scholar] [CrossRef]
- Ahmed, M.; Narain, R. Carbohydrate-based materials for targeted delivery of drugs and genes to the liver. Nanomedicine 2015, 10, 2263–2288. [Google Scholar] [CrossRef]
- Pranatharthiharan, S.; Patel, M.D.; Malshe, V.C.; Pujari, V.; Gorakshakar, A.; Madkaikar, M.; Ghosh, K.; Devarajan, P.V. Asialoglycoprotein receptor targeted delivery of doxorubicin nanoparticles for hepatocellular carcinoma. Drug Deliv. 2017, 24, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Kou, L.; Bhutia, Y.D.; Yao, Q.; He, Z.; Sun, J.; Ganapathy, V. Transporter-guided delivery of nanoparticles to improve drug permeation across cellular barriers and drug exposure to selective cell types. Front. Pharmacol. 2018, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Fan, W.; Chen, H.; Yao, N.; Tang, W.; Tang, J.; Yuan, W.; Kuai, R.; Zhang, Z.; Wu, Y.; et al. In Vitro and In Vivo investigation of glucose-mediated brain-targeting liposomes. J. Drug Target. 2010, 18, 536–549. [Google Scholar] [CrossRef]
- Dufes, C.; Gailard, F.; Uchegbu, I.; Schätzlein, A.; Olivier, J.-C.; Muller, J.-M. Glucose-targeted niosomes deliver vasoactive intestinal peptide (VIP) to the brain. Int. J. Pharm. 2004, 285, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, M.K.; Needham, C.N.; Neill, N.E.; White, M.C.; Kelly, C.B.; Mastro-Kishton, K.; Chauvigne-Hines, L.M.; Goodwin, T.J.; McIver, A.L.; Bartolotti, L.J.; et al. Glycoconjugated site-selective DNA-methylating agent targeting glucose transporters on glioma cells. Biochemistry 2017, 56, 421–440. [Google Scholar] [CrossRef] [PubMed]
- Sztandera, K.; Działak, P.; Marcinkowska, M.; Stańczyk, M.; Gorzkiewicz, M.; Janaszewska, A.; Klajnert-Maculewicz, B. Sugar modification enhances cytotoxic activity of PAMAM-doxorubicin conjugate in glucose-deprived MCF-7 cells—Possible role of GLUT1 transporter. Pharm. Res. 2019, 36, 140. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Cho, H.-J.; Kim, D.-D. Poly((D,L)lactic-glycolic)acid–star glucose nanoparticles for glucose transporter and hypoglycemia-mediated tumor targeting. Int. J. Nanomed. 2017, 12, 7453–7467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Huang, G.; Huang, H. Sugar ligand-mediated drug delivery. Future Med. Chem. 2020, 12, 161–171. [Google Scholar] [CrossRef]
- De Coen, R.; Vanparijs, N.; Risseeuw, M.D.P.; Lybaert, L.; Louage, B.; De Koker, S.; Kumar, V.; Grooten, J.; Taylor, L.; Ayres, N.; et al. pH-degradable mannosylated nanogels for dendritic cell targeting. Biomacromolecules 2016, 17, 2479–2488. [Google Scholar] [CrossRef]
- Asthana, G.S.; Asthana, A.; Kohli, D.V.; Vyas, S.P. Mannosylated chitosan nanoparticles for delivery of antisense oligonucleotides for macrophage targeting. BioMed Res. Int. 2014, 2014, 526391. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulos, V.; Thalhammer, T.; Tzakos, A.G.; Stojanovska, L. Targeting antigens to dendritic cell receptors for vaccine development. J. Drug Deliv. 2013, 2013, 869718. [Google Scholar] [CrossRef]
- Hockl, P.F.; Wolosiuk, A.; Pérez-Sáez, J.M.; Bordoni, A.V.; Croci, D.O.; Toum-Terrones, Y.; Soler-Illia, G.J.A.A.; Rabinovich, G.A. Glyco-nano-oncology: Novel therapeutic opportunities by combining small and sweet. Pharmacol. Res. 2016, 109, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Agarwal, A.; Majumder, S.; Lariya, N.; Khaya, A.; Agrawal, H.; Majumdar, S.; Agrawal, G.P. Mannosylated solid lipid nanoparticles as vectors for site-specific delivery of an anti-cancer drug. J. Control. Release Off. J. Control. Release Soc. 2010, 148, 359–367. [Google Scholar] [CrossRef]
- Jo, H.; Ban, C. Aptamer–nanoparticle complexes as powerful diagnostic and therapeutic tools. Exp. Mol. Med. 2016, 48, e230. [Google Scholar] [CrossRef] [Green Version]
- Aghanejad, A.; Babamiri, H.; Adibkia, K.; Barar, J.; Omidi, Y. Mucin-1 aptamer-armed superparamagnetic iron oxide nanoparticles for targeted delivery of doxorubicin to breast cancer cells. BioImpacts 2018, 8, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Aravind, A.; Varghese, S.H.; Veeranarayanan, S.; Mathew, A.; Nagaoka, Y.; Iwai, S.; Fukuda, T.; Hasumura, T.; Yoshida, Y.; Maekawa, T.; et al. Aptamer-labeled PLGA nanoparticles for targeting cancer cells. Cancer Nanotechnol. 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigum, S.; McIvor, E.; Munoz, C.; Feng, R.; Cantu, T.; Walsh, K.; Betancourt, T. Targeted therapy of hepatocellular carcinoma with aptamer-functionalized biodegradable nanoparticles. J. Nanoparticle Res. 2016, 18, 341. [Google Scholar] [CrossRef]
- Liu, J.; Wei, T.; Zhao, J.; Huang, Y.; Deng, H.; Kumar, A.; Wang, C.; Liang, Z.; Ma, X.; Liang, X.-J. Multifunctional aptamer-based nanoparticles for targeted drug delivery to circumvent cancer resistance. Biomaterials 2016, 91, 44–56. [Google Scholar] [CrossRef]
- Liang, C.; Guo, B.; Wu, H.; Shao, N.; Li, D.; Liu, J.; Dang, L.; Wang, C.; Li, H.; Li, S.; et al. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference-based bone anabolic strategy. Nat. Med. 2015, 21, 288–294. [Google Scholar] [CrossRef]
- Yu, L.; Hu, Y.; Duan, J.; Yang, X.-D. A novel approach of targeted immunotherapy against adenocarcinoma cells with nanoparticles modified by CD16 and MUC1 aptamers. J. Nanomater. 2015, 2015, 10. [Google Scholar] [CrossRef]
- Zhang, P.; Ye, J.; Liu, E.; Sun, L.; Zhang, J.; Lee, S.-J.; Gong, J.; He, H.; Yang, V.C. Aptamer-coded DNA nanoparticles for targeted doxorubicin delivery using pH-sensitive spacer. Front. Chem. Sci. Eng. 2017, 11, 529–536. [Google Scholar] [CrossRef]
- Catuogno, S.; Esposito, C.L.; de Franciscis, V. Aptamer-mediated targeted delivery of therapeutics: An update. Pharmaceuticals 2016, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farokhzad, O.C.; Karp, J.M.; Langer, R. Nanoparticle-aptamer bioconjugates for cancer targeting. Expert Opin. Drug Deliv. 2006, 3, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Dixit, S.; Novak, T.; Miller, K.; Zhu, Y.; Kenney, M.E.; Broome, A.-M. Transferrin receptor-targeted theranostic gold nanoparticles for photosensitizer delivery in brain tumors. Nanoscale 2015, 7, 1782–1790. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Wang, Y.; Koenig, L.; Gjyrezi, A.; Giannakakou, P.; Shin, E.H.; Tighiouart, M.; Chen, Z.G.; Nie, S.; et al. A folate receptor-targeting nanoparticle minimizes drug resistance in a human cancer model. ACS Nano 2011, 5, 6184–6194. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Swami, R.; Pooja, D.; Jeengar, M.K.; Khan, W.; Sistla, R. Lactoferrin bioconjugated solid lipid nanoparticles: A new drug delivery system for potential brain targeting. J. Drug Target. 2016, 24, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Xiao, X.; Ao, Y. Low density lipoprotein modified silica nanoparticles loaded with docetaxel and thalidomide for effective chemotherapy of liver cancer. Braz. J. Med. Biol. Res. 2018, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.A.; Nikolaev, B.P.; Yakovleva, L.Y.; Dobrodumov, A.V.; Zhakhov, A.V.; Mikhrina, A.L.; Pitkin, E.; Parr, M.A.; Rolich, V.I.; Simbircev, A.S.; et al. Recombinant interleukin-1 receptor antagonist conjugated to superparamagnetic iron oxide nanoparticles for theranostic targeting of experimental glioblastoma. Neoplasia 2015, 17, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Abdellatif, A.A.H.; Aldalaen, S.M.; Faisal, W.; Tawfeek, H.M. Somatostatin receptors as a new active targeting sites for nanoparticles. Saudi Pharm. J. 2018, 26, 1051–1059. [Google Scholar] [CrossRef]
- Nikanjam, M.; Blakely, E.A.; Bjornstad, K.A.; Shu, X.; Budinger, T.F.; Forte, T.M. Synthetic nano-low density lipoprotein as targeted drug delivery vehicle for glioblastoma multiforme. Int. J. Pharm. 2007, 328, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaxton, C.S.; Daniel, W.L.; Giljohann, D.A.; Thomas, A.D.; Mirkin, C.A. Templated spherical high density lipoprotein nanoparticles. J. Am. Chem. Soc. 2009, 131, 1384–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutjahr, A.; Phelip, C.; Coolen, A.-L.; Monge, C.; Boisgard, A.-S.; Paul, S.; Verrier, B. Biodegradable polymeric nanoparticles-based vaccine adjuvants for lymph nodes targeting. Vaccines 2016, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Marques Neto, L.M.; Kipnis, A.; Junqueira-Kipnis, A.P. Role of metallic nanoparticles in vaccinology: Implications for infectious disease vaccine development. Front. Immunol. 2017, 8, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetab Boushehri, M.A.; Abdel-Motalleb, M.M.; Beduneau, A.; Pellequer, Y.; Lamprecht, A. A nanoparticle-based approach to improve the outcome of cancer immunotherapy with lipopolysaccharides. Drug Deliv. 2018, 25, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Zolnik, B.S.; González-Fernández, A.; Sadrieh, N.; Dobrovolskaia, M.A. Nanoparticles and the immune system. Endocrinology 2010, 151, 458–465. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, R.; Nie, G. Applications of nanomaterials as vaccine adjuvants. Hum. Vaccines Immunother. 2014, 10, 2761–2774. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.-J.; Choi, I.-H. Immunostimulatory effects of silica nanoparticles in human monocytes. Immune Netw. 2013, 13, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Baron, L.; Gombault, A.; Fanny, M.; Villeret, B.; Savigny, F.; Guillou, N.; Panek, C.; Le Bert, M.; Lagente, V.; Rassendren, F.; et al. The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis. 2015, 6, e1629. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Zhao, F.; Guo, F.; Wang, C.; Fu, Z. Polymeric nanoparticles induce NLRP3 inflammasome activation and promote breast cancer metastasis. Macromol. Biosci. 2017, 17, 1700273. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.K.; Ramer-Tait, A.E.; Broderick, S.R.; Kong, C.-S.; Ulery, B.D.; Rajan, K.; Wannemuehler, M.J.; Narasimhan, B. Activation of innate immune responses in a pathogen-mimicking manner by amphiphilic polyanhydride nanoparticle adjuvants. Biomaterials 2011, 32, 6815–6822. [Google Scholar] [CrossRef]
- Camacho, A.I.; Da Costa Martins, R.; Tamayo, I.; de Souza, J.; Lasarte, J.J.; Mansilla, C.; Esparza, I.; Irache, J.M.; Gamazo, C. Poly(methyl vinyl ether-co-maleic anhydride) nanoparticles as innate immune system activators. Vaccine 2011, 29, 7130–7135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uto, T.; Akagi, T.; Yoshinaga, K. The induction of innate and adaptive immunity by biodegradable poly(g-glutamic acid) nanoparticles via a TLR4 and MyD88 signaling pathway. Biomaterials 2011, 32, 5206–5212. [Google Scholar] [CrossRef] [PubMed]
- Kedmi, R.; Ben-Arie, N.; Peer, D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 2010, 31, 6867–6875. [Google Scholar] [CrossRef]
- Shetab Boushehri, M.A.; Stein, V.; Lamprecht, A. Cargo-free particles of ammonio methacrylate copolymers: From pharmaceutical inactive ingredients to effective anticancer immunotherapeutics. Biomaterials 2018, 166, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.M.; Thanki, K.; Gangolff, M.; Falkenberg, E.; Zeng, X.; van Bijnen, D.C.; van Eden, W.; Franzyk, H.; Nielsen, H.M.; Broere, F.; et al. Immunogenicity testing of lipidoids in vitro and in silico: Modulating lipidoid-Mediated TLR4 activation by nanoparticle design. Mol. Ther. Nucleic Acids 2018, 11, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Legat, A.; Adam, E.; Steuve, J.; Gatot, J.-S.; Vandenbranden, M.; Ulianov, L.; Lonez, C.; Ruysschaert, J.-M.; Muraille, E.; et al. DiC14-amidine cationic liposomes stimulate myeloid dendritic cells through Toll-like receptor 4. Eur. J. Immunol. 2008, 38, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Lonez, C.; Irvine, K.L.; Pizzuto, M.; Schmidt, B.I.; Gay, N.J.; Ruysschaert, J.-M.; Gangloff, M.; Bryant, C.E. Critical residues involved in Toll-like receptor 4 activation by cationic lipid nanocarriers are not located at the lipopolysaccharide-binding interface. Cell. Mol. Life Sci. 2015, 72, 3971–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, G.; Liu, S.; Zhang, S.; Wang, L.; Wang, X.; Sun, B.; Yin, N.; Gao, X.; Xia, T.; Chen, J.-J.; et al. Graphene oxide induces Toll-like Receptor 4 (TLR4)-dependent necrosis in macrophages. ACS Nano 2013, 7, 5732–5745. [Google Scholar] [CrossRef]
- Bianchi, M.G.; Allegri, M.; Costa, A.L.; Blosi, M.; Gardini, D.; Del Pivo, C.; Prina-Mello, A.; Di Cristo, L.; Bussolati, O.; Bergamaschi, E. Titanium dioxide nanoparticles enhance macrophage activation by LPS through a TLR4-dependent intracellular pathway. Toxicol. Res. 2015, 4, 385–398. [Google Scholar] [CrossRef]
- Huang, C.; Sun, M.; Yang, Y.; Wang, F.; Ma, X.; Li, J.; Wang, Y.; Ding, Q.; Ying, H.; Song, H.; et al. Titanium dioxide nanoparticles prime a specific activation state of macrophages. Nanotoxicology 2017, 11, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Demento, S.L.; Eisenbarth, S.C.; Foellmer, H.G.; Platt, C.; Caplan, M.J.; Mark Saltzman, W.; Mellman, I.; Ledizet, M.; Fikrig, E.; Flavell, R.A.; et al. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine 2009, 27, 3013–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunshine, J.C.; Green, J.J. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine 2013, 8, 1173–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, J.W.; Vicente, F.P.; Howard, G.P.; Mao, H.-Q.; Schneck, J.P. Biologically inspired design of nanoparticle artificial antigen-presenting cells for immunomodulation. Nano Lett. 2017, 17, 7045–7054. [Google Scholar] [CrossRef] [PubMed]
- Neal, L.R.; Bailey, S.R.; Wyatt, M.M.; Bowers, J.S.; Majchrzak, K.; Nelson, M.H.; Haupt, C.; Paulos, C.M.; Varela, J.C. The basics of artificial antigen presenting cells in T cell-based cancer immunotherapies. J. Immunol. Res. Ther. 2017, 2, 68–79. [Google Scholar] [PubMed]
- Eggermont, L.J.; Paulis, L.E.; Tel, J.; Figdor, C.G. Towards efficient cancer immunotherapy: Advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014, 32, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Meyer, R.A.; Sunshine, J.C.; Perica, K.; Kosmides, A.K.; Aje, K.; Schneck, J.P.; Green, J.J. Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation. Small Weinh. Bergstr. Ger. 2015, 11, 1519–1525. [Google Scholar] [CrossRef] [Green Version]
- Perica, K.; De León Medero, A.; Durai, M.; Chiu, Y.L.; Bieler, J.G.; Sibener, L.; Niemöller, M.; Assenmacher, M.; Richter, A.; Edidin, M.; et al. Nanoscale artificial antigen presenting cells for T cell immunotherapy. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Agrahari, V.; Agrahari, V. Facilitating the translation of nanomedicines to a clinical product: Challenges and opportunities. Drug Discov. Today 2018, 23, 974–991. [Google Scholar] [CrossRef]
- Sarmento, B. Have nanomedicines progressed as much as we’d hoped for in drug discovery and development? Expert Opin. Drug Discov. 2019, 14, 723–725. [Google Scholar] [CrossRef]
- Agrahari, V.; Hiremath, P. Challenges associated and approaches for successful translation of nanomedicines into commercial products. Nanomedicine 2017, 12, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetab Boushehri, M.A.; Lamprecht, A. Nanoparticles as drug carriers: Current issues with In Vitro testing. Nanomedicine 2015, 10, 3213–3230. [Google Scholar] [CrossRef] [PubMed]
- Lorscheidt, S.; Lamprecht, A. Safety assessment of nanoparticles for drug delivery by means of classic in vitro assays and beyond. Expert Opin. Drug Deliv. 2016, 13, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.M.; Braydich-Stolle, L.K.; Schrand, A.M.; Murdock, R.C.; Yu, K.O.; Mattie, D.M.; Schlager, J.J.; Terrones, M. Toxicity evaluation for safe use of nanomaterials: Recent achievements and technical challenges. Adv. Mater. 2009, 21, 1549–1559. [Google Scholar] [CrossRef]





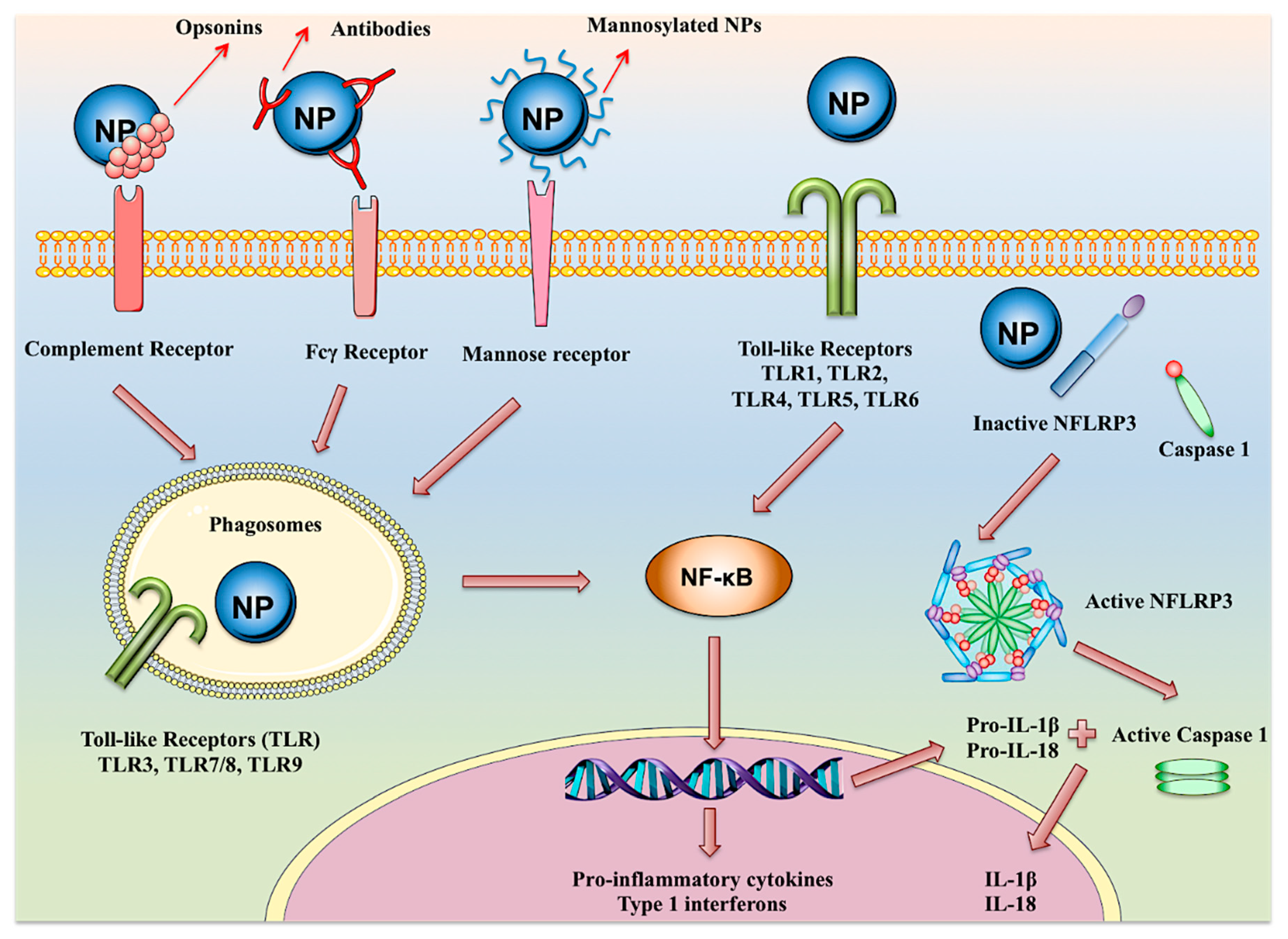

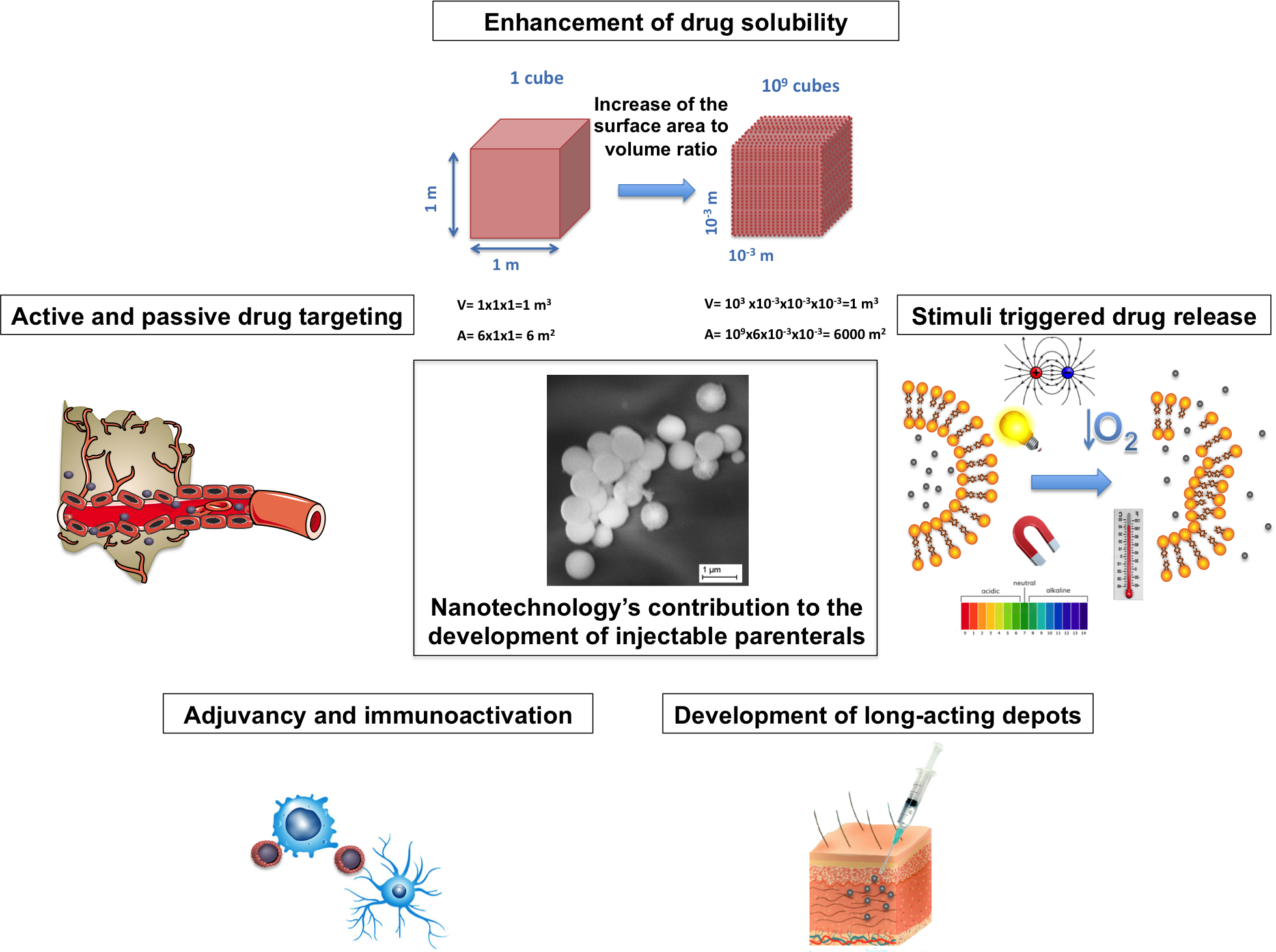{kind=link}
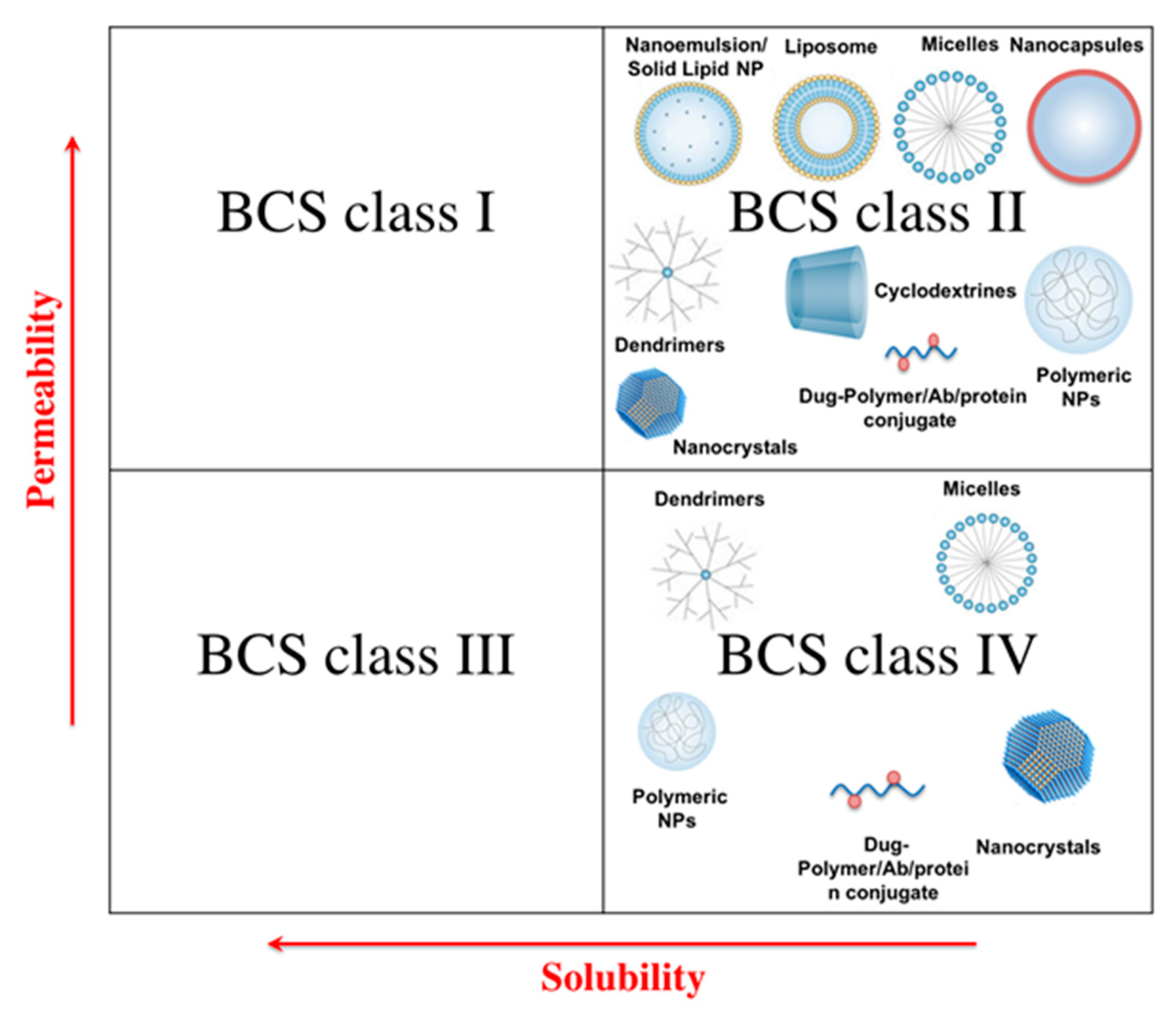{kind=link}
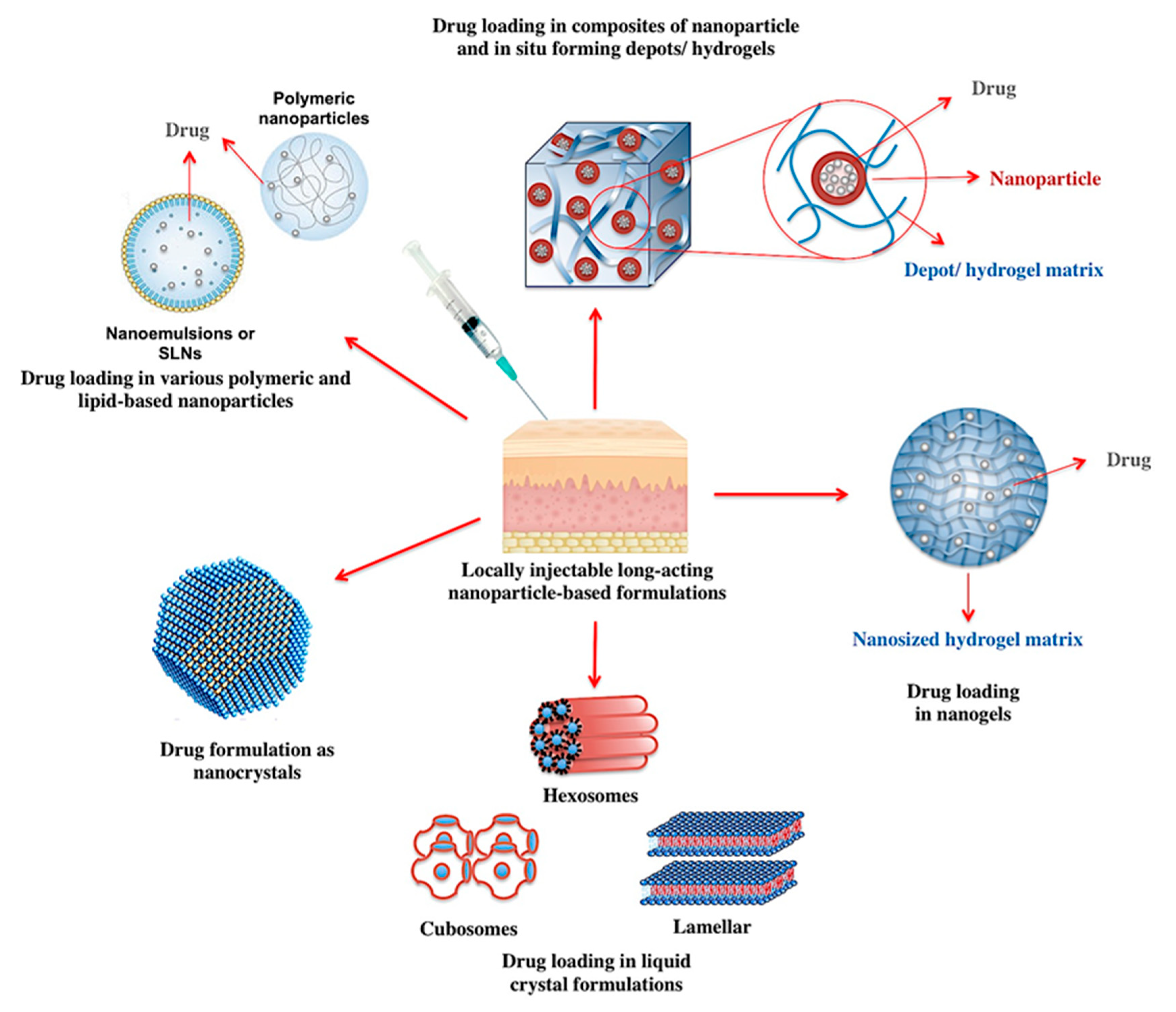{kind=link}
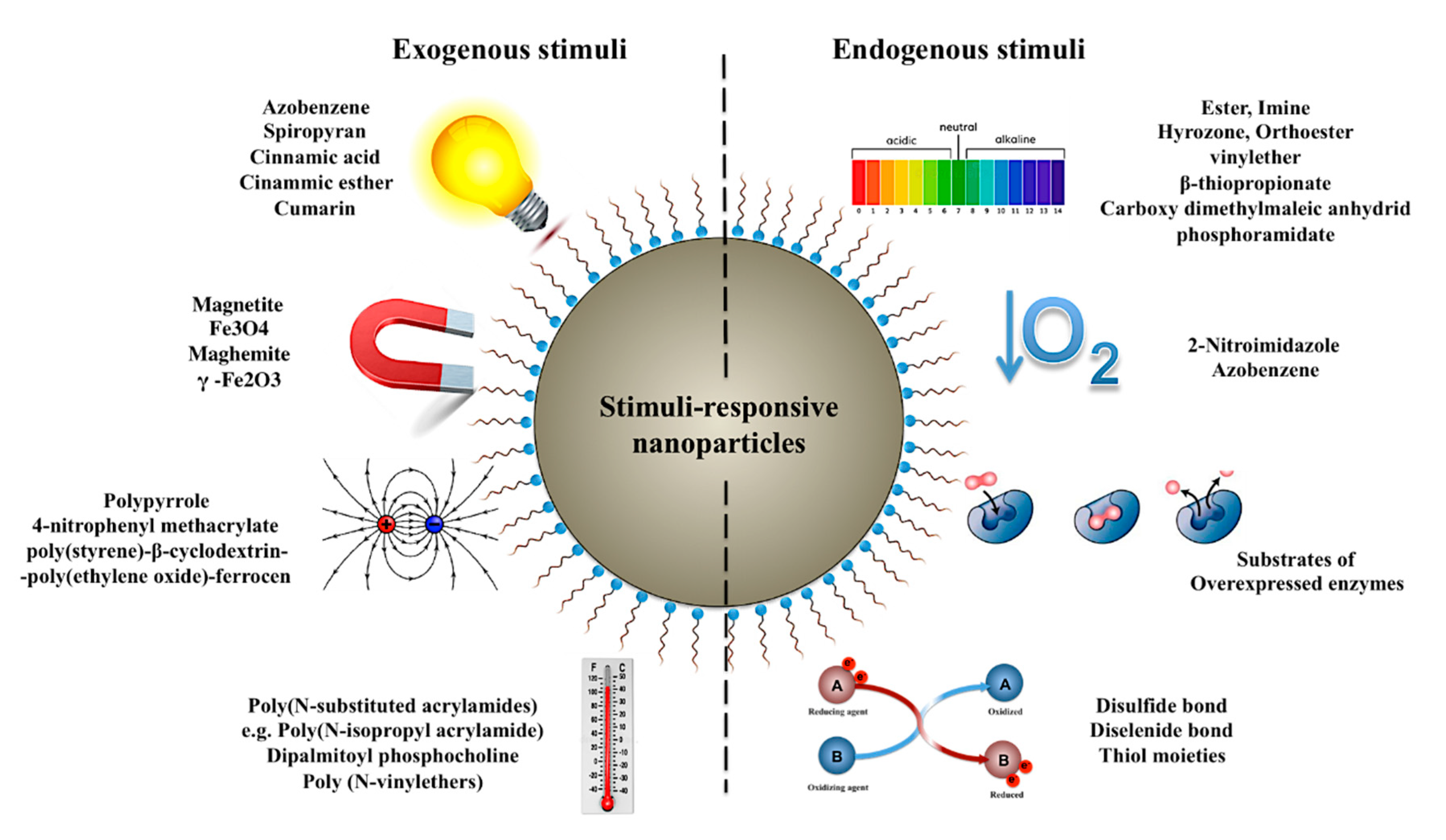{kind=link}
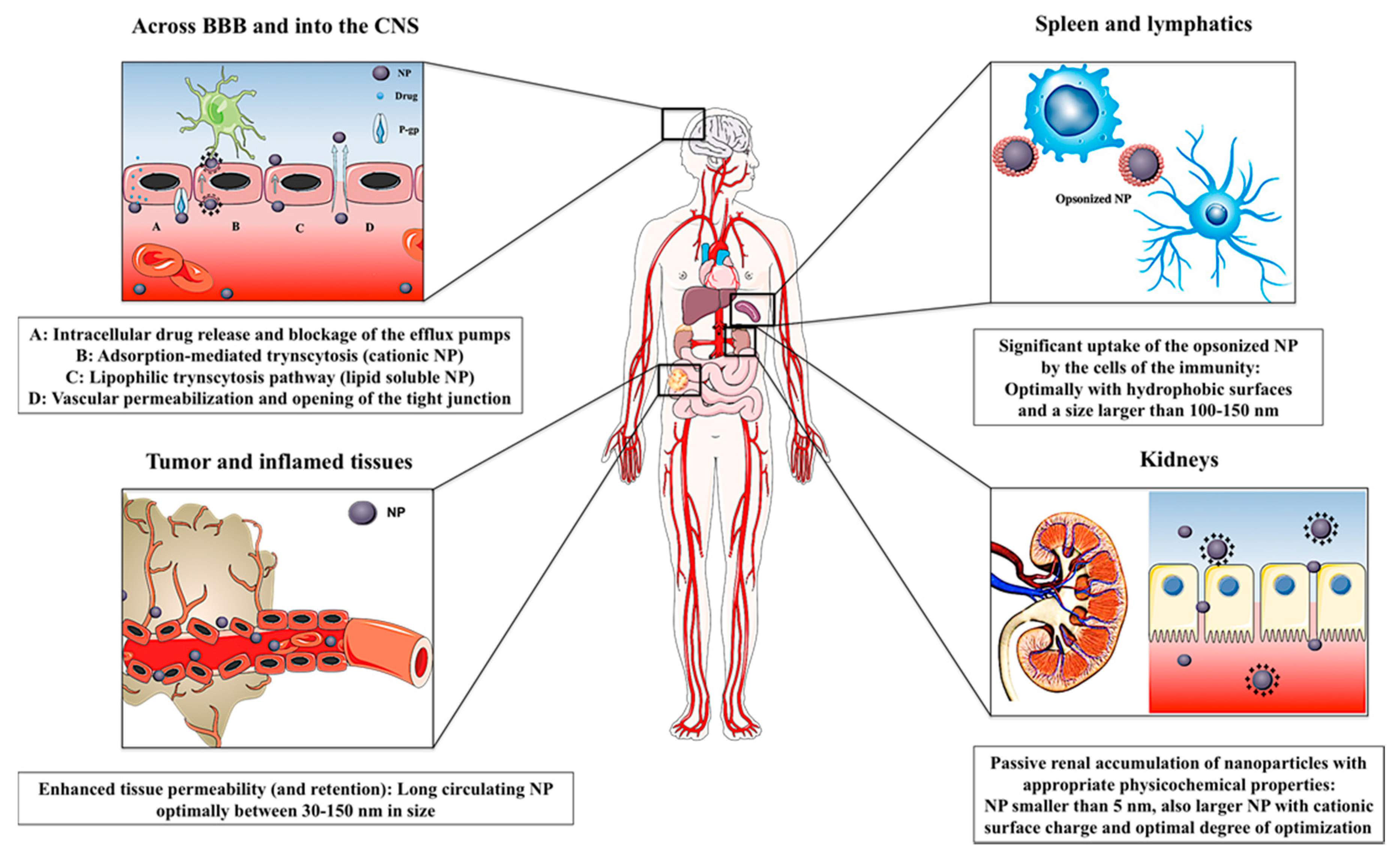{kind=link}
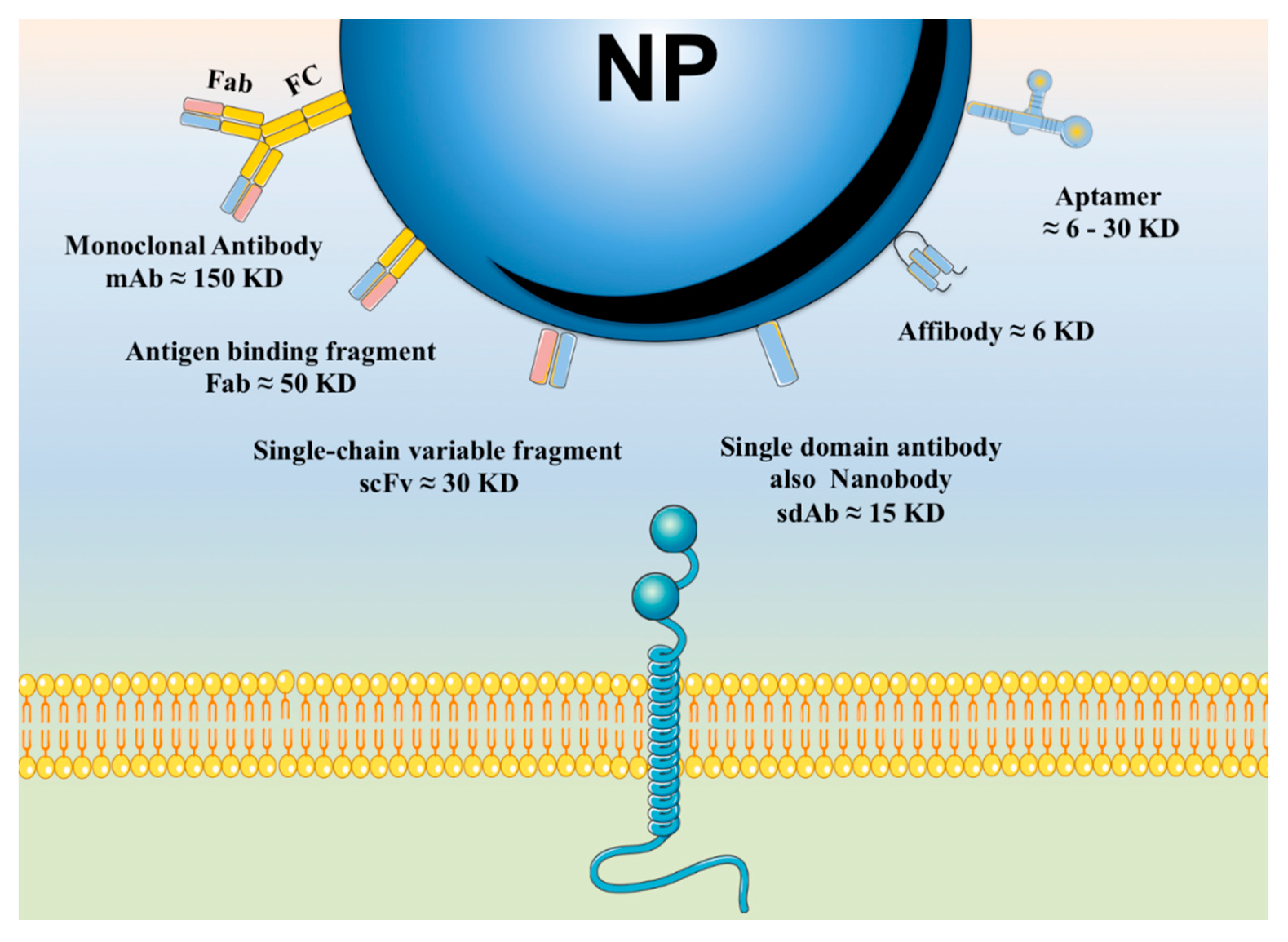{kind=link}
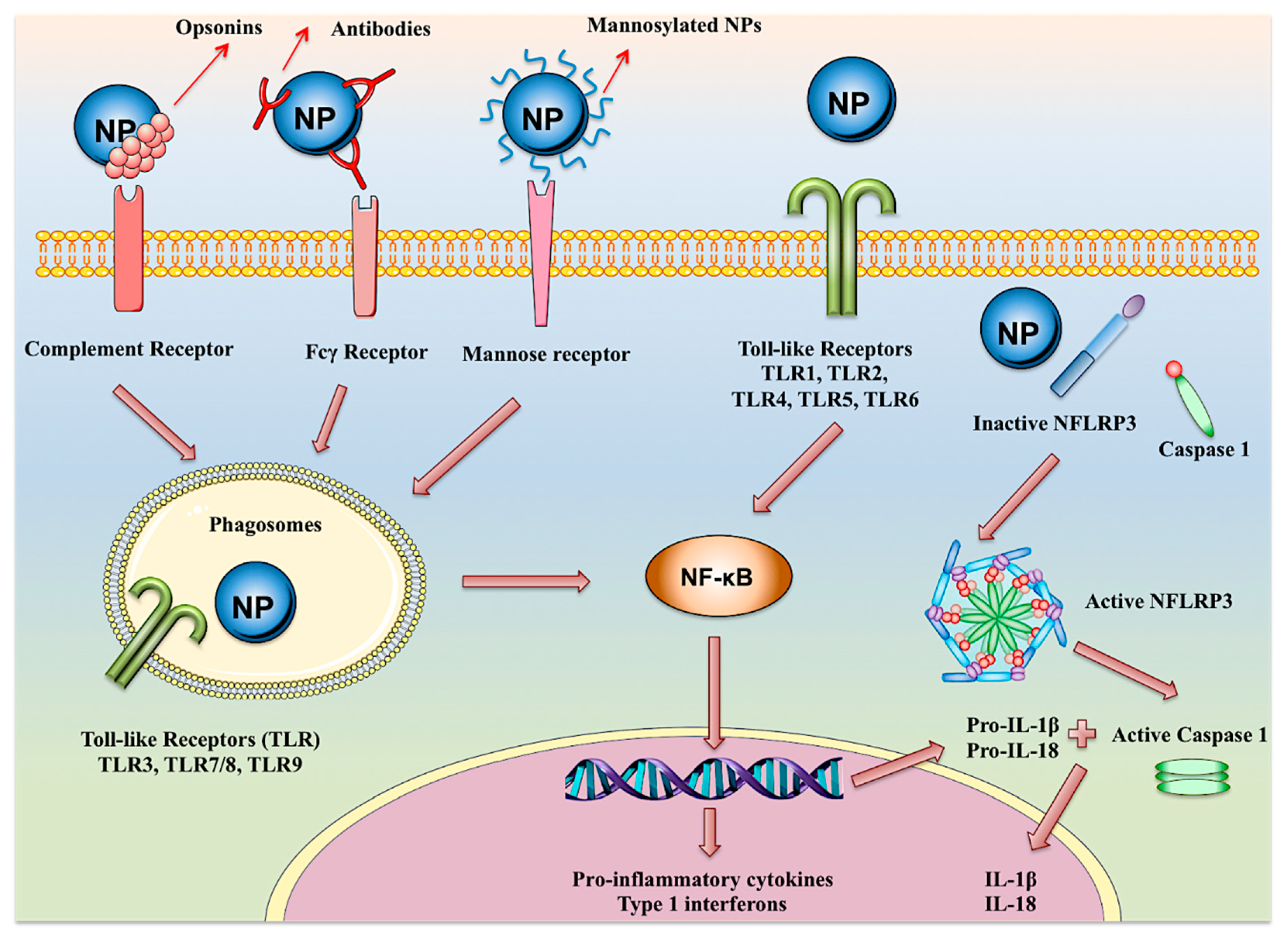{kind=link}
| Product | Nanocarrier | API | Indication | Function of the Carrier | Approval | Route of Injection |
|---|---|---|---|---|---|---|
| Abelcet® Amphocil® (Markted name outside USA) | Ribbon-like structures of a bilayered membrane and amphotericin B | Amphotericin B | Systemic fungal infection | MPS targeting | FDA 1995–1996 | IV |
| Abraxane® | Albumin-paclitaxel conjugates | Paclitaxel | Metastatic breast cancer, non-small-cell lung cancer | Passive tumor targeting | FDA 2005 | IV |
| Adagen® | Monomethoxypolyethylene glycol (PEG) covalently attached to the adenosine deaminase | Adenosine deaminase derived from bovine intestine | Enzyme replacement therapy for the treatment of severe combined immunodeficiency disease associated with adenosine deaminase deficiency | Increase of circulation time and reduction of immunogenicity | FDA 1990 | IM |
| Adynovate® | PEG-drug conjugate | Recombinant antihemophilic factor | Hemophilia A | Increase of the drug half life and stability | FDA 2016 | IV |
| AmBisome® | Liposome | Amphotericin B | Systemic fungal infections, cryptococcal meningitis and visceral leishmaniasis | MPS targeting | FDA 1997 | IV |
| Amphotec® | Colloidal dispersion of disc-like particles of amphotericin B and cholesteryl sulfate | Amphotericin B | Invasive aspergillosis in patients with kidney problems or unresponsive to conventional therapy | MPS targeting | FDA 1996 | IV |
| Cimzia® | PEGylated antibody | Fab’ fragment of a humanized anti-TNF-alpha antibody | Rheumatoid arthritis, active psoriatic arthritis, active ankylosing spondylitis, moderate-to-severe plaque psoriasis, Crohn’s disease | Increase of circulation time and reduction of immunogenicity | FDA 2008 | IV |
| Copaxone® | Polypeptide (average MW 6.4 kDa) composed of four amino acids (glatiramer) | Glatiramer acetate | Relapsing forms of multiple sclerosis | No mechanism attributable to nanosize | FDA 1996 | SC |
| DaunoXome® | Liposome | Daunorubicin citrate | AIDS-related Kopsi’s sarcoma | Passive tumor targeting | FDA 1996 | IV |
| DepoCyt® | Liposome | Cytarabine | Lymphomatous malignant meningitis | Sustained drug release | FDA 1999 | Intraventricular/intrathecal |
| DepoDur® | Liposome | Morphine sulfate | Pain relief | Sustained drug release | FDA 2004 | Epidural |
| Dexferrum® | Iron-dextran conjugate | Iron | Iron deficiency in patients with chronic kidney disease | MPS targeting, increase of dosage | FDA 1996 | IV |
| Diprivan® | Nanoemulsion | Propofol | Induction and maintenance of anesthesia | Solubility enhancement | FDA 1989 | IV |
| Eligard® | Polymeric nanoparticles | Leuprolide acetate | Advanced prostate cancer | Sustained drug release | FDA 2002 | SC |
| Exparel® | Liposome | Bupivacaine | Postsurgical analgesia | MPS targeting | FDA 2011 | IV |
| Feridex® | Dextran coated supramagenetic oxide nanoparticles | Diagnostic system | Liver and spleen lesion MRI | MPS targeting | FDA 1996 | IV |
| Feraheme™ (Ferumoxytol) | Dextran coated supramagenetic oxide nanoparticles | Iron | Treatment of iron deficient adults with chronic kidney disease | MPS targeting | FDA 2009 | IV |
| Ferrlecit® | Sodium ferric gluconate complex in sucrose injection | Iron | Treatment of iron deficient adults with chronic kidney disease | MPS targeting, increase of dosage | FDA 1999 | IV |
| Fungizone® | Micellar dispesion (following reconstitution) | Amphotericin B | Systemic fungal infections | Solubility enhancement | FDA 1966 | IV |
| Gendicine® | Virosome | p53 gene | Head and neck squamous cell carcinoma | Intracellular and nucleus targeting | People’s Republicof China 2003 | Intratumoral injection/Intravascular infusion |
| Genexol® | Micellar dispersion | Paclitaxel | Metastatic breast cancer, pancreatic cancer | Passive tumor targeting | South Korea 2001 | IV |
| Infed® | Iron-dextran complex | Iron | Treatment of iron deficient adults with chronic kidney disease | MPS targeting, increase of dosage | FDA 2009 | IV/IM |
| Inflexal® V | Liposome | Influenza virus antigens | Influenza prophylaxis | Intracellular targeting to the cells of the immunity | Switzerland 1997 | IV |
| Invega Sustenna® | Nanocrystal | Paliperidone palmitate | Schizophrenia, schizoaffective disorder | Sustained drug release, solubility enhancement | FDA 2009 | IM |
| Kadcyla® | Monoclonal antibody-drug conjugate | DM1 | Metastatic breast cancer | Passive and active tumor targeting (antibody against human epidermal growth factor receptor-2), redox responsiveness | FDA 2013 | IV |
| Krystexxa® | PEG-aptamer conjugate | Pegloticase | Chronic gout | Increase of circulation time and stability, active targeting | FDA 2010 | IV |
| Macugan® | Conjugate of PEG and anti vascular epidermal growth factor aptamer | Pegaptinib | Neovascular age related macular degredation | Increase of circulation time and stability, active targeting | FDA 2004 | Intravitreal |
| Marqibo® | Liposome | Vincristine sulfate | Acute lymphoid leukemia, relapsed or progressed Philadelphia chromosome-negative, | Passive tumor targeting | FDA 2012 | IV |
| Mepact™ | Liposomes | Mifamurtide | Non-metastasizing resectable osteosarcoma | MPS targeting | Europe 2009 | IV |
| Mircera® | methoxy polyethylene glycol-epoetin beta conjugate | Epoetin beta | Treatment of iron deficient adults with chronic kidney disease | Increase of stability | FDA 2007 | IV |
| MM-398 | Liposomes | Irinotecan | Treatment of iron deficient adults with chronic kidney disease | Passive tumor targeting | FDA 2015 | IV |
| Myocet® | Liposomes | Doxorubicin | Metastatic breast cancer | MPS targeting and formation of MPS depots for slow drug release | Europe 2000 | IV |
| NanoTherm® | Aminosilane-coated superparamagnetic iron oxide nanoparticles | Supramagnetic iron oxide nanoparticles | Glioblastoma, prostate and pancreatic cancer | Local tumor ablation under exposure to alternating magnetic field | Europe 2013 | Intratumoral |
| Neulasta® | PEG-filgrastim conjugate | Filgrastim (granulocyte colony-stimulating factor) | Febrile neutropenia, In patients with nonmyeloid malignancies; prophylaxis | Increase of protein stability | FDA 2002 | SC |
| Oncaspar® | PEG-L-asparaginase conjugate | L-asparaginase | Acute lymphoblastic leukemia | Increase of protein stability and circulation time | FDA 1994 | IV/IM |
| Onivyde® | Liposome | Irinotecan | Pancreatic cancer | Passive tumor targeting | FDA 2015 | IV |
| Ontak® | Protein (denileukin)-drug conjugate | Recombinant fusion protein of fragment A of diphtheria toxin (diftitox) | Primary cutaneous T-cell lymphoma, CD25-positive, persistent or recurrent disease | Intracellular targeting and lysosomal escape | FDA 1994/2006 | IV |
| Opaxio® | Drug conjugated polymeric nanoparticles | Paclitaxel | Glioblastoma | Passive tumor targeting | FDA 2012 | IV |
| Pegasys® | PEG-interferon alpha-2a conjugate | Interferon alpha-2a | Hepatitis B and C | Increase of circulation time and stability | FDA 2002 | IV |
| PegIntron® | PEG-interferon alphaa-2b conjugate | Interferon alpha-2b | Hepatitis C | Increase of circulation time and stability | FDA 2001 | IV |
| Plegridy® | PEG-interferon beta-1a conjugate | Interferon beta-1a | Multiple sclerosis | Increase of circulation time and stability | FDA 2014 | IV |
| Rebinyn® | GlycoPEG-recombinant coagulation factor IX conjugate | Recombinant coagulation factor IX | Hemophilia B | Increase of drug half life and Cmax | FDA 2017 | IV |
| Rexin-G® | Virosome | Gene for dominant-negative mutant form of human cycline G1 | Solid tumors | Intracellular and nucleus targeting | Philippines 2007 | IV |
| Ryanodex® | Nanocrystal | Dantrolene sodium | Acromegaly | Increase of the administration rate and dosis | FDA 2003 | IV |
| Somavert® | PEG-human growth hormone receptor antagonist conjugate | Pegvisomant | Acute lymphoblastic leukemia | Increase of protein stability and circulation time | FDA 1994 | IV/IM |
| Venofer t® | Iron-sucrose complex | Iron | Treatment of iron deficient adults with chronic kidney disease | Increase of administrable dose | FDA 2000 | IV |
| Visudyne® | Liposome | Verteporfin | Osteoarthritis knee pain | Solubility enhancement | FDA 2000 | IV |
| Vyxeos® | Liposome | Daunorubicin and cytarabine | Acute myeloid leukemia | Passive tumor targeting | FDA 2017 | IV |
| Zilretta® | Polymeric microparticles with nanosized pores | Triamcinolone acetonide | Primary cutaneous T-cell lymphoma, CD25-positive, persistent or recurrent disease | Sustained drug release | FDA 2017 | Intra-arterial |
| Zinostatin stimalamer® | Protein/copolymer of styrene-maleic acid-NCS conjugate | Antitumor protein NCS | Primary unresectable hepatocellular carcinoma | Passive tumor targeting | Japan 1994 | IV |
| Intervention | Nanosystem | Associated API | Function of the Nanosystem | Condition | Route of Injection | Stage of Evaluation | Status | Identifier |
|---|---|---|---|---|---|---|---|---|
| ABI-007 + Gemcitabin | Albumin stabilized NPs | Paclitaxel | Tumor targeting, drug solubility enhancement | - Metastatic breast cancer | IV | Phase II | Completed | NCT00110084 |
| ABI-007 | Albumin stabilized NPs | Paclitaxel | Tumor targeting, drug solubility enhancement | - Non-small cell lung cancer | IV | Phase I and II | Completed | NCT00073723 |
| BIND-014 | PEG-coated, PSMA targeted poly lactic NPs | Docetaxel | Passive and active tumor targeting, drug solubility enhancment |
- Metastatic cancer - Cancer - Solid tumors | IV | Phase I | Completed | NCT01300533 |
| BIND-014 | PEG-coated, PSMA targeted poly lactic NPs | Docetaxel | Passive and active tumor targeting, drug solubility enhancment |
- KRAS positive patients with non-small cell lung cancer - Squamous cell non-small cell lung cancer | IV | Phase II | Completed | NCT02283320 |
| BIND-014 | PEG-coated, PSMA targeted poly lactic NPs | Docetaxel | Passive and active tumor targeting, drug solubility enhancment |
- Urothelial carcinoma - Cholangiocarcinoma - Cervical cancer - Squamous cell carcinoma of head and neck | IV | Phase II | Terminated | NCT02479178 |
| BIND-014 | PEG-coated, PSMA targeted poly lactic NPs | Docetaxel | Passive and active tumor targeting, drug solubility enhancment | - Non-small cell lung cancer | IV | Phase II | Completed | NCT01792479 |
| BIND-014 | PEG-coated, PSMA targeted poly lactic NPs | Docetaxel | Passive and active tumor targeting, drug solubility enhancement | - Metastatic Castration-Resistant Prostate Cancer | IV | Phase II | Completed | NCT01812746 |
| C19-A3 GNP | Gold NPs | C19-A3 peptide | Enhanced APC uptake | Type 1 diabetes | Intradermal | Phase I | Active/not recruiting | NCT02837094 |
| Ceramide nanoliposome | Nanoliposome | Ceramide (non-conventional) | Passive tumor targeting |
- Cancer - Carcinoma - Solid tumors | IV | Phase I | Recruiting | NCT02834611 |
| CRLX101 | Drug-linear cyclodextrin–PEG copolymer conjugate | Camptothecin | Tumor targeting, drug solubility enhancement |
- Extensive stage small cell lung cancer - Recurrent small cell lung cancer | IV | Phase II | Terminated | NCT01803269 |
| CRLX101 + Olaparib | Drug-linear cyclodextrin–PEG copolymer conjugate | Camptothecin | Tumor targeting, drug solubility enhancement |
- Solid tumors - Small cell lung carcinoma - Non-small-cell lung carcinoma - Lung neoplasms - Small cell lung cancer - Lung cancer | IV | Phase I and II | Recruiting | NCT02769962 |
| ND-L02-s0201 (BMS-986263) | Vitamin A-moieties conjugated lipid NP | HSP47siRNA | Passive and active hepatic targeting, intracellular delivery, increase of the cargo’s stability | - Moderate to extensive hepatic fibrosis | IV | Phase I | Completed | NCT02227459 |
| Nab-paclitaxel | Albumin stabilized NPs | Paclitaxel | Passive tumor targeting, drug solubility enhancement | - Intraocular melanoma | IV | Phase II | Completed | NCT00738361 |
| Nab-paclitaxel | Albumin stabilized NPs | Paclitaxel | Tumor targeting, drug solubility enhancement | - Metastatic Breast Cancer | IV | Phase II | Terminated | NCT01416558 |
| Nab-paclitaxel | Albumin stabilized NPs | Paclitaxel | Ttumor targeting, drug solubility enhancement |
- Vascular Disease - Peripheral | IV | Phase II | Terminated | NCT00518284 |
| Nab-paclitaxel + Durvalumab + Carboplatin + Cisplatin | Albumin stabilized NPs | Paclitaxel | Tumor targeting, drug solubility enhancment |
- Carcinoma - Squamous cell - Oral cancer - Oropharynx cancer - Larynx cancer - Lip cancer - Esophageal cancer | IV | Phase II | Recruiting | NCT03174275 |
| Nab-paclitaxel + Pembrolizumab + Epirubicin + Cyclophosphamide | Albumin stabilized NPs | Paclitaxel | Tumor targeting, drug solubility enhancement | - Malignant neoplasm of breast | IV | Phase II | Active/not recruiting | NCT03289819 |
| Nab-paclitaxel + Pembrolizumab + Carboplatin | Albumin stabilized NPs | Paclitaxel | Tumor targeting, drug solubility enhancement | - Non-small cell lung cancer | IV | Phase III | Active/not recruiting | NCT02775435 |
| Nab-paclitaxel + Sargramostim | Albumin stabilized NPs | Paclitaxel | Tumor targeting, drug solubility enhancement |
- Brenner tumor - Fallopian tube cancer - Ovarian clear cell cystadenocarcinoma - Ovarian endometrioid adenocarcinoma - Ovarian mixed epithelial carcinoma - Ovarian mucinous cystadenocarcinoma - Ovarian serous cystadenocarcinoma - Ovarian undifferentiated adenocarcinoma - Peritoneal cavity cancer - Recurrent ovarian epithelial cancer - Stage III ovarian epithelial cancer - Stage IV ovarian epithelial cancer | IV | Phase II | Completed | NCT00466960 |
| NanoFlu | Recombinant hemagglutinin NPs | Recombinant hemagglutinin (antigen), Matrix-M™ adjuvant | Passive immune cell targeting, increase of immune stimulation | - Influenza prophylaxis | IM | Phase I and II | Completed | NCT03293498 |
| Nanoliposomal irinotecan | Nanoliposomes | Irinotecan | Tumor targeting | -High Grade Glioma | IV | Phase I | Enrolling by invitation | NCT02022644 |
| RXDX-107 | Albumin NPs | Bendamustine derivative (dodecanol alkyl ester) | Tumor targeting, Macropinocytosis mediated intracellular delivery | - Solid tumors | IV | Phase I | Terminated | NCT02548390 |
| STP705 | Polypeptide nanoparticle | Anti-fibrosis and anti-inflammatory siRNA | Enhanced targeted intracellular delivery | - Hypertrophic scar | Intradermal | Phase I and II | Recruiting | NCT02956317 |
| Type of the Nanocarrier | pH Responsive Moiety | Incorporated Cargo | Application | Reference |
|---|---|---|---|---|
| Layer-by-layer assembled nanoparticles | Neutravidin-iminobiotin bond | Quantum Dots | Stealth coating shedding, cancer therapy | [181] |
| Lipid core nanoparticles | polyethylene glycol-b-polyaspartic acid | Docetaxel | Stealth coating shedding, cancer therapy | [182] |
| Polymeric nanospheres | poly- (1,4-phenyleneacetone dimethylene ketal) | Dexamethasone | Intracellular drug release | [184] |
| Drug-polymer conjugate | Cleavable amide bond | Doxorubicin | Intracellular drug release, cancer therapy | [187] |
| Drug-polymer conjugate | Hydrazone bond | Doxorubicin | Intracellular drug release, cancer therapy | [188] |
| Cyclodextrin-derived nanoparticles | Poly(cyclohexane-1, 4-diyl acetone dimethylene ketal) | Paclitaxel | Intracellular drug release, cancer therapy | [189] |
| Drug-polymer conjugate | Hydrozone bond | Cisplatin | Intracellular drug release, cancer therapy | [190] |
| Mesoporpous silica nanoparticle | Hydrozone bond | Doxorubicin | Intracellular drug release, cancer therapy | [191] |
| Polymeric micelles | Poly(β-amino ester) | Doxorubicin | Intracellular drug release, cancer therapy | [192] |
| Nanogels | Amino groups | Oridonin | Intracellular drug release, Drug release in tumor extracellular environment, cancer therapy | [193] |
| Polymeric micelles | N-Boc-histidine | Doxorubicin | Drug release in tumor extracellular environment, cancer therapy | [179] |
| Polymeric micelles | Poly(β-amino ester) | Doxorubicin | Drug release in tumor extracellular environment, cancer therapy | [180] |
| Polymeric nanoparticles | Chitosan | Camptothecin | Drug release in tumor extracellular environment, cancer therapy | [194] |
| polymeric micelles | poly(beta-amino ester) | Doxorubicin | Drug release in tumor extracellular environment, cancer therapy | [195] |
| Flower-like polymeric micelle | poly(DEAP-Lys) | Doxorubicin | Drug release in tumor extracellular environment, cancer therapy | [196] |
| Micelle-like nanoparticles | Poly(N- methacryloyl-l-valine) and poly(N-methacryloyl-L-phenylala- nine) | - | - | [197] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shetab Boushehri, M.A.; Dietrich, D.; Lamprecht, A. Nanotechnology as a Platform for the Development of Injectable Parenteral Formulations: A Comprehensive Review of the Know-Hows and State of the Art. Pharmaceutics 2020, 12, 510. https://doi.org/10.3390/pharmaceutics12060510
Shetab Boushehri MA, Dietrich D, Lamprecht A. Nanotechnology as a Platform for the Development of Injectable Parenteral Formulations: A Comprehensive Review of the Know-Hows and State of the Art. Pharmaceutics. 2020; 12(6):510. https://doi.org/10.3390/pharmaceutics12060510
Chicago/Turabian StyleShetab Boushehri, Maryam A., Dirk Dietrich, and Alf Lamprecht. 2020. "Nanotechnology as a Platform for the Development of Injectable Parenteral Formulations: A Comprehensive Review of the Know-Hows and State of the Art" Pharmaceutics 12, no. 6: 510. https://doi.org/10.3390/pharmaceutics12060510
APA StyleShetab Boushehri, M. A., Dietrich, D., & Lamprecht, A. (2020). Nanotechnology as a Platform for the Development of Injectable Parenteral Formulations: A Comprehensive Review of the Know-Hows and State of the Art. Pharmaceutics, 12(6), 510. https://doi.org/10.3390/pharmaceutics12060510