Abstract
This paper describes a modified method of preparation of a number of α-aryl-α-(pyridazin-3-yl)-acetonitriles via the C-arylation reaction of the corresponding carbanions of phenylacetonitriles using 3-chloropyridazine derivatives. KOH and DMSO were used in the deprotonation process, which made the reaction very simple and safe to perform. Nitriles were obtained in the hydrolysis reaction to the corresponding α-aryl-α-(pyridazin-3-yl)-acetamide derivatives, which were next subjected to cyclization to afford the final products. A number of new derivatives of 7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione were synthesized in the cyclocondensation reaction of respective α-aryl-α-(pyridazin-3-yl)-acetamides with diethyl carbonate in the presence of EtONa. The structure and composition of the new compounds were confirmed by IR, 1H- and 13C- NMR analyses and by elemental C, H and N analysis.
Introduction
The imide moiety, which is present in various heterocyclic systems, may have a significant effect on the biological activity of their derivatives. This functionality is a significant structural element of an important group of compounds belonging to the so-called long-chain arylpiperazines (LCAPs), ligands of 5-HT1A receptors of high affinity (see Figure 1) [1,2,3,4,5].
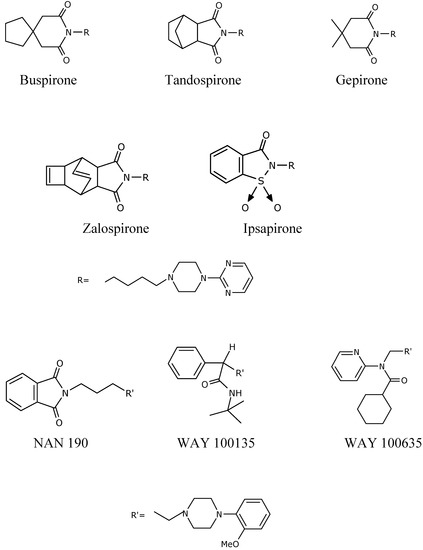
Figure 1.
LCAPs with an amide and imide moiety.
First Buspirone and then Tandospirone were introduced for medicinal purposes; both of them are prominent representatives of the LCAPs group. These drugs, as 5-HT1A receptor agonists, revolutionized the treatment of anxiety disorder by the mechanism of serotonergic neurotransmission [2,4,5,6,7,8,9,10]. As shown by the investigations to date, the imide group in Buspirone is an element of the nonpharmacophore part and plays an important role in stabilization of the ligand – 5-HT1A receptor complex. Moreover, it also affects the lipophility of the ligand, which in turn has a significant effect on selectivity of the ligand to 5-HT1A receptor, with respect to other receptors such as 5-HT2A or α1 [2,7,9]. Our earlier investigations also dealt with the preparation of new heterocyclic systems, such as for instance the derivatives of pyrido[1,2-c]pyrimidine and pyrrole[1,2-a]pyrazine, which have the afore-mentioned grouping [11,12,13]. We have now focused our studies on a little known pyrimido[1,6-b]pyridazine system, the derivatives of which had already been described by Bemis et al. Our investigations were concerned with Kinase p38 inhibitors which possess antitumor activity [14]. The aim of our investigations was to synthesize the new 7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione derivatives 4a-f, 4f′ and 4g′, which contain the imide moiety in their structure. These derivatives should be important substrates for further synthesis of the potential ligands of 5-HT1A receptors of higher selectivity in the LCAPs group.
Results and Discussion
The compounds described in this paper were obtained according to Scheme 1
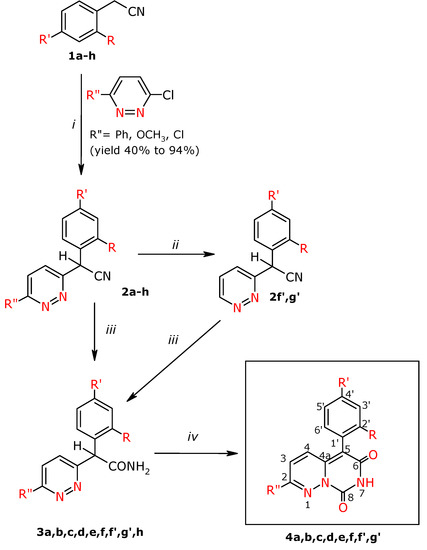
Scheme 1.
Synthesis of the title compounds and substituents.
The starting α-aryl-α-(pyridazin-3-yl)-acetonitriles 2a-h, which were necessary for the synthesis, were obtained in a C-arylation reaction of stabilized carbanions of appropriate arylacetonitriles using 3-chloropyridazine derivatives. A modified method of deprotonation of the above-mentioned arylacetonitriles was applied, using KOH in DMSO. This method was elaborated by us earlier and was used for the synthesis of a number of derivatives of α-aryl-α-(2-pyridyl)-acetonitriles in a C-arylation reaction using 2-bromopyridine [11,12]. The aim of the present studies was first of all to simplify the process of C-arylation to obtain a method which gives reproducible yields and is safe to perform.
The deprotonation processes used to obtain stabilized carbanions in C-arylation, were previously carried out using NaH, NaNH2 in anhydrous solvents such as THF or benzene [14,15,16,17]. Yamada et al. described the preparation of α-phenyl-α-(6-phenyl-pyridazin-3-yl)-acetonitrile 2a in the C-arylation reaction of phenylacetonitrile using 3-chloro-6-phenylpyridazine in the presence of NaNH2 in benzene [15]. The derivative 2f′ was obtained by Yamada et al. in a C-arylation of phenylacenitrile using 3-chloropyridazine in the presence of NaNH2 in benzene [15]. The same derivative was obtained by C-arylation of the above-mentioned phenylacetonitrile using 3-methoxypyridazine in the presence of NaH in THF [17]. Abboto et al. described synthesis of 2f′ in a C-arylation reaction of phenylacetonitrile using 3,6-dichloropyridazine in presents of NaH in THF [16].
The modified method used in the present work allowed us to obtain a number of new nitriles 2b-e, 2g, 2h, 2g′ in good yields. During the next stage of our studies the nitriles 2g and 2f, possessing chlorine in position 6 of the pyridazine moiety, were subjected to dehalogenation using ammonium formate and Pd/C in MeOH, instead of the hydrogenolysis process which had been used by Abbotto et al. during preparation of the derivative 2f′ [16].
A number of new derivatives of α-aryl-α-(pyridazin-3-yl)-acetamide 3a-f, 3h, 3f′ and 3g′, were obtained by hydrolyzing the above-obtained nitriles 2a-f, 2h, 2g′, 2f′ in acidic medium. The hydrolysis process was performed under different conditions. The nitriles 2a-f, 2h, 2f′ and 2g′ were hydrolyzed with sulphuric acid at the temperature of 50 °C. Hydrolysis of the nitriles 2c and 2d was performed in the sulphuric acid – acetic acid mixture at the temperature of 100 °C for 1 h for compound 3c, whereas the derivative 3d was obtained at 50 °C and the process was continued for 10 h.
The final compounds, the derivatives of 7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione 4a-f, 4f′ and 4g′ were obtained in good yields in an intermolecular cyclocondensation reaction of the respective amides 3a-f, 3h, 3f′ and 3g′ with diethyl carbonate in the presence of EtONa. During the cyclization process of the derivatives 3f and 3h the formation of one cyclization product 4f was observed as a result of simultaneous chlorine substitution in position 2 of the cyclic compound on the -OEt group in amide 3f, whereas in the case of amide 3h the –OMe group was exchanged into –OEt (see Scheme 2).
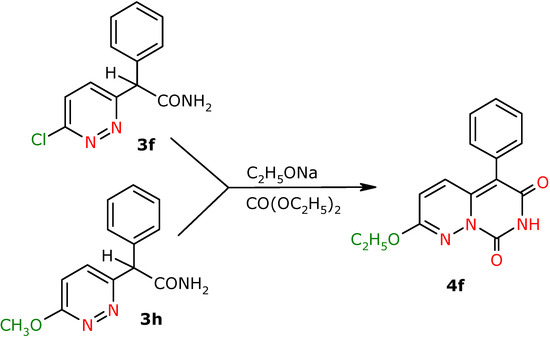
Scheme 2.
The structures and composition of the new intermediate and final compounds were confirmed by analysis of their IR, 1H- and 13C-NMR spectra, and also by elemental C, H and N analyses. The NMR spectra are mutually correlated and were in agreemeent with the literature data for similar systems, as well as with the theoretical spectra calculated according to ACD/NMR Predictor v. 8.09 program. In the amide proton spectra (compounds of series 3) we observed two NH proton signals from the NH2 group, which could indicate that there is nonequivalence of magnetic surroundings of these protons due to inhibited rotation about the C-N bond.
Conclusions
Applying a modified method (KOH, DMSO) for the deprotonation of phenylacetonitrile derivatives, we obtained a number of α-aryl-α-(pyridazin-3-yl)acetonitriles 2b-e, 2g, 2h, 2g′ that were not described previously in a C-arylation reaction of the carbanions obtained. Next, the nitriles were hydrolyzed in an acidic medium and thus we obtained another group of new compounds, the α-aryl-α-(pyridazin-3-yl)-acetamide derivatives 3a-f, 3h, 3f′ and 3g′. The title compounds, the 7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione derivatives 4a-f, 4f′, 4g′ were obtained in the cyclocondensation reaction of amides 3a-f, 3h, 3f′, 3g′ with diethyl carbonate in the presence of EtONa. All the intermediate and title compounds described in this paper were obtained in good yields.
Experimental
General
Melting points of the substances were determined on a Mel-Temp® 3.0 (Barnsted/Thermolyne, USA) apparatus and are uncorrected. Elemental analysis was performed on a Perkin-Elmer CHN model 2400 analyzer. The IR spectra (KBr tablets) were performed on a Shimadzu FT IR-8300 apparatus. 1H- and 13C-NMR spectra were recorded on the following spectrometers: a Bruker Avance DMX WB of basic frequency for the protons: 400.133 MHz, for nuclei 13C: 100.623 MHz and a Varian type Unity Plus of basic frequency for the protons: 500.605 MHz, for nuclei 13C: 125.877 MHz at room temperature. CDCl3 was used as solvent and tetramethylsilane as internal standard. The chemical shifts of resonance signals are given in ppm, and the coupling constants are in Hz. Thin-layer chromatography (TLC) was performed on Merck DC-Platten Kiesegel 60 F254 plates, developed in the dioxane, toluene, ethanol systems, 25% NH4OH (6.0:3.2:0.5:0.2, v/v) or chloroform, methanol, diethyl ether, 25% NH4OH (6.0:2.0:1.8:0.2, v/v) and visualized on a UV lamp. Analytical samples of the compounds were obtained by purification on the chromatographic column using the flash technique and Merck Kieselgel 60 (230-400 mesh) filling. They were eluted with methylene chloride/methanol mixtures (99:1; 97:3; 95:5, v/v). The starting phenylacetonitrile derivatives, 3,6-dichloropyridazine and 3-chloro-6-methoxypyridazine were commercial products purchased from Aldrich and used without further purification. The other starting reagent 3-chloro-6-phenylpyridazine was prepared by the reported procedure [18,19].
General procedure for the preparation of α-aryl-α-(3-pyridazin-3-yl)acetonitriles 2a-h
To DMSO (32.5 mL) was added KOH (16.5 g, 0.29 mol) and the mixture was stirred for 0.5 h at room temperature. Next an appropriate phenylacetonitrile derivative (0.11 mol) in DMSO (10 mL) was added dropwise and stirring was continued for another 0.5 h at room temperature. Next the appropriate 3-chloropyridazine derivative (0.07 mol) was added portionwise to the mixture, which was stirred at a temperature of 50 °C for 12 h. Next the post-reaction mixture was poured into ice water (1000 mL). The separated precipitates were filtered off. The obtained crude products 2c, 2d, 2e, 2f, 2f′ were purified by flash chromatography using CH2Cl2/MeOH (97:3 v/v), and then CH2Cl2/MeOH (99:1 v/v) as eluents, and then compounds 2c crystallized from absolute EtOH, 2d and 2f′ from EtOH. Compounds 2a, 2h, 2g′ were macerated with the ethyl acetate – hexane (1:1 v/v) mixture, and were then crystallized from EtOH. Compound 2b was crystallized from EtOH, 2g with ethyl acetate.
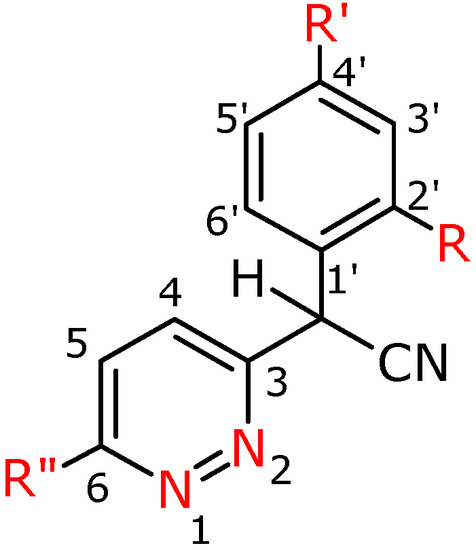
Figure 2.
Numbering system for compounds 2a-h.
α-Phenyl-α-(6-phenylpyridazin-3-yl)-acetonitrile (2a). Yield 65%; white crystals; m.p. 206.0-207.0ºC; {lit. [15], 201-203ºC}; IR (cm-1): 2246 (CN); 1H-NMR (500 MHz) δ: 5.73 (s, 1H, CH), 7.36 (tt, 3J=7.0, 4J=1.0, 1H, C4’H), 7.41 (td, 3J=7.5, 4J=1.5, 2H, C3’H, C5’H), 7.49- 7.56 (m, 5H, C2”H-C6”H), 7.63 (d, 3J=9.0, 1H, C4H), 7.88 (d, 1H, C5H), 8.07 (dd, 3J=7.5, 4J=2.0, 2H, C2’H, C6’H); 13C-NMR (125 MHz) δ: 43.6 (CH), 118.1 (CN), 125.1 (C5), 125.8 (C4), 127.2 (C3”, C5”), 127.6 (C2’, C6’), 128.9 (C4’), 129.2 (C3’, C5’), 129.5 (C2”, C6”), 130.6 (C4”), 133.5 (C1’), 135.4 (C1”), 157.0 (C3), 159.1 (C6); Anal. calcd. for C18H13N3: C, 79.68; H, 4.83, N, 15.49; found: C, 79.62; H, 4.83;N, 15.44.
α-(4-Fluorophenyl)-α-(6-phenypyridazin-3-yl)-acetonitrile (2b). Yield 40%; white crystals; m.p. 200.8-201.2 ºC; IR (cm-1): 2248 (CN); 1H-NMR (500 MHz) δ: 5.70 (s, 1H, CH), 7.10 (m, 2H, C3’H, C5’H), 7.49-7.56 (m, 5H, C2”H-C6”H), 7.64 (d, 3J=9.0, 1H, C4H), 7.90 (d, 1H, C5H), 8.08 (m, 2H, C2’H, C6’H); 13C-NMR (125 MHz) δ: 42.9 (CH), 116.6 (d*,2J=22, C3’,C5’), 117.9 (CN), 125.2 (C4), 125.6 (C5), 127.2 (C2”, C6”), 129.2 (C3”, C5”), 129.4 (d*, 4J=3.6, C1’), 129.4 (d*, 3J=8.7, C2’,C6’), 130.6 (C4”), 135.3 (C1”), 156.7 (C3), 159.1 (C6), 162.9 (d*, 1J=249.1, C4’); Anal. calcd. for C18H12N3F: C, 74.73; H, 4.18, N, 14.52; found: C, 74.64; H, 4.08; N, 14.56.
α-(4-Tolyl)-α-(6-phenylpyridazin-3-yl)-acetonitrile (2c). Yield 71%; white crystals; m.p. 143.9-144.6 ºC; IR (cm-1): 2250 (CN); 1H-NMR (400 MHz) δ: 2.34 (s, 3H, CH3), 5.67 (s, 1H, CH), 7.20 (d, 2H, C3’H, C5’H), 7.40 (d, 3J=7.6, 2H, C2’H, C6’H), 7.51 (m, 3H, C3”H, C4”H, C5”H), 7.60 (d, 1H, C4H), 7.86 (d, 3J=8.8, 1H, C5H), 8.06 (m, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 21.3 (CH3), 43.4 (CH), 118.5 (CN), 125.3 (C5), 125.9 (C4), 127.3 (C2’, C6’), 127.6 (C3”, C5”), 129.3 (C2”, C6”), 130.3 (C3’, C5’), 130.7 (C4”), 131.8 (C1’), 135.6 (C4’), 139.0 (C1”), 157.3 (C6), 159.1 (C3); Anal. calcd. for C19H15N3: C, 79.98; H, 5.30, N, 14.72; found: C, 79.20; H, 5.26; N, 15.54.
α-(4-Methoxyphenyl)-α-(6-phenylpyridazin-3-yl)-acetonitrile (2d). Yield 94%; white crystals; m.p. 206.8-209.4 ºC; IR (cm-1): 2250 (CN); 1H-NMR (400 MHz) δ: 3.80 (s, 3H, OCH3), 5.66 (s, 1H, CH), 6.92 (d, 2H, C3’H, C5’H), 7.44 (d, 3J=8.4, 2H, C2’H, C6’H), 7.52 (m, 3H, C3”H, C4”H, C5”H), 7.61 (d, 1H, C4H), 7.88 (d, 3J=8.8, 1H, C5H), 8.08 (m, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 42.1 (CH), 55.6 (OCH3), 114.4 (C3’, C5’), 116.2 (C4), 117.9 (CN), 124.8 (C1’), 127.9 (C5), 127.9 (C2’, C6’), 128.4 (C6),129.1 (C2”, C6”), 129.6 (C3”, C5”), 131.7 (C4”), 133.7 (C1”), 157.3 (C3), 159.2 (C4’); Anal. calcd. for C19H15N3O: C, 75.73; H, 5.02, N, 13.94; found: C, 75.77; H, 5.42; N, 13.95.
α-(2-Methoxyphenyl)-α-(6-phenylpyridazin-3-yl)-acetonitrile (2e). Yield 85%; white crystals; m.p. 151.0-152.0 ºC; IR (cm-1): 2245 (CN); 1H-NMR (400 MHz) δ: 3.84 (s, 3H, OCH3), 5.93 (s, 1H, CH), 6.92 (d, 3J=8.4, 1H, C3’H), 7.04 (t, 3J=7.2, 1H, C5’H), 7.36 (t, 3J=7.6, 1H, C4’H), 7.51 (ps, 3H, C3”H, C4”H, C5”H), 7.57 (pt, 2H, C4H, C6’H), 7.85 (d, 3J=8.8, 1H, C5H), 8.08 (pd, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 38.4 (CH), 55.9 (OCH3), 111.5 (C3’), 118.4 (CN), 121.6 (C5’), 122.3 (C1’), 124.7 (C5), 126.2 (C4), 127.3 (C2”, C6”), 129.3 (C3”, C5”), 129.8 (C6’), 130.6 (C4”), 130.7 (C4’), 135.8 (C1”), 156.4 (C3), 156.8 (C6), 158.8 (C2’); Anal. calcd. for C19H15N3O: C, 75.73; H, 5.02, N, 13.94; found: C, 75.47; H, 5.10; N, 13.71.
α-Phenyl-α-(6-chloropyridazin-3-yl)-acetonitrile (2f). Yield 69%; white crystals; m.p. 131.1-131.3 ºC; {lit. [16], 125-126ºC}; IR (cm-1): 2239 (CN); 1H-NMR (500 MHz) δ: 5.67 (s, 1H, CH), 7.38 (m, 1H, C4’H), 7.41 (m, 2H, C3’H, C5’H), 7.47 (m, 2H, C2’H, C6’H), 7.55 (m, 2H, C4H, C5H); 13C-NMR (125 MHz) δ: 43.1 (CH), 117.6 (CN), 127.5 (C2’, C6’), 127.7 (C5), 129.1 (C4’), 129.6 (C4), 129.7 (C3’, C5’), 132.8 (C1’), 157.0 (C3), 157.9 (C6); Anal. calcd. for C12H8N3Cl: C, 62.75; H, 3.51, N, 18.30; found: C, 62.85; H, 3.43; N, 18.46.
α-(4-Fluorophenyl)-α-(6-chloropyridazin-3-yl)-acetonitrile (2g). Yield 75%; white crystals; m.p. 133.0-133.5 ºC; IR (cm-1): 2251 (CN); 1H-NMR (500 MHz) δ: 5.65 (s, 1H, CH), 7.08-7.11 (m, 2H, C2’H, C6’H), 7.44-7.47 (m, 2H, C3’H, C5’H), 7.55-7.66 (m, 2H, C4H, C5H); 13C-NMR (125 MHz) δ: 42.4 (CH), 116.8 (C3’, C5’), 117.5 (CN), 127.7 (C5), 129.4 (C2’, C6’), 129.5 (C1’), 129.7 (C4), 157.6 (C3), 157.7 (C6), 163.9 (C4’); Anal. calcd. for C12H7N3FCl: C, 58.20; H, 2.85; N, 16.97; found: C, 58,07; H, 2.86; N, 16,77.
α-Phenyl-α-(6-methoxypyridazin-3-yl)-acetonitrile (2h). Yield 53%; white crystals; m.p. 153.0-156.0 ºC; IR (cm-1): 2245 (CN); 1H-NMR (500 MHz) δ: 4.14 (s, 3H, OCH3 ), 5.59 (s, 1H, CH), 6.99 (d, 1H, C5H), 7.37 (dd, 3H, C3’H, C4’H, C5’H), 7.42 (d, 3J=9.0, 1H, C4H), 7.47 (d, 2H, 3J=8.0, C2’H, C6’H); 13C-NMR (125 MHz) δ: 43.1 (CH), 55.1 (OCH3), 118.1 (CN), 118.9 (C5), 127.5 (C2’, C6’), 128.2 (C4), 128.8 (C4’), 129.5 (C3’, C5’), 133.6 (C1’), 153.8 (C3), 164.8 (C6); Anal. calcd. for C13H11N3O: C, 69.32; H, 4.92, N, 18.65; found: C, 69.01; H, 5.31; N, 18.52.
Synthesis of α-phenyl-α-(pyridazin-3-yl)-acetonitrile (2f′) and α-(4-fluorophenyl)-α-(pyridazin-3-yl)-acetonitrile (2g′)
The appropriate nitrile 2f or 2g (0.06 mol) was placed in MeOH (300 mL), along with ammonium formate (18.9 g, 0.3 mol) and Pd/C (10%, 3.4g). The combined mixture was brought to boiling while stirring, the reaction was performed under nitrogen. For compound 2f′ the reaction time was 2 h, for compound 2g′ it was 1.5 h (TLC). After cooling the whole was filtered off from the catalyst and the filtrate was evaporated to dryness. The obtained precipitate was dissolved in CHCl3 – H2O mixture (1:1 v/v, 100 mL) and then extracted with CHCl3 (3 x 100 mL). The combined chloroform extracts were dried with anhydrous MgSO4. After filtering off the drying agent the filtrate was evaporated to dryness. The obtained precipitate 2 f′ was purified on the chromatographic column by flash technique, using CH2Cl2/MeOH eluents (97:3 v/v), and then it was crystallized from ethanol. Compound 2g′ was purified by maceration with the hexane : ethyl acetate mixture (1:1 v/v).
α-Phenyl-α-(pyridazin-3-yl)-acetonitrile (2f′).Yield 57%; white crystals; m.p. 139.8-141.7 ºC; {lit. [15,16], 136-137 ºC; lit. [17], 138-140 ºC}; IR (cm-1): 2245 (CN); 1H-NMR (500 MHz) δ: 5.69 (s, 1H, CH), 7.37 (m, 1H, C4’H), 7.41 (m, 2H, C3’H, C5’H), 7.50 (m, 2H, C2’H, C6’H), 7.52 (m, 1H, C5H), 7.59 (m, 1H, C4H), 9.19 (m, 1H, C6H); 13C-NMR (125 MHz) δ: 44.2 (CH), 118.3 (CN), 125.7 (C4, C5), 127.9 (C2’, C6’), 129.3 (C4’), 129.9 (C3’, C5’), 133.7 (C1’), 151.4 (C6), 159.1 (C3); Anal. calcd. for C12H9N3: C, 73.83; H, 4.65, N, 21.52; found: C, 73.62; H, 4.87; N, 21.46.
α-(4-Fluorophenyl)-α-(pyridazin-3-yl)-acetonitrile (2g′). Yield 58%; white crystals; m.p. 127.0-128.0 ºC; IR (cm-1): 2248 (CN); 1H-NMR (500 MHz) δ: 5.66 (s, 1H, CH), 7.07-7.11 (m, 2H, C2’H, C6’H), 7.46-7.61 (m, 4H, C4H, C5H, C3’H, C5’H), 9.18-9.20 (dd, 1H, C6H); 13C-NMR (125 MHz) δ: 43.2 (CH), 116.7 (C3’, C5’), 117.8 (CN), 127.7 (C5), 129.2 (C2’, C6’), 129.4 (C1’), 129.5 (C4), 151.2 (C6), 158.6 (C3), 163.9 (C4’); Anal. calcd. for C12H8N3F: C, 67.60; H, 3.78; N, 19.71; found: C, 66.41; H, 3.75; N, 19.45.
General procedure for the preparation of α-aryl- α-(pyridazin-3-yl)-acetamides 3a-f, 3h, 3f′, 3g′
To concentrated sulphuric acid (50 mL) was added the appropriate nitrile 2a, b, c, f, h, f′, g′ (0.03 mol). The mixture was stirred for 12 h at the temperature of 50 °C. Compounds 2e and 2d were hydrolyzed in the mixture of concentrated sulphuric acid (10 mL) and glacial acetic acid (30 mL) for 1 h at the temperature of 100 °C (2e) and for 10 h at the temperature of 50 oC (2d).
Then the post-rection mixture was cooled and made alkaline at a temperature of 5 °C with concentrated NH4OH to pH~8. The separated reaction product was extracted with chloroform (3 x 150 mL). The combined chloroform extracts were dried with anhydrous MgSO4. After filtering the drying agent the solvent was distilled off and the crude product was macerated with acetonitrile 3c, 3d; hexane 3e and 3f, and the ethyl acetate- hexane mixture (1:1 v/v). After maceration for compounds 3d and 3e flash column chromatography was applied, using the following mixtures for elution: for 3d CHCl3-MeOH mixture (99:1 v/v), whereas for 3e CH2Cl2:CHOH (98:2 v/v). Next the compounds were purified by crystallization from EtOH 3a, 3b, 3c, 3d, 3f, 3h, 3f′, 3g′.
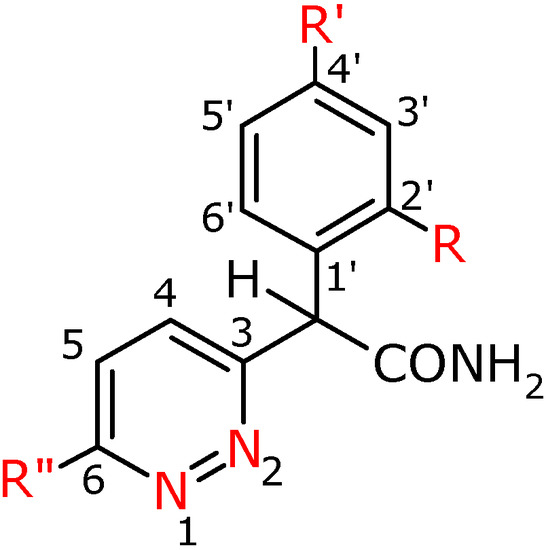
Figure 3.
Numbering system for compounds 3a-f, 3h, 3f′, 3g′.
α-Phenyl-α-(6-phenylpyridazin-3-yl)-acetamide (3a). Yield 74%; white crystals; m.p. 177.7-179.8 ºC; IR (cm-1): 1655 (CO-NH2); 1H-NMR (500 MHz) δ: 5.36 (s, 1H, CH), 5.77 (bs, 1H, NH(2)), 7.17 (bs, 1H, NH (1)), 7.29 (tt, 3J=7.5, 4J=1.0, 1H, C4’H), 7.36 (td, 3J=7.5, 4J=1.5, 2H, C3’H, C5’H), 7.47 – 7.55 (m, 5H, C2”H-C6”H), 7.62 (d, 3J=8.5, 1H, C4H), 7.83 (d, 1H, C5H), 8.05 (dd, 3J=7.5, 4J=1.5, 2H, C2’H, C6’H); 13C-NMR (125 MHz) δ: 58.8 (CH), 124.7 (C5), 127.1 (C2’,C6’), 127.9 (C4), 128.2 (C4’), 128.5 (C3”, C5”), 129.0 (C3’, C5’), 129.1 (C2”, C6”), 130.2 (C4”), 135.9 ( C1’), 137.4 (C1”), 158.4 (C3), 159.8 (C6), 172.2 (CO); Anal. calcd. for C18H15N3O: C, 74.72; H, 5.23, N, 14.52; found: C, 74.74; H, 5.24; N, 14.48.
α-(4-Fluorophenyl)-α-(6-phenylpyridazin-3-yl)-acetamide (3b). Yield 63%; beige crystals; m.p. 199.5-200.7 ºC; IR (cm-1): 1686 (CO-NH2); 1H-NMR (500 MHz) δ: 5.31(s, 1H, CH), 6.82 (bs, 1H, NH (2)), 7.05 (m, 2H, C3’H, C5’H), 7.23 (bs, 1H, NH(1)), 7.46-7.56 (m, 5H, C2”H-C6”H), 7.60 (d, 3J=9.0 Hz, 1H, C4H), 7.85 (d, 1H, C5H), 8.05 (m, 2H, C2’H, C6’H); 13C-NMR (125 MHz) δ: 57.9 (CH), 116.0 (d*, 2J=22.0, C3’, C5’), 124.9 (C4), 127.1 (C2”, C6”), 128.1 (C5), 129.1 (C3”, C5”), 130.2 (d*, 3J=8.2, C2’, C6’), 130.3 (C4”), 133.2 (d*, 4J=3.3 ,C1’), 135.8 (C1”), 158.5 (C3), 159.6 (C6), 162.4 (d*, 1J=249.5, C4’), 171.9 (CO); Anal. calcd. for C18H14N3OF: C, 70.35; H, 4.59, N, 13.67; found: C, 70.30; H, 4.58; N, 13.77.
α-(4-Tolylo)-α-(6-phenylpyridazin-3-yl)-acetamide (3c). Yield 46 %; white crystals; m.p. 208.5-209.4 ºC; IR (cm-1): 1683 (CO-NH2); 1H-NMR (400 MHz) δ: 2.33 (s, 3H, CH3), 5.33 (s, 1H, CH), 5.64 (bs, 1H, NH(2)), 7.12 (bs, 1H, NH(1)), 7.18 (d, 2H, C3’H, C5’H), 7.40 (d, 3J=8.0, 2H, C2’H, C6’H), 7.53 (m, 3H, C3”H, C4”H, C5”H), 7.63 (d, 3J=9.2, 1H, C4H), 7.85 (d, 1H, C5H), 8.06 (dd, 3J=7.6, 4J=2.4, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 21.3 (CH3), 58.4 (CH), 125.3 (C5), 127.3 (C2’,C6’), 128.6 (C3”, C5”), 128.8 (C4), 129.4 (C3’, C5’), 130.1 (C2”, C6”), 130.6 (C4”), 134.5 (C1’), 136,0 (C1”), 138.2 (C4’), 158.6 (C3), 160.0 (C6), 172.4 (CO); Anal. calcd. for C19H17N3O: C, 75.22; H, 5.65, N, 13.85; found: C, 74.32; H, 5.51; N, 14.16.
α-(4-Methoxyphenyl)-α-(6-phenylpyridazin-3-yl)-acetamide (3d). Yield 70%; white crystals; m.p. 168.2-170.7 ºC; IR (cm-1): 1662 (CO-NH2); 1H-NMR (400 MHz) δ: 3.67 (s, 3H, OCH3), 5.32 (s, 1H, CH), 6.17 (s, 2H, NH2), 6.77 (m, 2H, C3’H, C5’H), 7.19 (m, 2H, C2’H, C6’H), 7.40 (m, 3H, C3”H, C4”H, C5”H), 7.57 (m, 1H, C4H), 7.71 (m, 1H, C5H), 7.93 (m, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 55.4 (OCH3), 57.7 (CH), 114.6 (C3’,C5’), 124.9 (C4), 127.2 (C2”, C6”), 128.2 (C5), 128.5 (C1”), 129.2 (C2’, C6’), 129.8 (C3”, C5”), 130.6 (C4”), 136.1 (C1’), 158.4 (C6), 159.3 (C3), 160.4 (C4’), 173.2 (CO); Anal. calcd. for C19H17N3O2: C, 71.46; H, 5.37, N, 13.16; found: C, 71.33; H, 5.57; N, 13.01.
α-(2-Methoxyphenyl)-α-(6-phenylpyridazin-3-yl)-acetamide (3e). Yield 16%; white crystals; m.p. 183.0-184.0 ºC; IR (cm-1): 1686 (CO-NH2); 1H-NMR (400 MHz) δ: 3.82 (s, 3H, OCH3), 5.78 (bs, 1H, NH(1)), 5.88 (s, 1H, CH), 6.91 (d, 3J=8.4, 1H, C3’H), 6.97 (bs, 1H, NH(2)), 6.99 (t, 3J=7.6, 1H, C5’H), 7.31 (t, 3J=7.6, 1H, C4’H), 7.52 (m, 4H, C6’H, C3”H, C4”H, C5”H), 7.72 (d, 1H, C4H), 7.89 (d, 3J=8.8, 1H, C5H), 8.06 (dd, 3J=6.4, 4J=2.4, 2H, C2”H, C6”H); 13C-NMR (100 MHz) δ: 51.9 (CH), 55.8 (OCH3), 111.3 (C3’), 121.4 (C5’), 125.4 (C5), 125.7 (C1’), 127.3 (C2”, C6”), 129.3 (C4, C3”, C5”), 129.6 (C4’), 129.7 (C6’), 130.6 (C4”), 135.7 (C1”), 157.1 (C3), 158.3 (C6), 160.0 (C2’), 172.4 (CO); Anal. calcd. for C19H17N3O2: C, 71.46; H, 5.37, N, 13.16; found: C, 71.46; H, 5.45; N, 13.08.
α-Phenyl-α-(6-chloropyridazin-3-yl)-acetamide (3f). Yield 29 %; white crystals; m.p. 140.0-141.0 ºC; IR (cm-1): 1685 (CO-NH2); 1H-NMR (500 MHz) δ: 5.45 (s, 1H, CH), 6.21 (bs, 1H, NH(2)), 6.83 (bs, 1H, NH(1)), 7.23-7.45 (m, 5H, 3J=8.8, 5H, C2’H, C3’H, C4’H, C5’H, C6’H), 7.44 (d, 1H, 3J=8.8, C5H), 7.64 (d, 1H, C4H); 13C-NMR (125 MHz) δ: 57.8 (CH), 128.1 (C5), 128.3 (C2’, C6’), 128.7 (C4’), 129.2 (C3’, C5’), 130.2 (C4), 136.8 (C1’), 156.1 (C3), 160.9 (C6), 175.0 (CO); Anal. calcd. for C12H10N3OCl: C, 58.19; H, 4.07, N, 16.97; found: C, 58.01; H, 4.02; N, 16.88.
α-Phenyl-α-(pyridazin-3-yl)-acetamide (3f′). Yield 47%; white crystals; m.p. 177.0-178.0 ºC; IR (cm-1):1679 (CO-NH2); 1H-NMR (500 MHz) δ: 5.61 (bs, 1H, NH(2)), 5.29 (s, 1H, CH), 6.96 (bs, 1H, NH(1)), 7.28-7.38 (m, 3J0=7.0, 4Jm=1.5, 3H, C3’H, C4’H, C5’H), 7.43-7.48 (m, 3H, C5H, C2’H, C6’H), 7.56 (dd, 3J0=8.5, 4Jm=1.5, 1H, C4H), 9.11 (dd, 3J0=5.0, 4Jm=2.0, 1H, C6H); 13C-NMR (125 MHz) δ: 59.1 (CH), 127.1 (C5), 127.8 (C4), 128.0 (C4’), 128.4 (C2’, C6’), 129.2 (C3’, C5’), 137.2 (C1’), 150.4 (C6), 161.4 (C3), 171.9 (CO); Anal. calcd. for C12H11N3O: C, 67.59; H, 5.20, N, 19.71; found: C, 67.52; H, 4.91; N, 19.78.
α-(4-Fluorophenyl)-α-(pyridazin-3-yl)-acetamide (3g′). Yield 66%; white crystals; m.p. 157.5-158.5 ºC; IR (cm-1): 1690 (CO-NH2); 1H-NMR (500 MHz) δ: 5,37 (s, 1H, CH), 5,94 (bs, 1H, NH(2)), 7,03 (t, 3J=8,4, 2H, C3’H, C5’H), 7,24 (bs, 1H, NH(1)), 7,40-7,52 (m, 3H, C5H, C2’H, C6’H), 7,62 (d, 3J=8,4, 1H, C4H), 9,12 (d, 1H, C6H); 13C-NMR (125 MHz) δ: 57.9 (CH), 116.0 (d*, 2J=21,6, C3’, C5’), 127,4 (C4), 127.8 (C5), 130,2 (d*, 3J=8,2, C2’, C6’), 132,9 (C1’), 150.5 (C6), 161.4 (C3), 162.4 (d*, 1J=246,4, C4’), 171.9 (CO); Anal. calcd. for C12H10N3FO: C, 62.33; H, 4.36; N, 18.17; found: C, 61,99; H, 4,31; N, 17,94.
α-Phenyl-α-(6-methoxypyridazin-3-yl)-acetamide (3h). Yield 69%; white crystals; m.p. 165-168 ºC; IR (cm-1): 1688 (CO-NH2); 1H-NMR (500 MHz) δ: 4.10 (s, 3H, OCH3), 5.28 (s, 1H, CH), 6.10 (s, 1H, NH(2)), 6.94 (d, 1H, C5H), 7.15 (bs, 1H, NH(1)), 7.27 (m, 1H, C4’H), 7.32 (m, 2H, C3’H, C5’H), 7.42 (m, 2H, C2’H, C6’H), 7.46 (d, 3J=9.0, 1H, C4H); 13C-NMR (125 MHz) δ: 54.8 (OCH3, J=1.0), 58.2 (CH), 118.2 (C5), 127.8 (C4), 128.4 (C2’,C6’), 129.1 (C3’, C5’), 130.6 (C4’), 137.6 (C1’), 156.7 (C3), 164.5 (C6), 172.6 (CO); Anal. calcd. for C13H13N3O2: C, 64.19; H, 5.39, N, 17.27; found: C, 64.59; H, 5.37; N, 17.36.
General procedure for the preparation of 7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione derivatives 4a-f, 4f′, 4g′
To absolute ethanol (30 mL) was added sodium (0.18 mol) and the mixture was stirred until they had reacted. Next diethyl carbonate (0.17 mol) was added and the respective amide 3a-f, 3h, 3f′, 3g′ (0.1 mol) was added portionwise. The reaction was performed in boiling for 12 h. After cooling the post-reaction mixture was poured into distilled water (100 mL) and acidified with acetic acid to pH~3. The yellow precipitate separated and was filtered off. Compounds 4a, 4b, 4e were purified on a chromatographic column by the flash technique, using the CH2Cl2-MeOH (97:3 v/v) and CH2Cl2-MeOH (99:1 v/v) mixtures as eluents. Compound 4c was macerated with acetonitrile, 4d was crystallized from MeOH and 4f′ was crystallized from EtOH, and next was flash chromatographed using the CH2Cl2-MeOH (98:2 v/v) mixture as eluent. Compound 4g′ was crystallized from EtOH. Compound 4f was purified by flash chromatography with CHCl3-MeOH (97:3 v/v) and next was crystallized from MeOH.
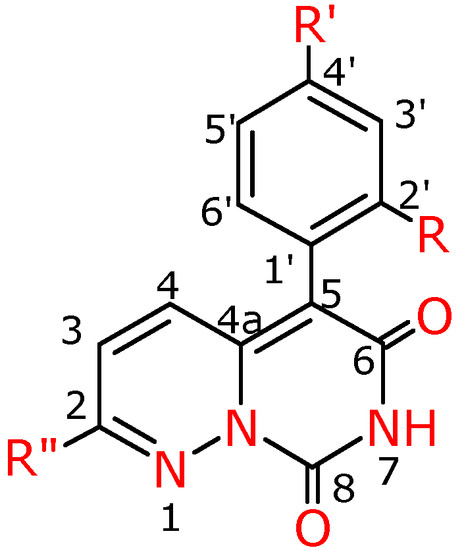
Figure 4.
Numbering system for compounds 4a-f, 4f′, 4g′.
2,5-Diphenyl-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4a). Yield 94%; yellow crystals; m.p. 302-303 ºC; IR (cm-1): 1660 (CO), 1725 (CO); 1H-NMR (500 MHz) δ: 7.17(d, 3J=10.0 , 1H, C3H), 7.34 (dd, 3J=8.0, 4J=1.5 , 2H, C2’H, C6’H), 7.40 (d, 1H, C4H), 7.42 (tt, 3J=7.0, 4J=1.0 , 1H, C4”H), 7.46-7.54 (m, 5H, C3’H, C4’H, C5’H, C3”H, C5”H,), 7.92 (dd, 3J=6.4, 4J=1.6, 2H, C2”H, C6”H) 8.51 (bs, 1H, NH); 13C-NMR (125 MHz) δ: 107.5 (C5), 122.8 (C4), 126.7 (C3”, C5”), 128.7 (C4’), 129.0 (C2”, C6”), 129.2 (C3’, C5’), 130.6 (C1’), 131.01 (C2’, C6’), 131.03 (C4”), 131.1 (C3), 133.5 (C1”), 141.3 (C2), 147.5 (C8), 150.0 (C4a), 160.2 (C6); Anal. calcd. for C19H13N3O2: C, 72.37; H, 4.15, N, 13.33; found: C, 71.61; H, 4.15;N, 13.20.
2-Phenyl-5-(4-fluorophenyl)-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4b). Yield 91%; yellow crystals; m.p. 292.8-293.2 ºC; IR (cm-1):1654 (CO), 1709 (CO); 1H-NMR (400 MHz) δ: 7.23-7.30 (m, 3H, C3H, C3’H, C5’H), 7.30-7.40 (m, 2H, C2’H, C6’H), 7.50-7.64 (m, 4H, C4H, C3”H, C4”H, C5”H), 7.92-8.20 (m, 2H, C2”H, C6”H), 12.03 (s, 1H, NH); 13C-NMR (100 MHz) δ: 107.1 (C5), 117.4 (d*,2J=21.4, C3’, C5’), 125.0 (C4), 128.4 (C3”, C5”), 130.1 (d*, 4J=3.0, C1’), 131.1 (C2”, C6”), 132.7 (C4”), 132.8 (C3), 135.3 (d*, 3J=8.0, C2’, C6’), 135.6 (C1”), 143.0 (C2), 149.7 (C8), 150.6 (C4a), 162.8 (C6), 163.7 (d*, 1J=244.8, C4’); Anal. calcd. for C19H12N3O2F: C, 68.46; H, 3.63, N, 12.61; found: C, 68.84; H, 4.01; N, 13.00.
2-Phenyl-5-(4-tolyl)-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4c). Yield 92%; yellow crystals; m.p. 321.0-321.4 ºC; IR (cm-1): 1659 (CO), 1728 (CO); 1H-NMR (400 MHz) δ: 2.35 (s, 3H, CH3), 7.19 (d, 3J=7.6, 2H, C3’H, C5’H), 7.26(m, 3H, C3H, C2’H, C6’H), 7.55 (m, 4H, C4H, C3”H, C4”H, C5”H), 7.97 (m, 2H, C2”H, C6”H), 11.97 (s, 1H, NH); 13C-NMR (100 MHz) δ: 22.9 (CH 3), 108.1 (C5), 124.8 (C4), 128.4 (C3”,C5”), 130.8 (C1’), 131.1 (C2’, C6’, C2”, C6”), 132.8 (C4”), 132.9 (C3), 133.0 (C3’, C5’), 135.6 (C1”), 139.1 (C4’), 142.8 (C2), 149.7 (C8), 150.5 (C4a), 162.8 (C6); Anal. calcd. for C20H15N3O2: C, 72.94; H, 4.59; N, 12.76; found: C, 72.56; H, 4.45; N, 13.06.
2-Phenyl-5-(4-methoxyphenyl)-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4d). Yield 98%; yellow crystals; m.p. 306.6-308.4 ºC; IR (cm-1): 1654 (CO), 1717 (CO); 1H-NMR (400 MHz) δ: 3.86 (s, 3H, OCH3), 7.02 (d, 3J=10.0 , 2H, C3’H, C5’H), 7.17 (d, 3J=10.0 , 1H, C3H), 7.27 (d, 2H, C2’H, C6’H), 7.42 (d, 1H, C4H), 7.52 (m, 3H, C3”H, C4”H, C5”H), 7.92 (m, 2H, C2”H, C6”H), 8.60 (bs, 1H, NH); 13C-NMR (100 MHz) δ: 55.6 (OCH 3), 107.4 (C5), 114.7 (C3’, C5’), 122.8 (C4), 125.0 (C1’), 126.9 (C3”, C5”), 129.4 (C2”, C6”), 131.3 (C4”), 131.4 (C3), 132.4 (C2’, C6’), 133.7 (C1”), 141.3 (C2), 148.4 (C8), 150.2 (C4a), 155.5 (C4’), 159.9 (C6); Anal. calcd. for C20H15N3O3: C, 69.56; H, 4.38, N, 12.17; found: C, 69.46; H, 4.68; N, 11.78.
2-Phenyl-5-(2-methoxyphenyl)-7H,8H- pyrimido[1,6-b]pyridazin-6,8-dione (4e).Yield 60%; yellow crystals; m.p. 275.0-277.0 ºC; IR (cm-1): 1643 (CO), 1747 (CO); 1H-NMR (400 MHz) δ: 3.79 (s, 3H, OCH3), 7.02 (d, 3J=8.4 , 1H, C3’H), 7.07 (t, 3J=7.6, 1H, C5’H), 7.11 (d, 3J=10.0 , 1H, C3H), 7.16 (d, 1H, C4H), 7.25 (d, 1H, C6’H), 7.42 (td, 3J=8.0 , 1H, C4’H), 7.50 (m, 3H, C3”H, C4”H, C5”H), 7.92 (m, 2H, C2”H, C6”H), 8.52 (bs, 1H, NH); 13C-NMR (100 MHz) δ: 55.2 (OCH3), 103.3 (C5), 111.1 (C3’), 118.7 (C1’), 120.6 (C5’), 121.9 (C4), 126.3 (C2”, C6”), 128.7 (C3”, C5”), 130.0 (C4’), 130.5 (C4”), 131.2 (C3), 132.4 ( C6’), 133.2 (C1”), 141.1 (C2), 147.3 (C8), 149.3 (C4a), 157.3 (C2’), 159.7 (C6); Anal. calcd. for C20H15N3O3: C, 69.56; H, 4.38, N, 12.17; found: C, 69.36; H, 4.57; N, 11.26.
2-Ethoxy-5-phenyl-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4f). Yield 31% (from 3f), 72%(from 3h); yellow crystals; m.p. 272.0-274.0 ºC; IR (cm-1): 1636 (CO), 1738 (CO); 1H-NMR (500 MHz) δ: 1.42 (t, 3H, 3J=7.0 , C2Het), 4.40 (k, 2H, C1Het), 6.48 (d, 3J=9.5 , 1H, C3H), 7.25 (d, 1H, C4H), 7.28-7.33 (m, 2H, C3’H ,C5’H), 7.37-7.42 (m, 1H, C4’H), 7.42-7.48 (m, 2H, C2’H, C6’H), 8.82 (bs, NH); 13C-NMR (125 MHz) δ: 14.0 (C2et), 64.1 (C1et), 108.5 (C5), 121.0 (C3), 128.6 (C4’), 128.9 (C2’,C6’), 130.9 (C1’), 131.1 (C3’, C5’), 132.6 (C4), 141.0 (C4a), 147.3 ( C8), 156.5 (C6), 160.4 (C2); Anal. calcd. for C15H13N3O3: C, 63.60; H, 4.62, N, 14.83; found: C, 63.26; H, 4.60; N, 14.74.
5-Phenyl-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4f′). Yield 27 %; yellow crystals; m.p. 265.5-266.3 ºC; IR (cm-1): 1651 (CO), 1751 (CO); 1H-NMR (500 MHz) δ: 6.85 (dd, 3J1=12.0 , 3J2=4.5 , 1H, C3H), 7.27 (dd, 3J=12.0 , 4J=2.0 , 1H, C4H), 7.32 (dd, 3J=10.5 , 4J=2.0 , 2H, C2’H, C6’H), 7.40 (tt, 3J=8.0 , 4J=2.5 , 1H, C4’H), 7.47 (t, 3J=8.0 , 2H, C3’H, C5’H), 8.06 (m, 1H, C2H), 9.5 (bs, 1H, NH); 13C-NMR (125 MHz) δ: 105.0 (C5), 125.2 (C4), 129.6 (C4’), 130.0 (C3’, C5’), 131.7 (C3), 132.3 (C1’), 132.5 (C2’,C6’), 144.5 (C2), 147.0 (C8), 150.3 (C4a), 163.5 (C6); Anal. calcd. for C13H9N3O2: C, 65.27; H, 3.79, N, 17.56; found: C, 65.58; H, 3.82 N, 17.14.
5-(4-Fluorophenyl)-7H,8H-pyrimido[1,6-b]pyridazin-6,8-dione (4g’). Yield 69%; yellow crystals; m.p. 278.0-278.5 ºC; IR (cm-1): 1625 (CO), 1756 (CO); 1H-NMR (500 MHz) δ: 6.67-6.69 (dd, 1H, C4H), 7.13-7.18 (m, 2H, C2’H, C6’H), 7.25-7.30 (m, 3H, C3H, C3’H, C5’H), 7.98 (d, 1H, C2H), 8.65 (s, 1H, NH); 13C-NMR (125 MHz) δ: 106.8 (C5), 116.4 (C3’, C5’), 123.3 (C1’), 126.2 (C4), 130.5 (C2’, C6’), 132.8 (C3), 142.3 (C8), 142.4 (C2), 147.4 (C4a), 161.9 (C6), 163.8 (C4’); Anal. calcd. for C13H8N3O2F: C, 60.70; H, 3.13; N, 16.34; found: C, 60.83; H, 3.37; N, 16.56.
References
- Olivier, B.; Soudijn, W.; van Wijngaarden, J. The 5-HT1A receptor and its ligands: structure and function. Prog. Drug Res. 1999, 52, 103–165. [Google Scholar]
- López-Rodriguez, M.L.; Ayala, D.; Benhamú, B.; Morcillo, M.J.; Viso, A. Arylpiperazine Derivatives Acting at 5-HT1A Receptors. Curr. Med. Chem. 2002, 9, 443–469. [Google Scholar] [CrossRef]
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1115. [Google Scholar] [CrossRef]
- Hoyer, D.; Hannon, J.P.; Martin, G.R. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav. 2002, 71, 533–554. [Google Scholar] [CrossRef]
- Jones, B.J.; Blackburn, T.P. The medical benefit of 5-HT research. Pharmacol. Biochem. Behav. 2002, 71, 555–568. [Google Scholar] [CrossRef]
- Ishizumi, K.; Kojima, A.; Antoku, F. Synthesis and anxiolytic activity of N-substituted cyclic imides (1R*, 2S*, 3R*, 4S*)-N-[4-[4-(2-pyrimidinyl)-1-piperazinyl]butyl]-2,3-bicyclo[2.2.1]-heptanedicarboximide (Tandospirone) and related compounds. Chem. Pharm. Bull (Tokio) 1991, 39, 2288–2300. [Google Scholar] [CrossRef]
- Abou-Gharbia, M.A.; Childres, W.A. Jr.; Fletcher, H.; McGaughey, G.; Patel, U.; Webb, M.B.; Yardley, J.; Andree, T.; Boast, C.; Kucharik, R.J.Jr.; Marquis, K.; Morris, H.; Scerni, R.; Moyer, J.A. Synthesis and SAR of adatanserin: novel adamantyl aryl- and heteroarylpiperazines with dual serotonin 5-HT1A and 5-HT2A activity as potential anxiolytic and antidepressant agents. J. Med. Chem. 1999, 42, 5077–5094. [Google Scholar] [CrossRef]
- Den Boer, J.A.; Bosker, F.J.; Slaap, B.R. Serotonergic drugs in the treatment of depressive and anxiety disorders. Hum. Psychopharmacol. 2000, 15, 315–356. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, M.L.; Morcillo, M.J.; Rovat, T.K.; Fernández, E.; Vicente, B.; Sanz, A.M.; Hernández, M.; Orensanz, L. Synthesis and structure-activity relationships of a new model of arylpiperazines. 4. 1-[ω-(4-Arylpiperazin-1-yl)alkyl]3-(diphenylmethylene)-2,5-pyrrolidinediones and -3-(9H-fluoren-9-ylidene)-2,5-pyrrolidinediones: study of the steric requirements of the terminal amide fragment on 5-HT1A affinity/selectivity. J. Med. Chem. 1999, 42, 36–49. [Google Scholar] [CrossRef]
- Caliendo, G.; Fiorino, F.; Grieco, P.; Perissutti, E.; Santagarda, V.; Albrizio, S.; Spadola, L.; Bruni, G.; Romeo, M.R. Sythesis and binding affinities for 5-HT1A, 5-HT2A and 5-HT2c receptors of a series of 1- and 2-(4-arylpiperazinylalkyl)-4-(benzoyl)-1,2,3-triazole derivatives. Eur. J. Med. Chem. 1999, 34, 719–727. [Google Scholar] [CrossRef]
- Herold, F.; Wolska, I.; Helbin, E.; Król, M.; Kleps, J. Synthesis and Structure of Novel 4-Aryl-hexahydro-1H,3H-pyrido[1,2-c]pyrimidine Derivatives. J.Heterocycl. Chem. 1999, 36, 389–396. [Google Scholar]
- Herold, F.; Kleps, J.; Anulewicz-Ostrowska, R.; Szczęsna, B. Synthesis and Molecular Structure of Novel 4-Aryloctahydropyrido[1,2-c]pyrimidine Derivatives. J. Heterocycl. Chem. 2002, 39, 773–782. [Google Scholar]
- Herold, F.; Dawidowski, M.; Wolska, I.; Chodkowski, A.; Kleps, J.; Turło, J.; Zimniak, A. The synthesis of new diastereomers of (4S,8aS)- and (4R,8aS)-4-phenyl-perhydropyrrole[1,2-a]pyrazine-1,3-dione. Tetrahedron: Asymmetr. 2007, 18, 2091–2098. [Google Scholar] [CrossRef]
- Bemis, G.W.; Salituro, F.G.; Duffy, J.P.; Cochran, J.E.; Harrington, E.M.; Murcko, M.A.; Wilson, K.P.; Galullo, V.P. Preparation of annelated pyrimidinones and analogs as p38 kinase inhibitors. WO 98/27098 1998. [Chem.Abstr. 1998, 129, 81749]. [Google Scholar]
- Yamada, T.; Nobuhara, Y.; Shimamura, H.; Yoshihara, K.; Yamaguchi, A.; Ohki, M. Studies on New Antiulcer Agents. I. Synthesis and Antisecretory Activity of Pyridazine Derivatives. Chem. Pharm. Bull. 1981, 29, 3433–3439. [Google Scholar] [CrossRef]
- Abbotto, A.; Alanzo, V.; Bradamante, S.; Pagani, G.A. Preparation of Heteroaryl † Phenylmethanes and a 13C and 15N NMR Spectroscopic Study of their Conjugate Carbanions. Rotational Isomerism and Charge Maps of the Anions and Ranking of the Charge Demands of the Heterocycles. J. Chem. Soc. Perkin Trans 2 1991, 481–488. [Google Scholar]
- Yamanaka, H.; Ohba, S. Reaction of Methoxy-N-Heteroaromatics with Phenylacetonitrile under Basic Conditions. Heterocycles 1990, 31, 895–909. [Google Scholar] [CrossRef]
- Gabriel, S.; Colman, J. Synthese des Pyridazins und seiner Derivate. Chem. Ber. 1899, 32, 395–409. [Google Scholar] [CrossRef]
- Libermann, D; Rouaix, A. Sur les hydrazino-dérivés de quelques series hétérocycliques. Bull. Soc. Chim. Fr. 1959, 1793–1798. [Google Scholar]
- Sample Availability: Samples of the compounds are available from authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.