Spatial Organization of the Gene Regulatory Program: An Information Theoretical Approach to Breast Cancer Transcriptomics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Gene Regulatory Program
1.2. Spatial Anomalies in Cancer-Associated GRPs
1.3. An Information Theoretical Approach to Gene Regulatory Programs
2. Analysis
2.1. Data
2.2. GRP Inference
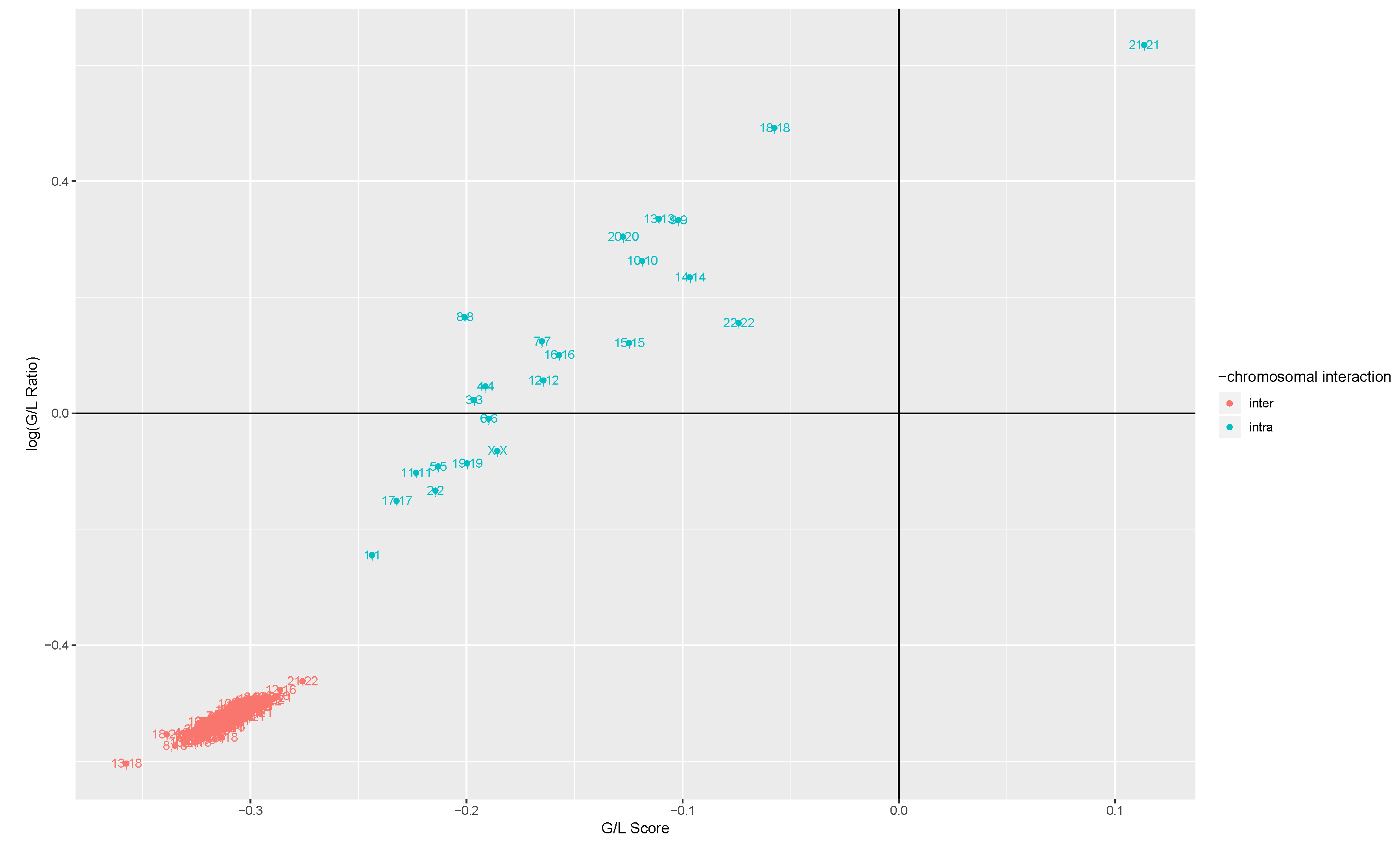
2.3. Measures of Change in MI between Health and Disease
2.3.1. Gain Loss Score
2.3.2. Gain Loss Ratio
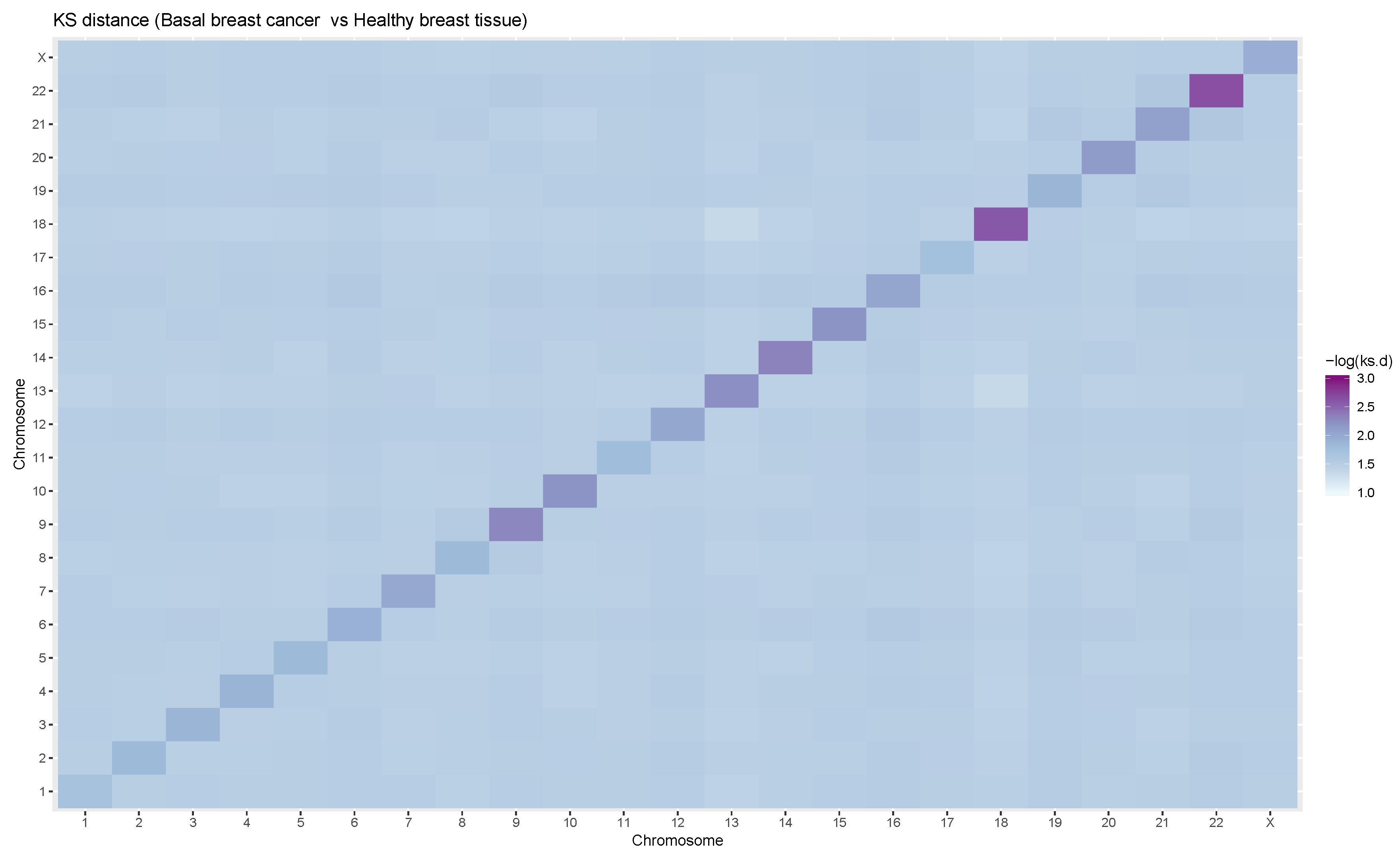
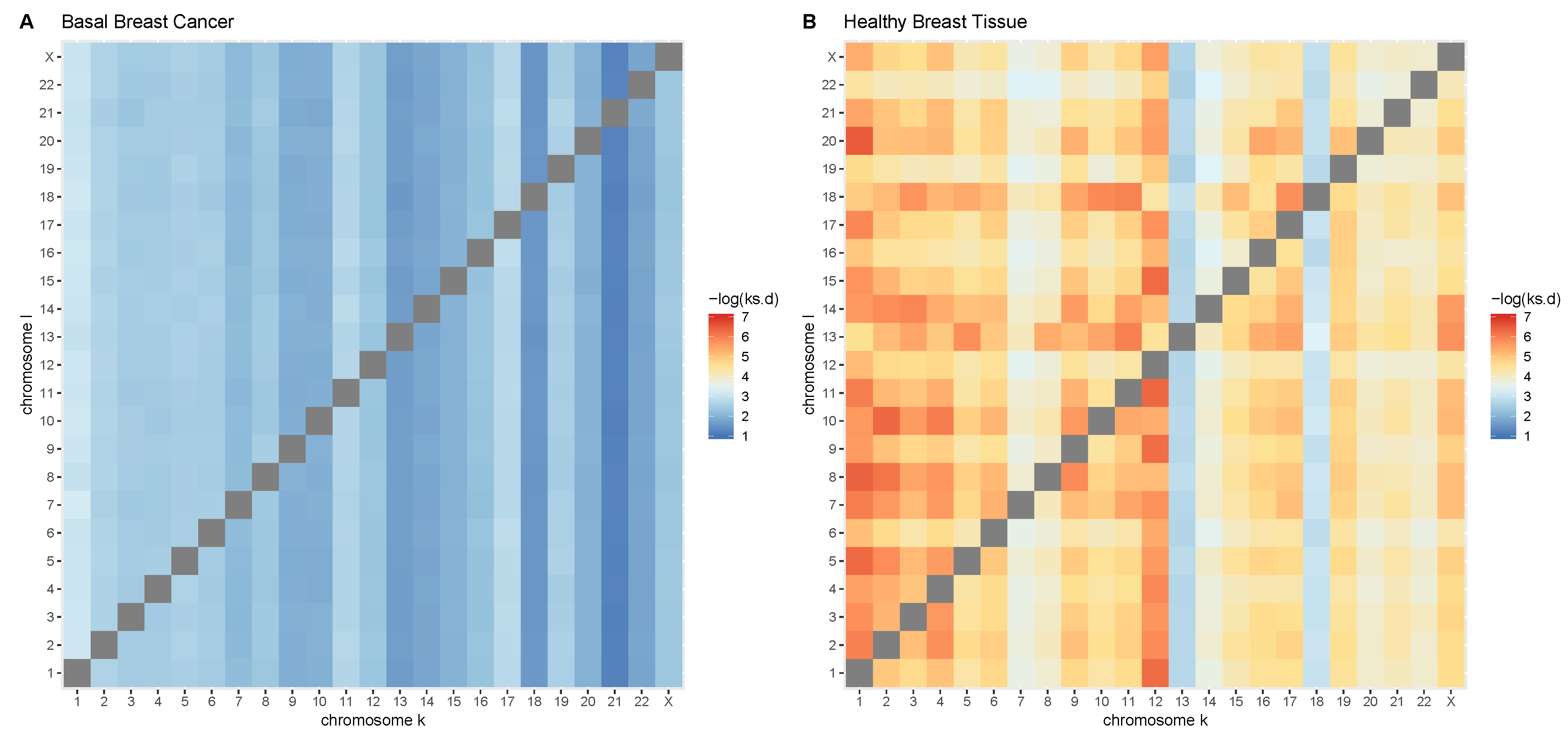
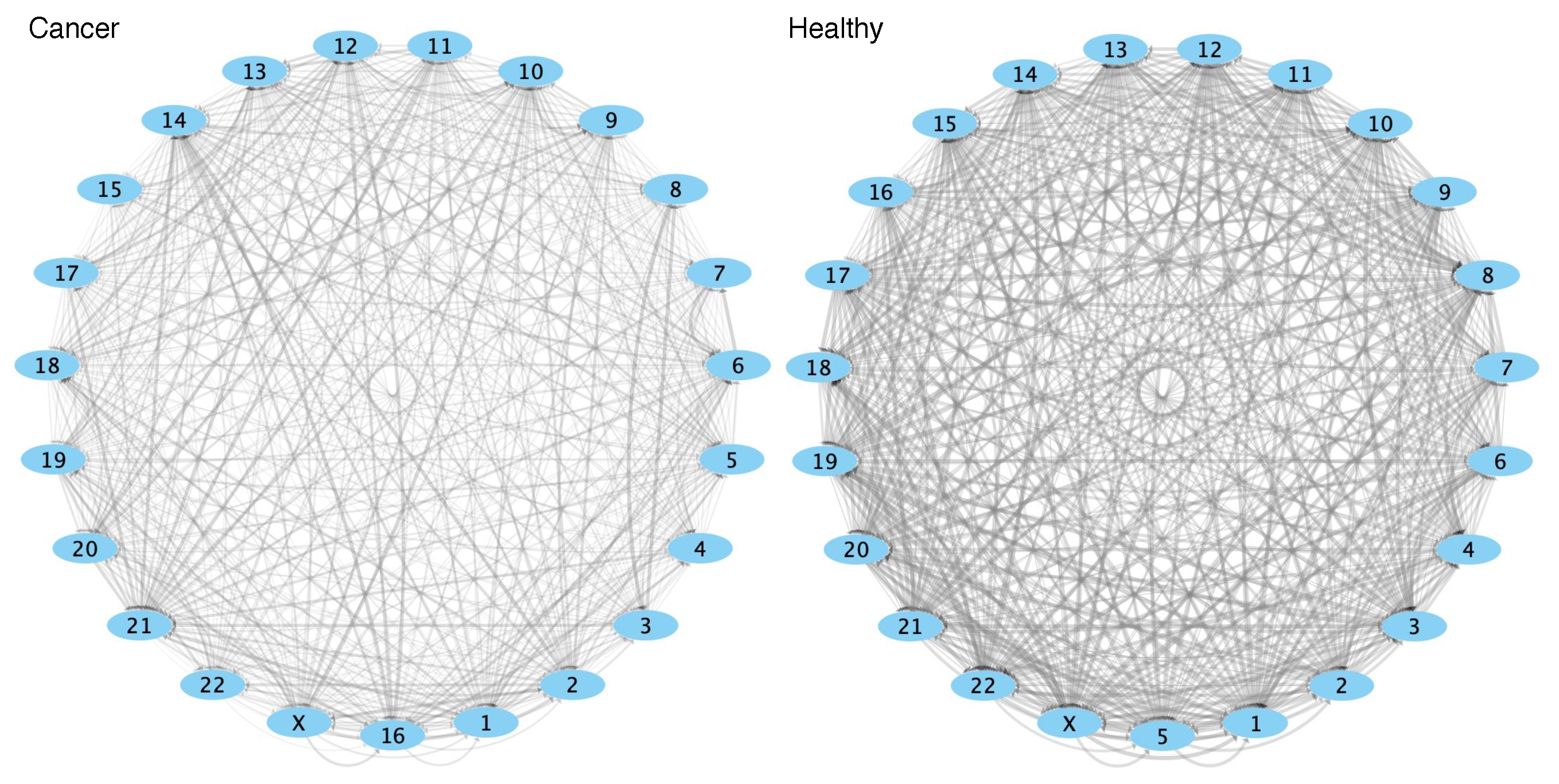
2.4. Comparison of GRPs between Control and Cancer Conditions
2.5. Comparison between cis- and trans-GRPs in Each Condition
3. Results and Discussion
3.1. Intra- and Inter-Chromosome Interactions Exhibit Differences in MI Changes
3.2. Cis-Patterns Depend on the Chromosome Size
3.3. Cis-GRPs Are More Similar in Health and Disease than Trans-GRPs
3.4. Differences in cis-and trans-GRPs in Health and Disease
Reconstructing a Spatial Dimension of Gene Regulation through Information Theoretic Approaches
4. Conclusions
- To what extent changes in gene regulation are relevant to breast cancer evolution?
- What are the possible consequences (functional or otherwise) of regulatory localization?
- Why different chromosomes behave differently? Including, but not limited to size effects.
- Are these patterns different in different cancers? Are they similar?
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GRP | Gene regulatory program |
| MI | Mutual information function |
| Probability distribution function | |
| TCGA | The Cancer Genome Atlas |
References
- Stergachis, A.B.; Neph, S.; Sandstrom, R.; Haugen, E.; Reynolds, A.P.; Zhang, M.; Byron, R.; Canfield, T.; Stelhing-Sun, S.; Lee, K.; et al. Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature 2014, 515, 365. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Arnal, L.; Sassone-Corsi, P. Chromatin dynamics of circadian transcription. Curr. Mol. Biol. Rep. 2015, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dryden, N.H.; Broome, L.R.; Dudbridge, F.; Johnson, N.; Orr, N.; Schoenfelder, S.; Nagano, T.; Andrews, S.; Wingett, S.; Kozarewa, I.; et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cremer, M.; Schmid, V.J.; Kraus, F.; Markaki, Y.; Hellmann, I.; Maiser, A.; Leonhardt, H.; John, S.; Stamatoyannopoulos, J.; Cremer, T. Initial high-resolution microscopic mapping of active and inactive regulatory sequences proves non-random 3D arrangements in chromatin domain clusters. Epigenet. Chromatin 2017, 10, 39. [Google Scholar] [CrossRef]
- Espinal-Enriquez, J.; Fresno, C.; Anda-Jauregui, G.; Hernandez-Lemus, E. RNA-Seq based genome-wide analysis reveals loss of inter-chromosomal regulation in breast cancer. Sci. Rep. 2017, 7, 1760. [Google Scholar] [CrossRef] [PubMed]
- De Anda-Jaúregui, G.; García-Cortés, D.; Fresno, C.; Espinal-Enríquez, J.; Hernández-Lemus, E. Intra-chromosomal regulation decay in breast cancer. Appl. Math. Nonlinear Sci. 2018, 4, 1–8. (in press). [Google Scholar]
- García-Cortés, D.; de Anda-Jáuregui, G.; Fresno, C.; Hernandez-Lemus, E.; Espinal-Enriquez, J. Loss of Trans Regulation in Breast Cancer Molecular Subtypes. bioRxiv 2018. [Google Scholar] [CrossRef]
- Hernández-Lemus, E.; Rangel-Escareño, C. The role of information theory in gene regulatory network inference. In Information: Theory New Research; Deloumeaux, P., Gorzalka, J.D., Eds.; Nova Science Publishing: New York, NY, USA, 2011; pp. 109–144. ISBN 978-1-62100-395-3. [Google Scholar]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Kindermann, R. Markov Random Fields and Their Applications; American Mathematical Society: Providence, RI, USA, 1980. [Google Scholar]
- Moussouris, J. Gibbs and Markov random systems with constraints. J. Stat. Phys. 1974, 10, 11–33. [Google Scholar] [CrossRef]
- Merchan, L.; Nemenman, I. On the sufficiency of pairwise interactions in maximum entropy models of networks. J. Stat. Phys. 2016, 162, 1294–1308. [Google Scholar] [CrossRef]
- TCGA. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61. [Google Scholar] [CrossRef]
- Tovar, H.; García-Herrera, R.; Espinal-Enríquez, J.; Hernández-Lemus, E. Transcriptional master regulator analysis in breast cancer genetic networks. Comput. Biol. Chem. 2015, 59, 67–77. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla Favera, R.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7 (Suppl. 1), S7. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Anda-Jáuregui, G.; Espinal-Enriquez, J.; Hernández-Lemus, E. Spatial Organization of the Gene Regulatory Program: An Information Theoretical Approach to Breast Cancer Transcriptomics. Entropy 2019, 21, 195. https://doi.org/10.3390/e21020195
de Anda-Jáuregui G, Espinal-Enriquez J, Hernández-Lemus E. Spatial Organization of the Gene Regulatory Program: An Information Theoretical Approach to Breast Cancer Transcriptomics. Entropy. 2019; 21(2):195. https://doi.org/10.3390/e21020195
Chicago/Turabian Stylede Anda-Jáuregui, Guillermo, Jesús Espinal-Enriquez, and Enrique Hernández-Lemus. 2019. "Spatial Organization of the Gene Regulatory Program: An Information Theoretical Approach to Breast Cancer Transcriptomics" Entropy 21, no. 2: 195. https://doi.org/10.3390/e21020195