Two Step Synthesis of a Non-symmetric Acetylcholinesterase Reactivator
by
Kamil Musilek
1,
Kamil Kuca
1,*, 
Vlastimil Dohnal
1,
Daniel Jun
1,
Jan Marek
2 and
Vit Koleckar
3 1
Department of Toxicology, Faculty of Military Health Sciences, Trebesska 1575, 500 01 Hradec Kralove, Czech Republic
2
Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Heyrovskeho 1203, 500 05 Hradec Kralove, Czech Republic
3
Center of Advanced Studies, Faculty of Military Health Sciences, Trebesska 1575, 500 01 Hradec Kralove, Czech Republic
4
Department of Food Technology, Mendel University of Agriculture and Forestry Brno, Zemedelska 1, 613 00 Brno, Czech Republic
*
Author to whom correspondence should be addressed.
Molecules 2007, 12(8), 1755-1761; https://doi.org/10.3390/12081755
Submission received: 25 June 2007
/
Revised: 1 August 2007
/
Accepted: 6 August 2007
/
Published: 7 August 2007
Abstract
:The newly developed and very promising acetylcholinesterase reactivator (E)-1-(2-hydroxyiminomethylpyridinium)-4-(4-hydroxyiminomethylpyridinium)-but-2-ene dibromide was prepared using two different pathways via a two-step synthesis involving the appropriate (E)-1-(4-bromobut-2-enyl)-2- or 4-hydroxyiminomethyl-pyridinium bromides. Afterwards, purities and yields of the desired product prepared by both routes were compared. Finally, its potency to reactivate several nerve agent-inhibited acetylcholinesterases was tested.
Introduction
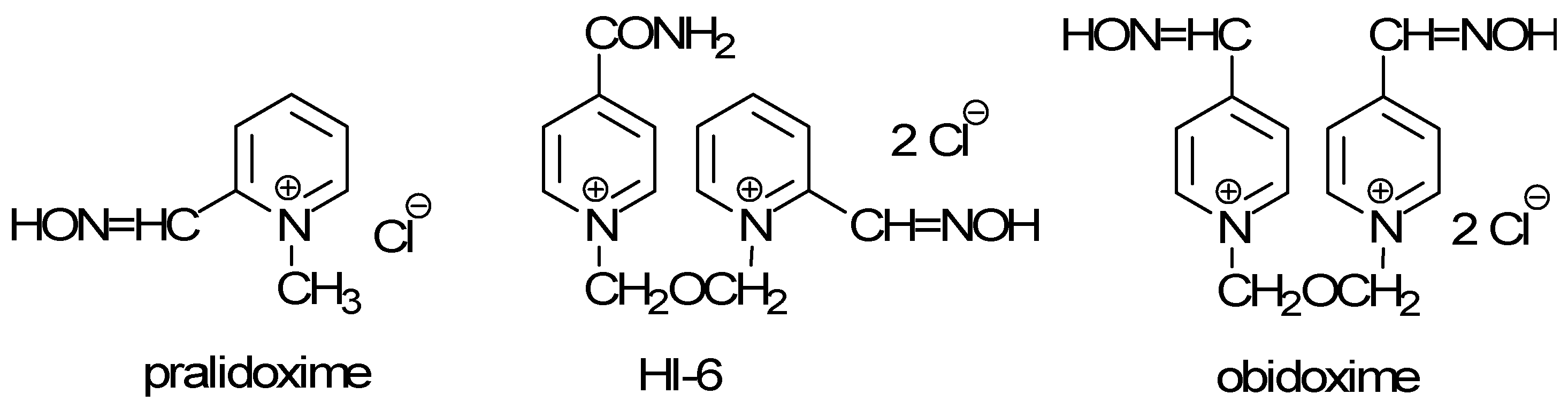
Organophosphorus nerve agents are probably the most toxic compounds prepared by the chemical industry. Their production began in Germany prior to World War II (the organophosphate Tabun, 1936). Since World War II, many other organophosphorus derivatives have been considered as more or less toxic compounds for military purposes. After the introduction of nerve agents, their antidotes were also extensively developed. General treatments for nerve agent intoxication consist of anticholinergics (mainly atropine) and acetylcholinesterase (AChE; EC 3.1.1.7) reactivators (pralidoxime, HI-6, obidoxime) [1,2,3] (Figure 1).
Figure 1.
Reactivators of acetylcholinesterase.
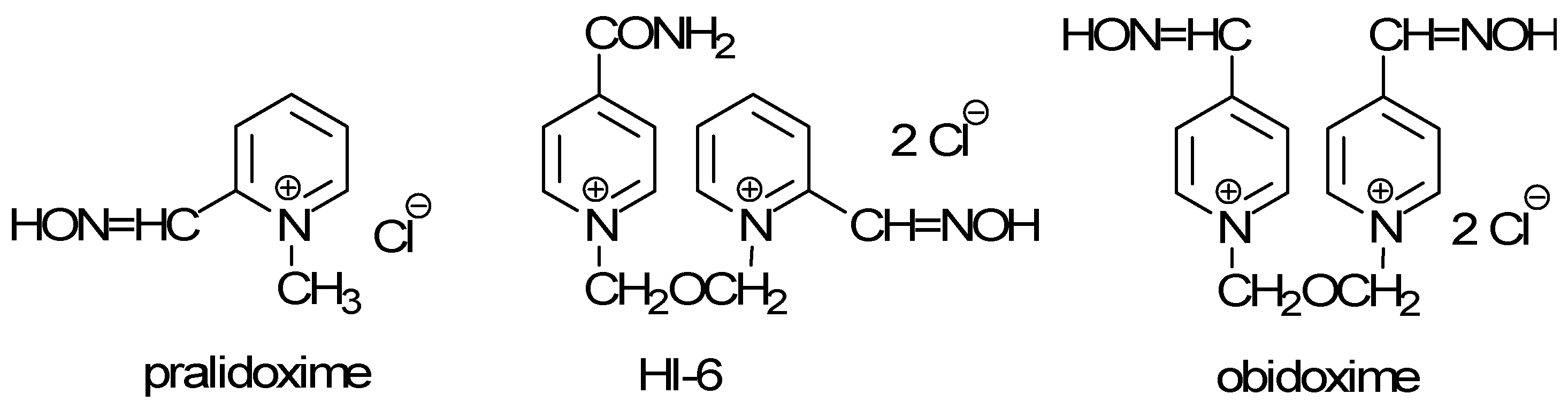
Unfortunately, no single compound is capable of acting as broad-spectrum AChE reactivator for treatment of intoxications by every nerve agent [3]. Due to this, many laboratories are focused on synthesis of such broad-spectrum reactivators [4,5,6,7,8]. In 2006 we prepared a promising non-symmetrical AChE reactivator: 1-(2-hydroxyiminomethylpyridinium)-4-(4-hydroxyiminomethylpyridinium)-but-2-ene dibromide (designated as K053) [5]. It was prepared using two-step synthetic pathway via 1-(4-bromobut-2-ene)-2-hydroxyiminomethylpyridinium bromide. Because of its very promising activity and to carry out further in vitro and in vivo investigations, it was necessary to increase the yield and purity of this compound. Owing to this fact, in this article we focus our attention on preparing this reactivator using a reverse sequence of reaction steps. A similar approach for reactivator K027 was previously published by our group [9]. This compound is currently under consideration as a very promising AChE reactivator with a broad reactivation potency and relatively low toxicity [10,11,12,13,14,15,16]. However, it was not applicable for our current synthesis of the new reactivator K053 (3) because of different connection chains between both the substituted pyridinium rings in each compound (a trimethylene linker in K027 versus an (E)-but-2-ene in K053).
Results and Discussion
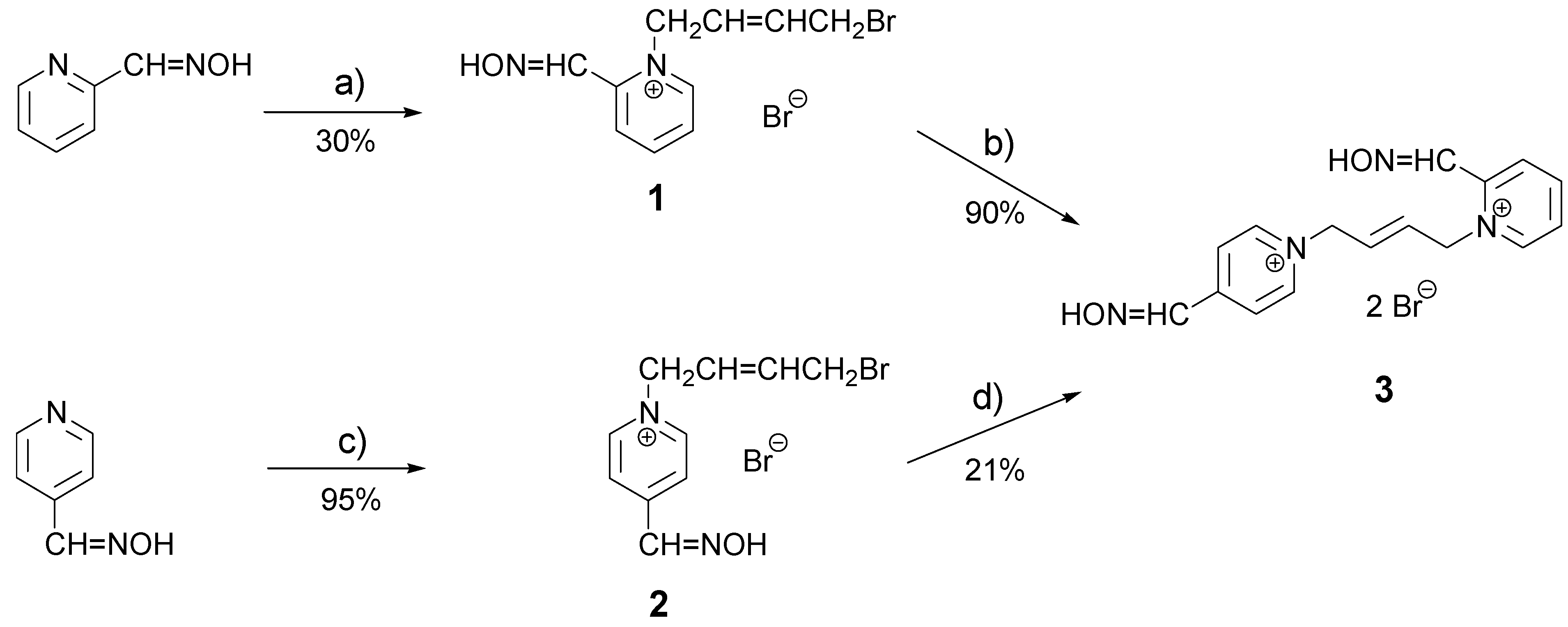
This work could be divided into two parts – a synthetic part (main part) and a biochemical part. The target bisquaternary pyridinium AChE reactivator (E)-1-(2-hydroxyiminomethylpyridinium)-4-(4-hydroxyiminomethylpyridinium)-but-2-ene dibromide (3; K053) can be prepared via two different synthetic pathways, each involving two steps. The first step – creation of the monoquaternary intermediate – is the overall yield limiting one because of the potential creation of byproducts. To decrease formation of the undesired byproduct, a 5 equivalent excess of the alkylating agent was added the solvent volume was increased (to 60 mL). The minor bisquaternary byproducts present in the mixture, could be removed by recrystallization from acetonitrile. Results of both monoquaternizations are summarized in Scheme 1.
Scheme 1.
Preparation of oxime K053.
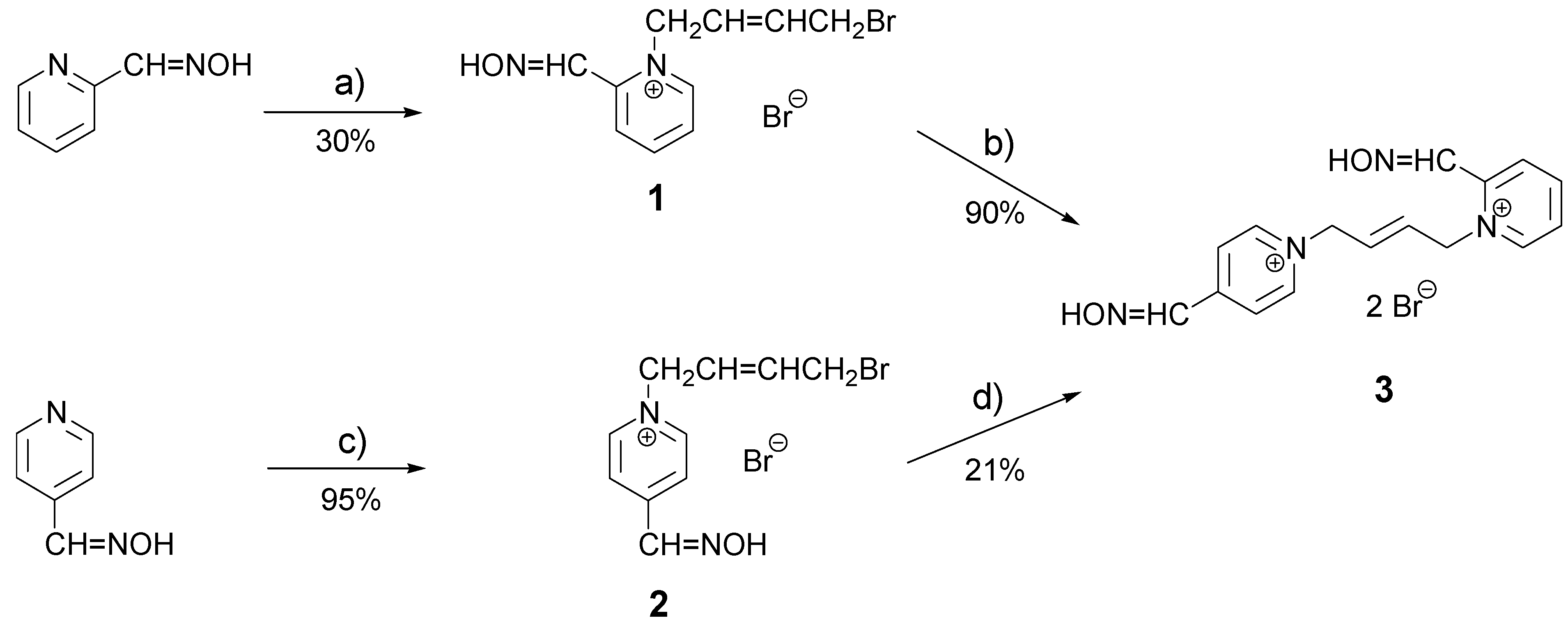
(a) (E)-1,4-dibromobut-2-ene (5 equiv), acetone, 50°C, 6.5 h; (b) 4-hydroxyiminomethylpyridine (2 equiv), DMF, 60°C, 6 h; (c) (E)-1,4-dibromobut-2-ene (5 equiv), acetone, reflux, 2 h; (d) 2-hydroxyiminomethylpyridine (2 equiv), DMF, 60°C, 10.5 h.
The second step of the complete synthetic pathway – the creation of the non-symmetrical product (3; K053) did not prove as difficult as in the case of the monoquaternary intermediates (no byproducts are possible and therefore, the desired transformation can be carried out under more forcing reaction conditions: 10 mL DMF, 60°C). Grey crystals were formed in the solution and then they were simply recrystallized from acetonitrile. Results obtained for both second steps are summarized in Scheme 1.
Comparing the yields of monoquaternary intermediates, the higher yield was obtained and almost no impurities were formed for the pyridine with an oxime group in the para-position. The lower quaternization yield obtained for an ortho-substituted pyridine was probably due to the steric hindrance of the oxime group, which hinders attack on the pyridine nitrogen. As mentioned, no byproducts are possible in the second step of each sequence, but a similar yield dependency on the substitution pattern of the pyridine was observed and a lower yield was noted (21% – presumably due to steric hindrance in the pyridine substituted at the ortho-position), compared to the para-substituted pyridine (90 % in the second step). If the overall yields of both synthetic pathways are compared, synthesis via an ortho-substituted pyridine in the first step gave higher yield.
Biochemistry
In our biochemical studies, the ability of the prepared compound 3 (K053) to split organophosphate-AChE complexes was tested using our in vitro reactivation test [19]. Our previous results (on chlorpyrifos-inhibited AChE reactivation) were updated with results for Tabun-, Cyclosarin- and VX-inhibited AChE reactivation (Table 1). As it is clearly presented, this oxime is also able to reactivate all other nerve agent-inhibited AChEs tested. Maximal reactivation activity was achieved for chlorpyrifos, the lowest for Tabun. These results are closely related to the splitting of an enzyme-inhibitor complex. While the complex of AChE with the pesticide chlorpyrifos is easily reactivated, the nerve agent complexes are degraded by a process called aging, during which the nerve agent moiety undergoes a change, whereby an alkyl or amidoyl group is replaced by a hydroxyl (negative charge) and consequently, becomes almost resistant to reactivation. Although Tabun does not have a fast aging halftime (t1/2 = 46 h), its reactivation is poor (Table 1) [17]. The poor reactivation can be explained by the free electron pair on the amidoyl group, which may easily interact with the other hydrogen bonds. In contrast, Cyclosarin (t1/2 = 40 h) and VX (t1/2 ~ 48 h) are reactivated satisfactorily, whereas Cyclosarin had the better reactivator profile with K053. The worse reactivation of VX was probably caused by presence of an oxime in the 2-position in K053 instead of an oxime in position four, which is required for VX poisoning [18]. The biochemical results should be further investigated by appropriate more sophisticated methods.

Table 1.
Reactivation of nerve agent-inhibited AChE by reactivator K053 (3) (source of the enzyme – rat brain acetylcholinesterase; time of inhibition by nerve agent – 30 min; time of reactivation – 10 min; pH – 7.6; 25°C).
| Chlorpyrifos |  | 63 | 48 |
| Tabun | 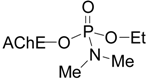 | 9 | 0 |
| Cyclosarin | 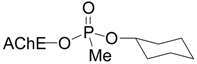 | 31 | 40 |
{kind=link}
{kind=link}
Reactivation data are expressed as mean form three independent measurements
* concentration of reactivator 10-3 M
** concentration of reactivator 10-5 M
Conclusions
In conclusion, a new promising AChE reactivator was prepared using two different synthetic pathways, via two different reactive monoquaternary intermediates. These intermediates could be simply used for preparation of other reactivators derived from the structure of the oxime K053. This procedure will be in future used for preparation of large amounts of this compound which will be needed for further in vitro and in vivo investigations. Moreover, splitting of the bond between nerve agents and AChE showed that this reactivator is able to reactivate not only chlorpyrifos (a pesticide), as previously known before, but also Cyclosarin-, Tabun- and VX-inhibited AChE.
Experimental
General
Solvents (acetone, DMF, MeCN) and reagents were purchased from Fluka and Sigma-Aldrich (Czech Republic) and used without further purification. Reactions were monitored by TLC using DC-Alufolien Cellulose F (Merck, Germany) and 5:1:2 BuOH-CH3COOH-H2O as mobile phase; detection was by a Dragendorff reagent solution (10 mL CH3COOH, 50 mL H2O and 5 mL of basic solution prepared by mixing of two fractions – fraction A: 850 mg Bi(NO3)3, 40 mL H2O, 10 mL CH3COOH; fraction B: 8 g KI, 20 mL H2O). Melting points were measured on a PHMK 05 (VEB Kombinat Nagema, Radebeul, Germany) micro heating stage and are uncorrected. NMR spectra were generally recorded on a Varian (Palo Alto CA, US) Gemini 300 instrument (1H at 300 MHz, 13C at 75 MHz). In all cases, the chemical shift values are reported in ppm (δ) relative to residual CHD2SO2CD3 (δ 2.50) or D2O (δ 4.79) for the 1H spectra and to the DMSO-d6 solvent peak (δ 39.43) for the 13C spectra, respectively. Signals are quoted as s (singlet), d (doublet), t (triplet) and m (multiplet). Mass spectra were recorded using combination of high performance liquid chromatography and mass spectrometry. A HP1100 HPLC system (Agilent Technologies, Waldbronn, Germany) was used. It consisted of vacuum degasser G1322A, quaternary pump G1311A, autosampler G1313A and quadrupole mass spectrometer MSD1456 VL equipped with electrospray ionization source. Nitrogen for the mass spectrometer was supplied by a Whatman 75-720 nitrogen generator. Data were collected in positive ion mode with an ESI probe voltage of 4000 V. The pressure of nebulizer gas was set up to 35 psig. Drying gas temperature was operated at 335 °C and flow at 13 L/min.
Synthesis
Preparation of monoquaternary salts – A solution of the corresponding hydroxyiminomethylpyridine (2.0 g, 16.4 mmol) and (E)-1,4-dibromobut-2-ene (17.51 g, 81.9 mmol) in acetone (60 mL) was stirred at reflux for 6-6.5 h. The reaction mixture was cooled to the room temperature; the crystalline crude product was collected by filtration, washed with acetone (3×20 mL) and recrystalized from MeCN.
Preparation of bisquaternary salts – A solution of the monoquaternary salt (0.50 g, 1.5 mmol) and corresponding hydroxyiminomethylpyridine (2.2 mmol) in DMF (10 mL) was stirred at 60 °C for 2-10.5 h. The reaction mixture was cooled to the room temperature and partitioned with acetone (50 mL); the crystalline crude product was collected by filtration, washed with acetone (3×20 mL) and recrystalized from MeCN.
Product characterization data
(E)-1-(4-brombut-2-enyl)-2-hydroxyiminomethylpyridinium bromide (1). Yield 30%; m.p. 132-135°C; 1H-NMR (DMSO d6): δ 9.11 (d, 1H, J = 6.0 Hz, H-6), 8.78-8.54 (m, 2H, H-5 + -CH=NOH), 8.42 (d, 1H, J = 6.6 Hz, H-3), 8.22-8.08 (m, 1H, H-4), 6.18-6.04 (m, 1H, N-CH2-CH=), 6.03-5.82 (m, 1H, =CH-CH2-Br), 5.53 (d, 2H, J = 5.8 Hz, N-CH2-), 4.14 (d, 2H, J = 7.4 Hz, -CH2-Br); 13C-NMR (DMSO d6): δ 147.06, 145.82, 145.68, 141.31, 132.05, 127.85, 125.66, 121.41, 60.13, 58.13; ESI-MS: m/z 254.9 [M]+ (calculated for [C10H12N2O]+ 255.01); EA: calculated 35.74% C, 3.60% H, 8.34% N; found 36.12% C, 3.89% H, 8.79% N.
(E)-1-(4-brombut-2-enyl)-4-hydroxyiminomethylpyridinium bromide (2). Yield 95%; m.p. 187-191°C; 1H-NMR (DMSO d6): δ 9.02 (d, 2H, J = 6.0 Hz, H-2 + H-6), 8.45 (s, 1H, -CH=NOH), 8.27 (d, 2H, J = 6.0 Hz, H-3 + H-5), 6.26-6.09 (m, 2H, -CH=CH-CH2Br), 5.32 (d, 2H, J = 4.9 Hz, N-CH2-), 4.17 (d, 2H, J = 6.0 Hz, -CH2-Br); 13C-NMR (DMSO d6): δ 148.67, 145.05, 144.85, 133.72, 127.69, 124.17, 60.25, 32.01; ESI-MS: m/z 254.9 [M]+ (calculated for [C10H12N2O]+ 255.01); EA: calculated 35.74% C, 3.60% H, 8.34% N; found 35.69% C, 3.72% H, 8.49% N.
(E)-1-(2-hydroxyiminomethylpyridinium)-4-(4-hydroxyiminomethylpyridinium)-but-2-ene dibromide (3). Yield 85%; m.p. 202-203°C; 1H-NMR (DMSO d6): δ 9.15 (d, 1H, J = 6.0 Hz, H-6), 9.02 (d, 2H, J = 6.0 Hz, H-2´ + H-6´), 8.72-8.55 (m, 2H, H-5 + -CH=NOH), 8.51-8.36 (m, 2H, H-3 + -CH=NOH´), 8.25 (d, 2H, J = 6.0 Hz, H-3´+ H-5´), 8.20-8.10 (m, 1H, H-4), 6.37-6.22 (m, 1H, N-CH2-CH=), 6.06-5.91 (m, 1H, =CH-CH2-N´), 5.57 (d, 2H, J = 4.7 Hz, N-CH2-), 5.32 (d, 2H, J = 6.0 Hz, -CH2-N´); 13C-NMR (DMSO d6): δ 148.64, 147.10, 146.03, 145.73, 145.04, 144.90, 141.48, 131.20, 127.95, 127.70, 125.72, 123.98, 60.23, 58.23; ESI-MS: m/z 149.1 [M]2+ (calculated for [C8H9N2O]2+ 149.07); EA: calculated 41.95% C, 3.96% H, 12.23% N; found 41.73% C, 4.15% H, 12.41% N.
Acknowledgements
The authors would like to thank Mrs Martina Hrabinova, Mrs. Petra Hanusova and Mr. Petr Stodulka for their excellent technical help. This work was supported by the grant agency of Ministry of Defence (Czech Republic) – grant no. FVZ0000604.
References
- Marrs, T.C. Organophosphate poisoning. Pharmacol. Therap. 1993, 58, 51–66. [Google Scholar] [CrossRef]
- Bajgar, J. Organophosphates/nerve agent poisoning: mechanism of action, diagnosis, prophylaxis, and treatment. Adv. Clin. Chem. 2004, 38, 151–216. [Google Scholar] [PubMed]
- Kuca, K.; Jun, D.; Musilek, K. Structural requirements of acetylcholinesterase reactivators. Mini-Rev. Med. Chem. 2006, 6, 269–277. [Google Scholar]
- Pang, YP.; Kollmeyer, TM.; Hong, F.; Lee, JC.; Hammond, PI.; Haugabouk, SP.; Brimijoin, S. Rational design of alkylene-linked bis-pyridiniumaldoximes as improved acetylcholinesterase reactivators. Chem. Biol. 2003, 10, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Kuca, K.; Jun, D.; Dohnal, V.; Dolezal, M. Synthesis of the novel series of bispyridinium compounds bearing (E)-but-2-ene linker and evaluation of their reactivation activity against chlorpyrifos-inhibited acetylcholinesterase. Biorg. Med. Chem. Lett. 2006, 16, 622–627. [Google Scholar]
- Kim, T.H.; Kuca, K.; Jun, D.; Jung, Y.S. Design and synthesis of new bis-pyridinium oximes as cyclosarin-inhibited acetylcholinesterase reactivators. Bioorg. Med. Chem. Lett. 2005, 15, 2914–2917. [Google Scholar] [CrossRef] [PubMed]
- Musilek, K.; Holas, O.; Hambalek, J.; Kuca, K.; Jun, D.; Dohnal, V.; Dolezal, M. Synthesis of Bispyridinium Compounds bearing Propane Linker and Evaluation of their Reactivation Activity against Tabun- and Paraoxon-Inhibited Acetylcholinesterase. Lett. Org. Chem. 2006, 3, 831–835. [Google Scholar] [CrossRef]
- Musilek, K.; Kuca, K; Jun, D.; Dolezal, M. Progress in synthesis of new acetylcholinesterase reactivators during the period 1990-2004. Curr. Org. Chem. 2007, 11, 229–238. [Google Scholar]
- Kuca, K.; Bielavsky, J.; Cabal, J.; Bielavska, M. Synthesis of a potential reactivator of acetylcholinesterase 1-(4-hydroxyiminomethylpyridinium)-3-(carbamoylpyridinium)-propane dibromide. Tetrahedron Lett. 2003, 44, 3123–3125. [Google Scholar]
- Kuca, K.; Kassa, J. In vitro reactivation of acetylcholinesterase using of the oxime K027. Vet. Hum. Toxicol. 2004, 46, 15–18. [Google Scholar] [PubMed]
- Kassa, J.; Kuca, K.; Cabal, J.; Paar, M. A comparison of the efficacy of new asymmetric bispyridinium oximes (K027, K048) with currently available oximes against tabun by in vitro and in vivo methods. J. Toxicol. Env. Health 2006, 69, 1875–1882. [Google Scholar]
- Calic, M.; Lucic-Vrdoljak, A.; Radic, B.; Jelic, D.; Jun, D.; Kuca, K.; Kovarik, Z. In vitro and in vivo evaluation of pyridinium oximes: mode of interaction with acetylcholinesterase, effect on tabun- and soman-poisoned mice and their cytotoxicity. Toxicology 2006, 219, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Petroianu, GA.; Arafat, K.; Kuca, K.; Kassa, J. Five oximes (K-27, K-33, K-48, BI-6 and methoxime) in comparison with pralidoxime: in vitro reactivation of red blood cell acetylcholinesterase inhibitied by paraoxon. J. Appl. Toxicol. 2006, 26, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Petroianu, GA.; Nurulain, SM.; Nagelkerke, N.; Al-Sultan, MAH.; Kuca, K.; Kassa, J. Five oximes (K-27, K-33, K-48, BI-6 and methoxime) in comparison with pralidoxime: survival in rats exposed to the organophosphate paraoxon. J. Appl. Toxicol. 2006, 26, 262–268. [Google Scholar]
- Tekes, K.; Hasan, MY.; Sheen, R.; Kuca, K.; Petroianu, G. HPLC determination of the serum concentration of K-27, a novel oxime-type cholinesterase reactivator. J. Chromatogr. A. 2006, 1122, 84–87. [Google Scholar] [CrossRef]
- Lucic-Vrdoljak, A.; Calic, M.; Radic, B.; Berend, S.; Kuca, K.; Kovarik, Z. Pre-treatment with pyridinium oximes improves antidotal therapy against tabun poisoning. Toxicology 2006, 228, 41–50. [Google Scholar]
- Gupta, R. Toxicology of organophosphate & carbamate compounds. In Cl Cholinesterase inhibitors as chemical warfare agents: Community preparedness guidelines; Elsevier Academic Press: London, 2006; pp. 47–68. [Google Scholar]
- Kuca, K.; Jun, D.; Musilek, K.; Bajgar, J. Structure-activity relationship for the reactivators of acetylcholinesterase inhibited by nerve agent VX. Toxicol. Lett. 2006, 164, S51–S52. [Google Scholar] [CrossRef]
- Kuca, K.; Cabal, J. Evaluation of newly synthesized reactivators of the brain cholinesterase inhibited by sarin-nerve agent. Toxicol. Mech. Meth. 2005, 15, 247–252. [Google Scholar]
- Sample Availability: Samples of the prepared compounds are available from authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Musilek, K.; Kuca, K.; Dohnal, V.; Jun, D.; Marek, J.; Koleckar, V. Two Step Synthesis of a Non-symmetric Acetylcholinesterase Reactivator. Molecules 2007, 12, 1755-1761. https://doi.org/10.3390/12081755
AMA Style
Musilek K, Kuca K, Dohnal V, Jun D, Marek J, Koleckar V. Two Step Synthesis of a Non-symmetric Acetylcholinesterase Reactivator. Molecules. 2007; 12(8):1755-1761. https://doi.org/10.3390/12081755
Chicago/Turabian StyleMusilek, Kamil, Kamil Kuca, Vlastimil Dohnal, Daniel Jun, Jan Marek, and Vit Koleckar. 2007. "Two Step Synthesis of a Non-symmetric Acetylcholinesterase Reactivator" Molecules 12, no. 8: 1755-1761. https://doi.org/10.3390/12081755