Studies with Azinylacetonitriles: 2-Pyridylacetonitrile as a Precursor to Functionally Substituted Pyridines

Abstract
:Introduction
Results and Discussion



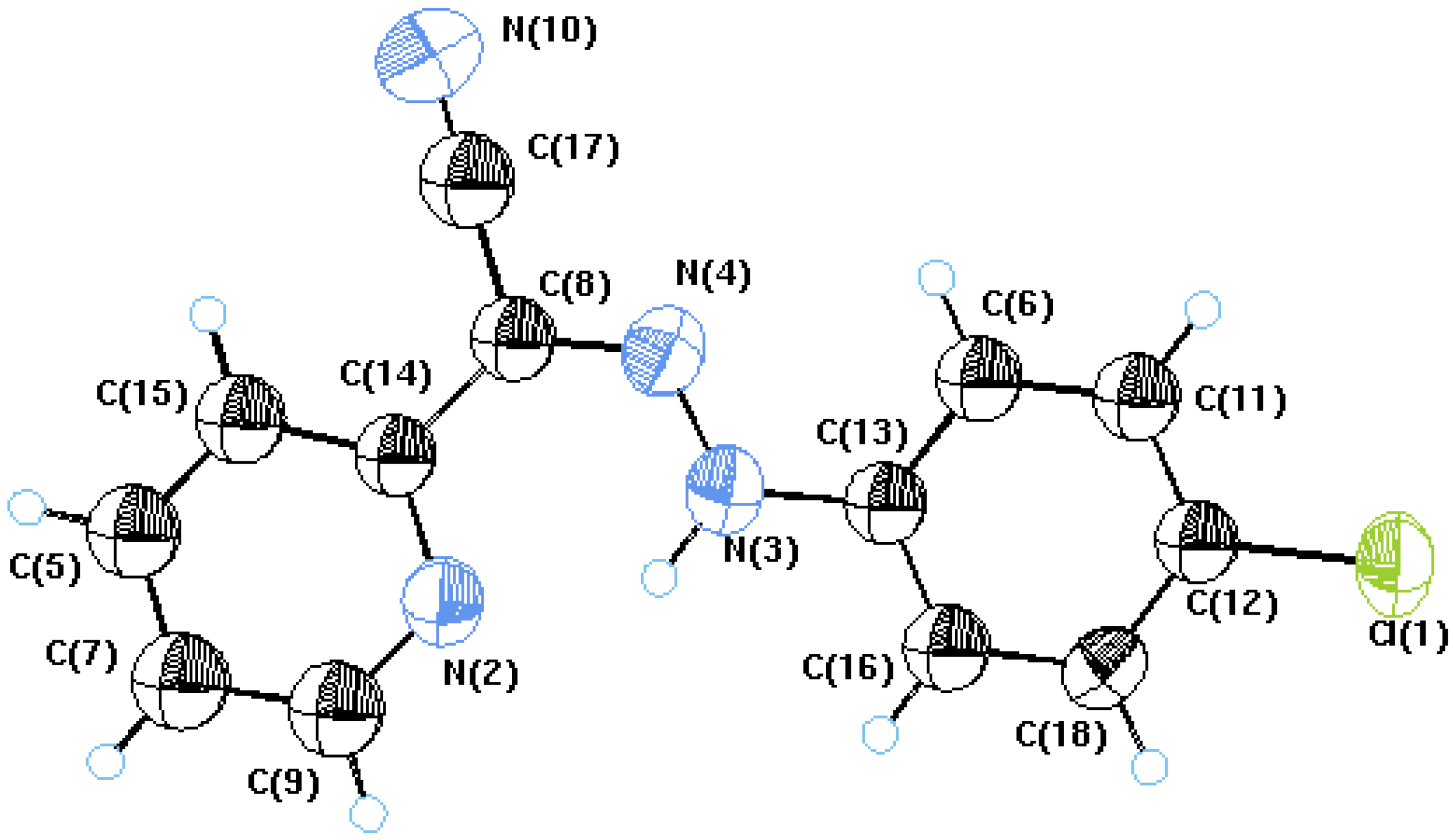{kind=link}
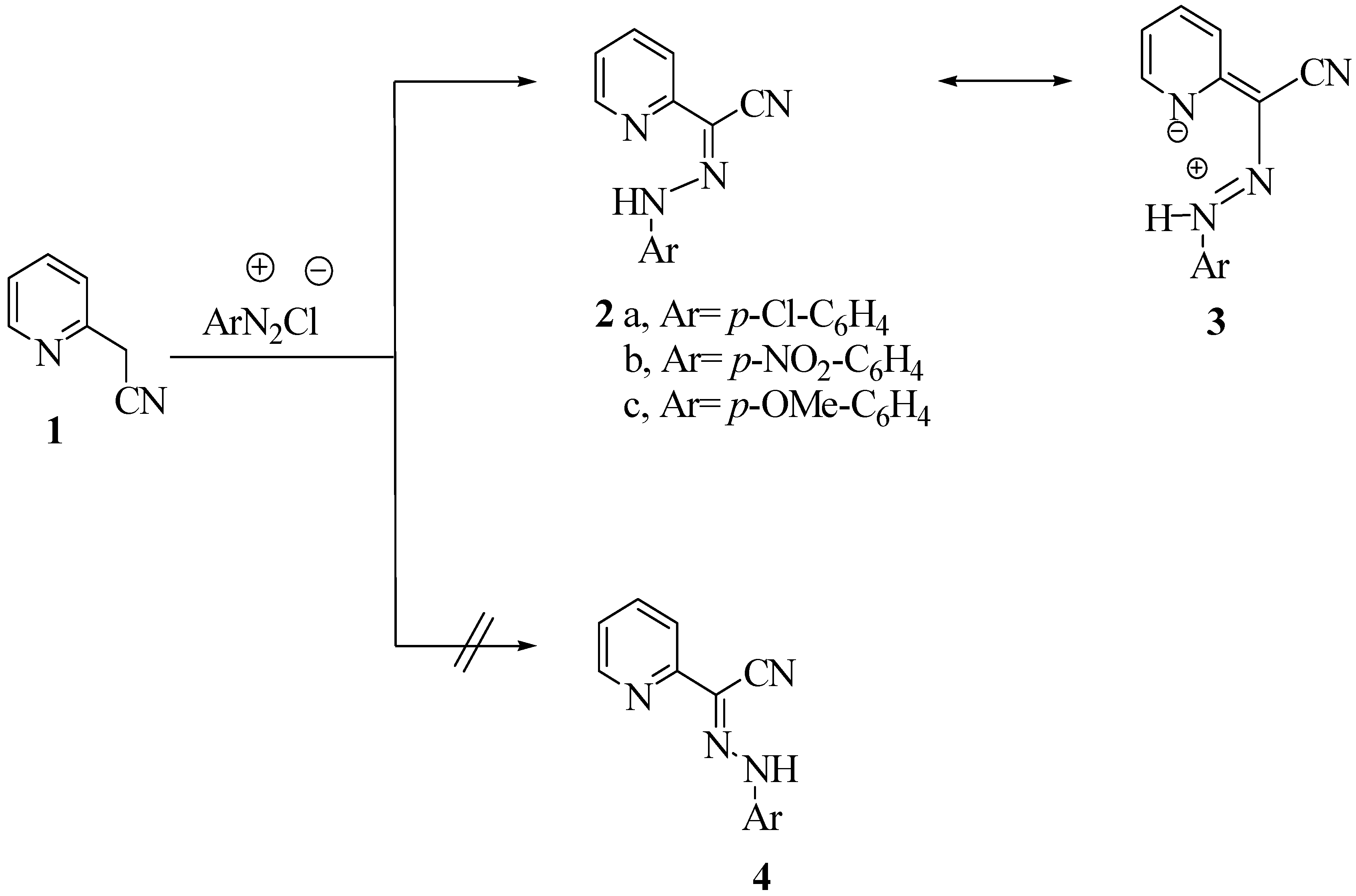{kind=link}
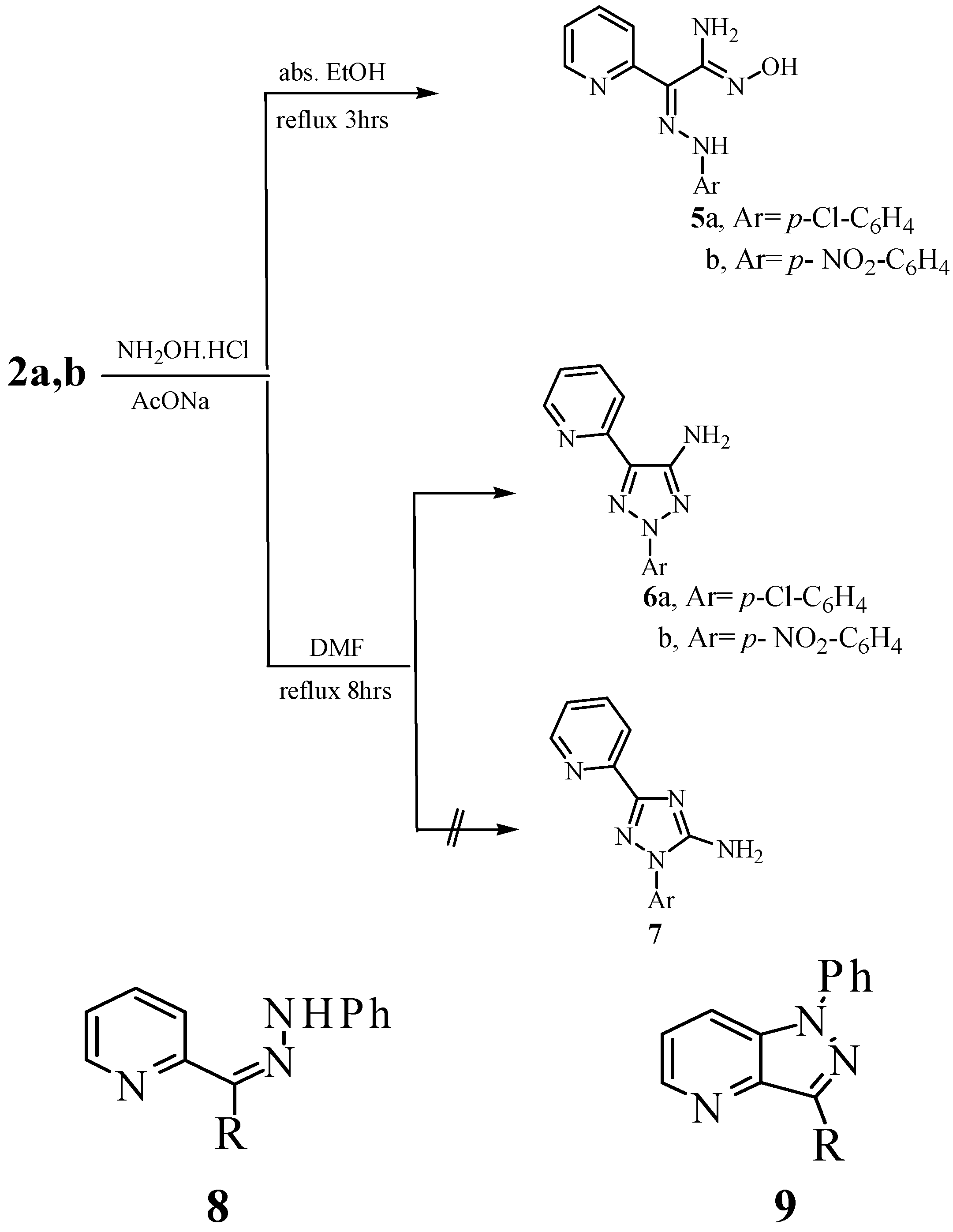{kind=link}
{kind=link}
| Parameter | 2a | ||
| Empirical Formula | C13H9N4Cl | ||
| Formula weight | 256.696 | ||
| Crystal System | Monoclinic | ||
| Space group | P21/c | ||
| Unit cell parameters | |||
| Unit cell volumeZ | 1228.62(9)4 | ||
| Temperature (K) | 298 | ||
| Radiation type | Mo Ka | ||
| Dx Mg/m3 | 1.388 | ||
| F(000) | 528 loop | ||
| Absorption coefficient (mm-1) | 0.30 | ||
| Parameters | 103 | ||
| R factor | 0.061 | ||
| Bond lengths | Bond lengths | ||
| N4 C8 | 1.320(4) | N3 N4 | 1.326(4) |
| N2 C9 | 1.326(5) | N3 C13 | 1.412(5) |
| N2 H3 | 1.906(3) | C6 C13 | 1.363(5) |
| C8 C17 | 1.449(5) | Cl 1 C12 | 1.742(3) |
| N2 C14 | 1.349(4) | N10 C17 | 1.143(4) |
| N4 H3 | 1.985(3) | C13 C16 | 1.377(4) |
| Bond angles | Bond angles | ||
| N3 N4 C8 | 118.6(3) | N2 C14 C8 | 116.9(3) |
| C14 C8 C17 | 118.5(3) | N2 H3 N3 | 132.7(2) |
| N2 C9 C7 | 124.3(4) | N3 H3 N4 | 35.4(2) |
| C8C17 N10 | 179.3(4) | C8 C14C15 | 122.1(3) |
| N3C13 C6 | 122.2(3) | N4 C8 C14 | 130.4(3) |
| N4 N3 C13 | 118.4(3) | N3 C13C16 | 117.6(3) |


Experimental
General
General procedure for the synthesis of arylhydrazones 2a-c
General procedure for the synthesis of compounds 5a,b
General procedure for the synthesis of compounds 6a,b
Conclusions
References
- Elnagdi, M.H.; Selim, M.A.; Abd El Latif, F.M.; Samia, S. Studies on azolylacetonitriles: The reactivity of thiazol-2-yl, thiadiazol-2-yl acetonitriles toward electrophilic reagents. Phosphor. Sulfur Silicon 2002, 177, 1175–1182. [Google Scholar] [CrossRef]
- Klimko, Y.E.; Pisanenko, D.A. Addition of acetonitrile to 5-methylene- and 5-ethylidenebicyclo[2.2.1]hept-2-enes in the Ritter reaction. Ukr. Khim. Zh. 2008, 74, 104–108. [Google Scholar]
- Ghozlan, S.A.S.; Abdelhamid, I.A.; Ibrahim, H.M.; Elnagdi, M.H. Studies with 2-aryl-hydrazononitriles: A new convenient synthesis of 2,4-disubstituted-1,2,3-triazole-5-amines. ARKIVOC 2006, XV, 53–60. [Google Scholar]
- Bey, E.; Marchais-Oberwinkler, S.; Werth, R.; Negri, M.; Al-Soud, Y.A.; Kruchten, P.; Oster, A.; Frotscher, M.; Birk, B.; Hartmann, R.W.D. Synthesis, Biological evaluation and pharmacokinetics of bis(hydroxyphenyl) substituted azoles, thiophenes, benzenes, and aza-benzenes as potent and selective nonsteroidal inhibitors of β-hydroxysteroid dehydrogenase type 1 (17b -HSD1). J. Med. Chem. 2008, 51, 6725–6739. [Google Scholar]
- Al-Matar, H.M.; Riyadh, S.M.; Elnagdi, M.H. 2-Arylhydrazononitriles in heterocyclic synthesis: A novel route to 1,3-diaryl-1,2,4-triazol-5-amines via a Tiemann rearrangement of arylhydrazono-amidoximes. ARKIVOC 2007, XIII, 53–62. [Google Scholar]
- Itoh, T.; Matsuzaki, K.; Suzuki, S.; Kishida, K.; Ogura, H.; Kawahara, N.; Nakajima, T. Photochemical reactions of condensed azoles with dimethyl acetylenedicarboxylate. IV. Heterocycles 1983, 20, 1321–4132. [Google Scholar] [CrossRef]
- Riyadh, S.M.; Al-Matar, H.M.; Elnagdi, M.H. Studies with 2-Arylhydrazononitriles: Further Investigations on reactivity of 2-Arylhydrazononitriles towards hydroxylamine. J. Heterocycl. Chem. 2008, 45, 1–5. [Google Scholar] [CrossRef]
- Volovenko, Y.M.; Nemazanyi, A.G.; Vesel'skaya, G.L.; Babichev, F.S. Synthesis of condensed azines with a backbone nitrogen atom under intramolecular nucleophilic substitution in the series of thiophene derivatives. Ukr. Khim. Zh. 1987, 53, 1085–1088. [Google Scholar]
- Ghozlan, S.A.S.; Abdelhamid, I.A.; Elnagdi, M.H. Functionally substituted arylhydrazones as building blocks in heterocyclic synthesis: routes to pyridazinesand pyridazinoquinazolines. ARKIVOC 2006, XIII, 147–157. [Google Scholar]
- Crystal data for 2a (ref. CCDC 743498) can be obtained on request from the Director, Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK.
- Altomare, A.; Cascarano, G.; Giacorazzo, C.; Guaghardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. [Google Scholar]
- Johnson, C.K. ORTEP-11; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1976. [Google Scholar]
- Filak, L; Rokob, T.A; Vasko, G.A.; Egyed, O.; Gomory, A.; Riedl, Z.; Hajos, G. A new cyclization to fused pyrazoles tunable for pericyclic or pseudopericyclic route: An experimental and theoretical study. J. Org. Chem. 2008, 73, 3900–3906. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1-13 are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Sheikh, M.A.; Elnagdi, M.H. Studies with Azinylacetonitriles: 2-Pyridylacetonitrile as a Precursor to Functionally Substituted Pyridines. Molecules 2009, 14, 4406-4413. https://doi.org/10.3390/molecules14114406
Al-Sheikh MA, Elnagdi MH. Studies with Azinylacetonitriles: 2-Pyridylacetonitrile as a Precursor to Functionally Substituted Pyridines. Molecules. 2009; 14(11):4406-4413. https://doi.org/10.3390/molecules14114406
Chicago/Turabian StyleAl-Sheikh, Mariam Abdullah, and Mohamed Hilmy Elnagdi. 2009. "Studies with Azinylacetonitriles: 2-Pyridylacetonitrile as a Precursor to Functionally Substituted Pyridines" Molecules 14, no. 11: 4406-4413. https://doi.org/10.3390/molecules14114406
APA StyleAl-Sheikh, M. A., & Elnagdi, M. H. (2009). Studies with Azinylacetonitriles: 2-Pyridylacetonitrile as a Precursor to Functionally Substituted Pyridines. Molecules, 14(11), 4406-4413. https://doi.org/10.3390/molecules14114406