Diaroyl Tellurides: Synthesis, Structure and NBO Analysis of (2-MeOC6H4CO)2Te – Comparison with Its Sulfur and Selenium Isologues. The First Observation of [MgBr][R(C=Te)O] Salts

Abstract
:Introduction
Results and Discussion
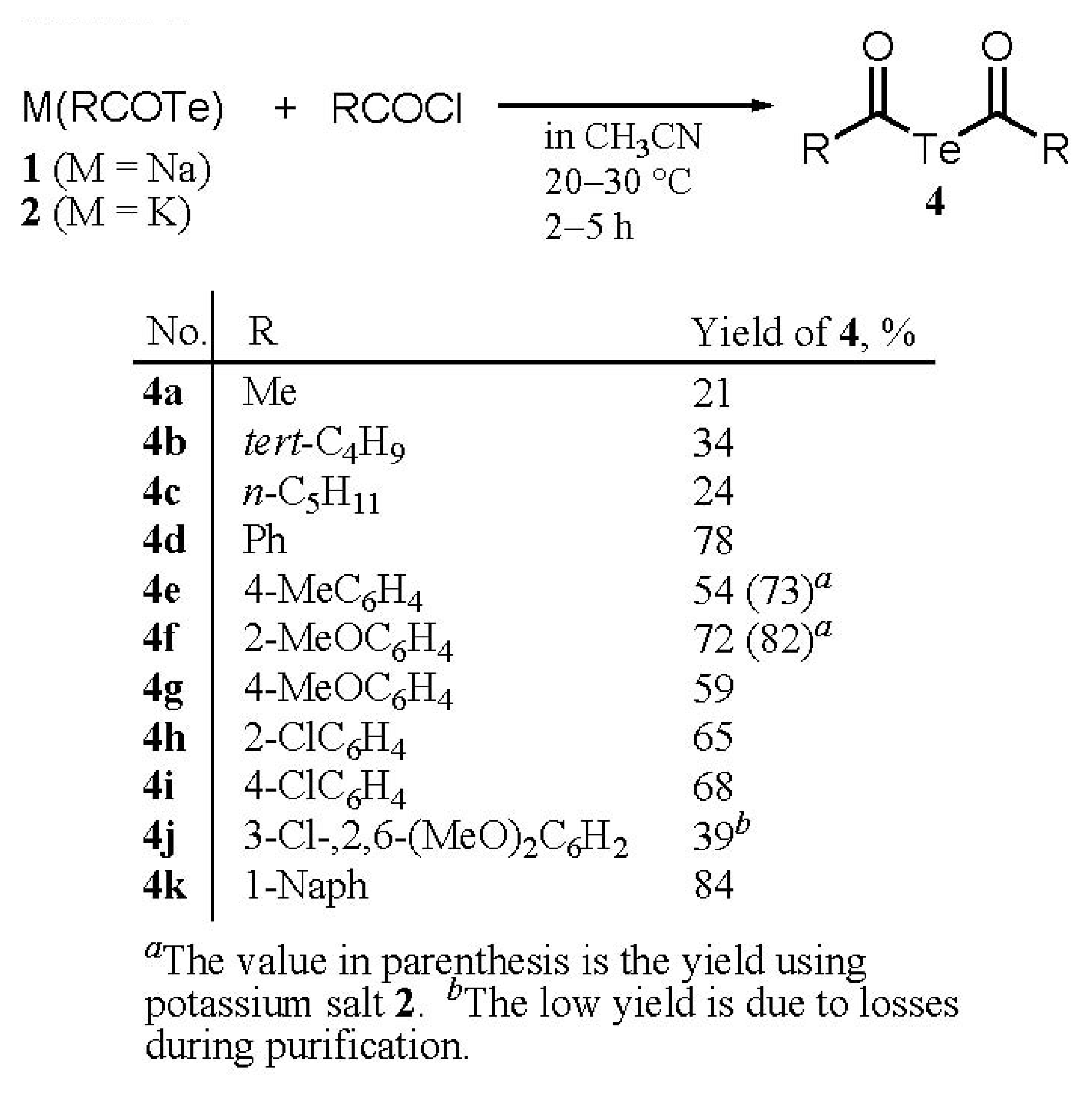
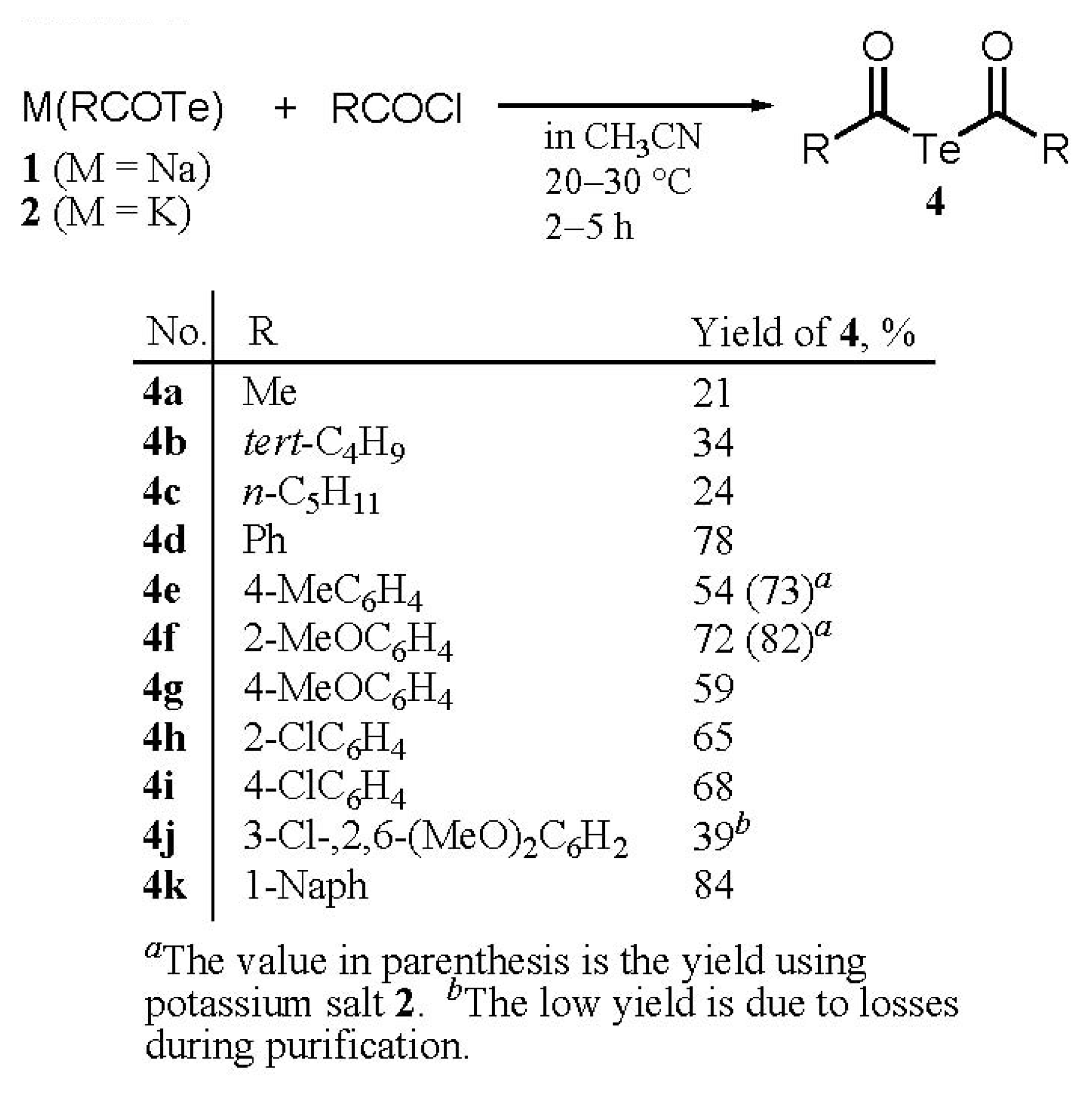
Synthesis
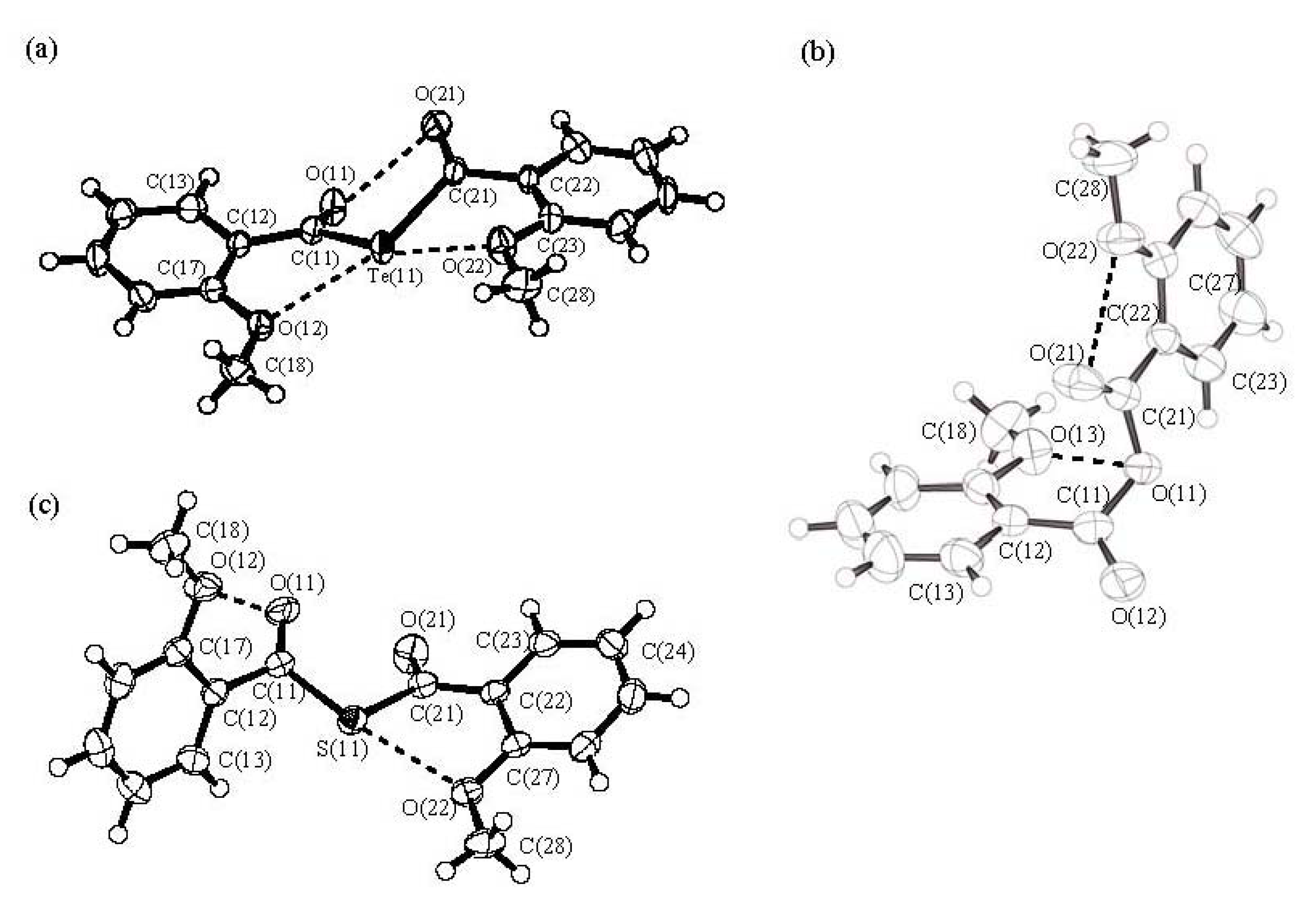
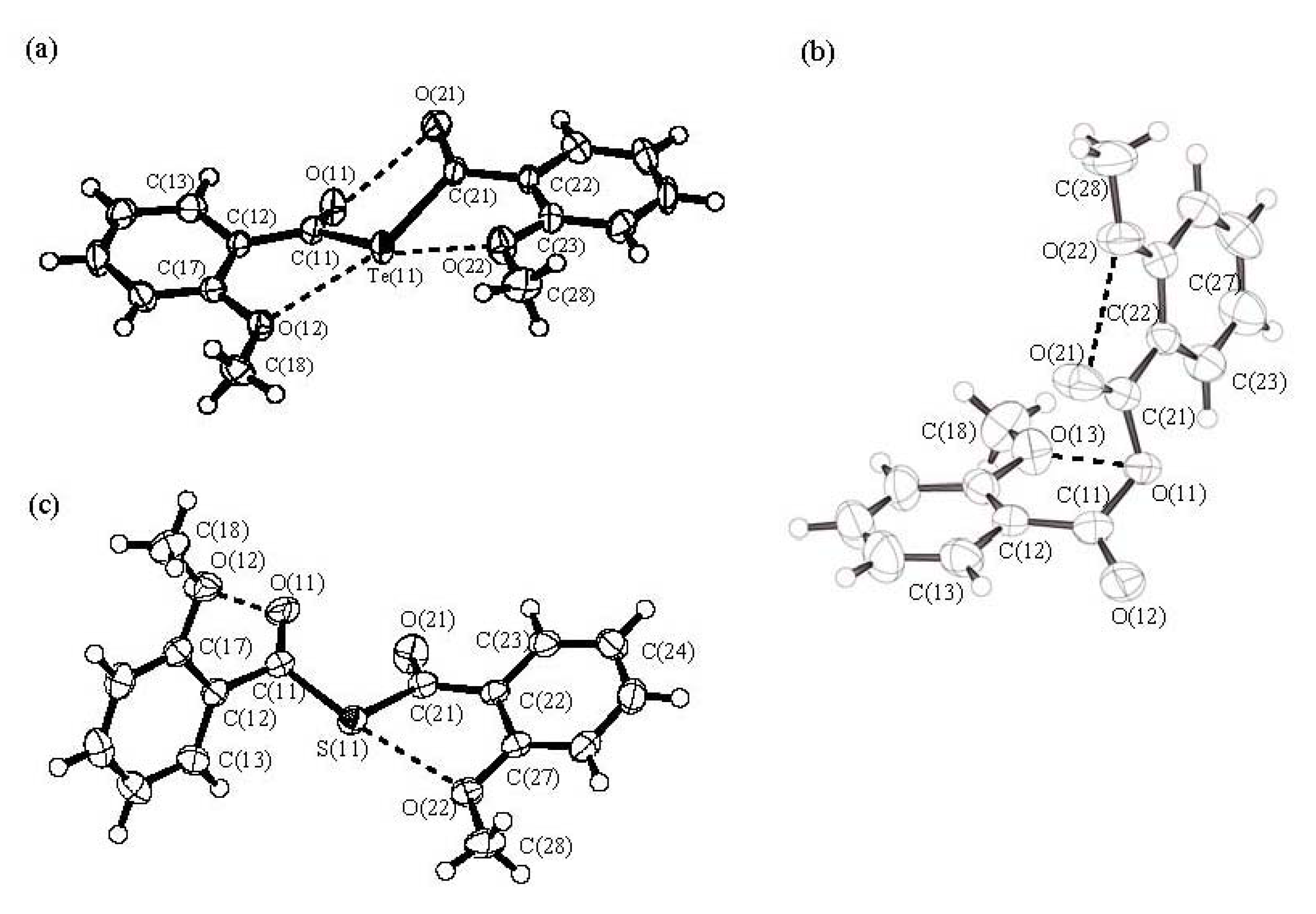
Molecular structures of bis(2-methoxybenzoyl) chalcogenides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
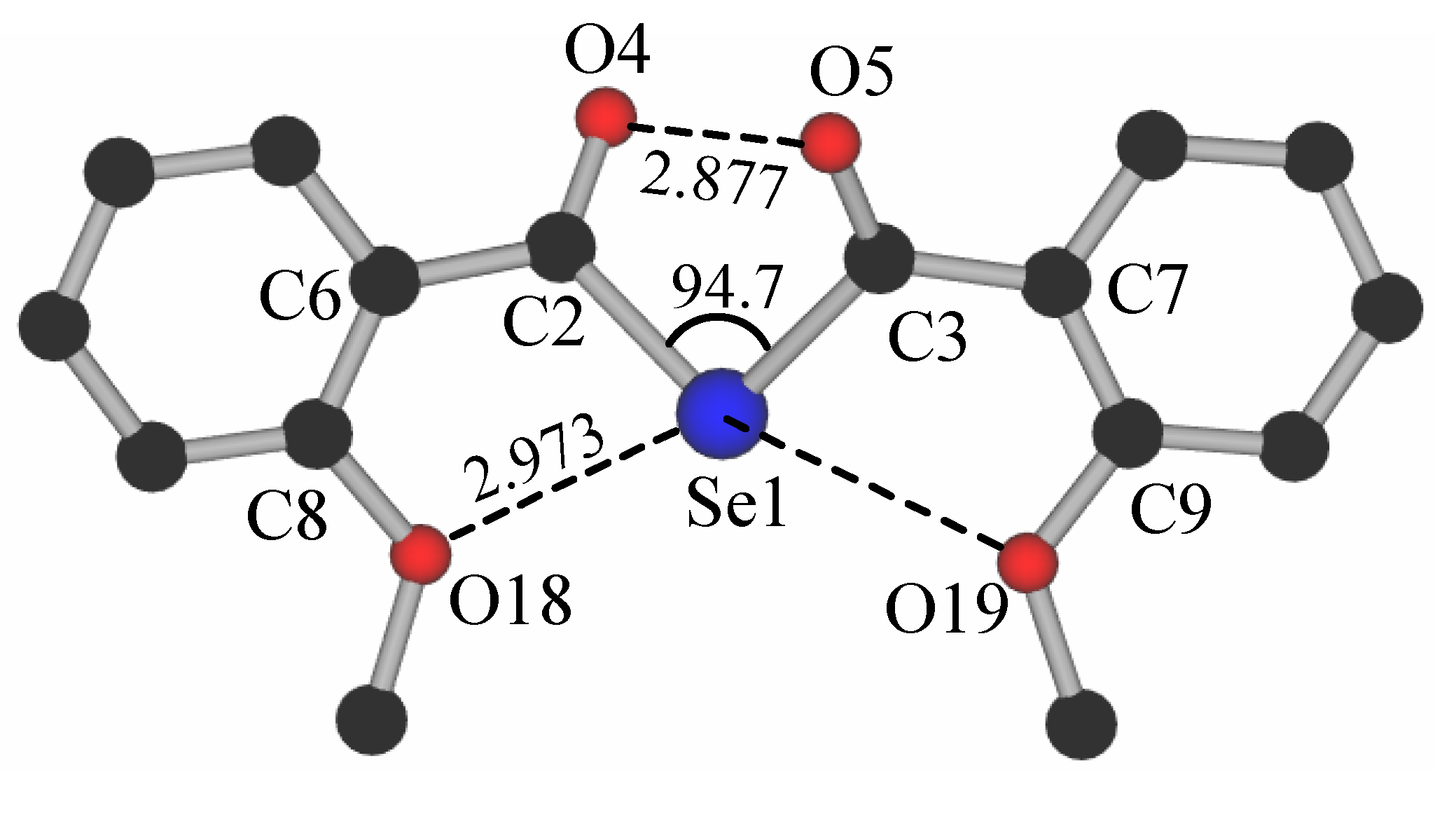
{kind=link}
{kind=link}
{kind=link}
 |

 |
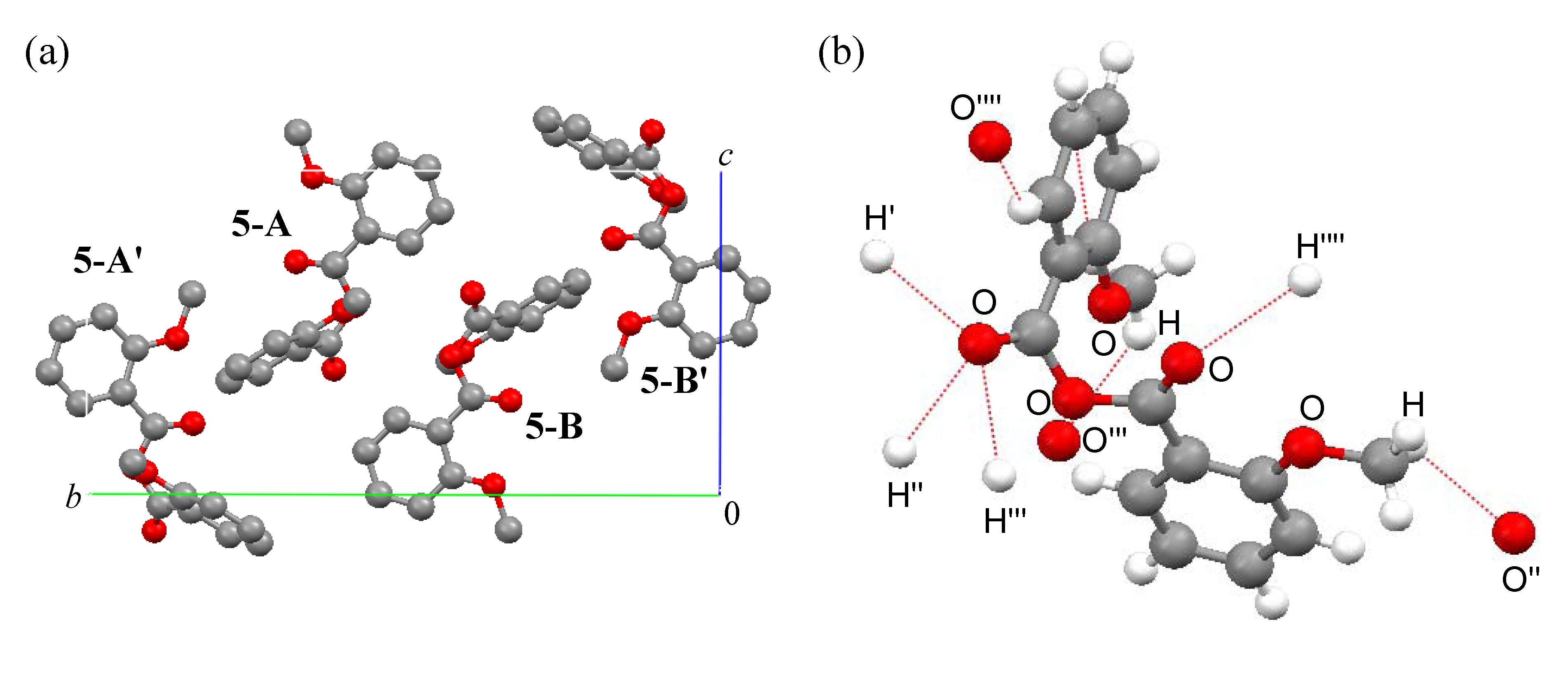
 |
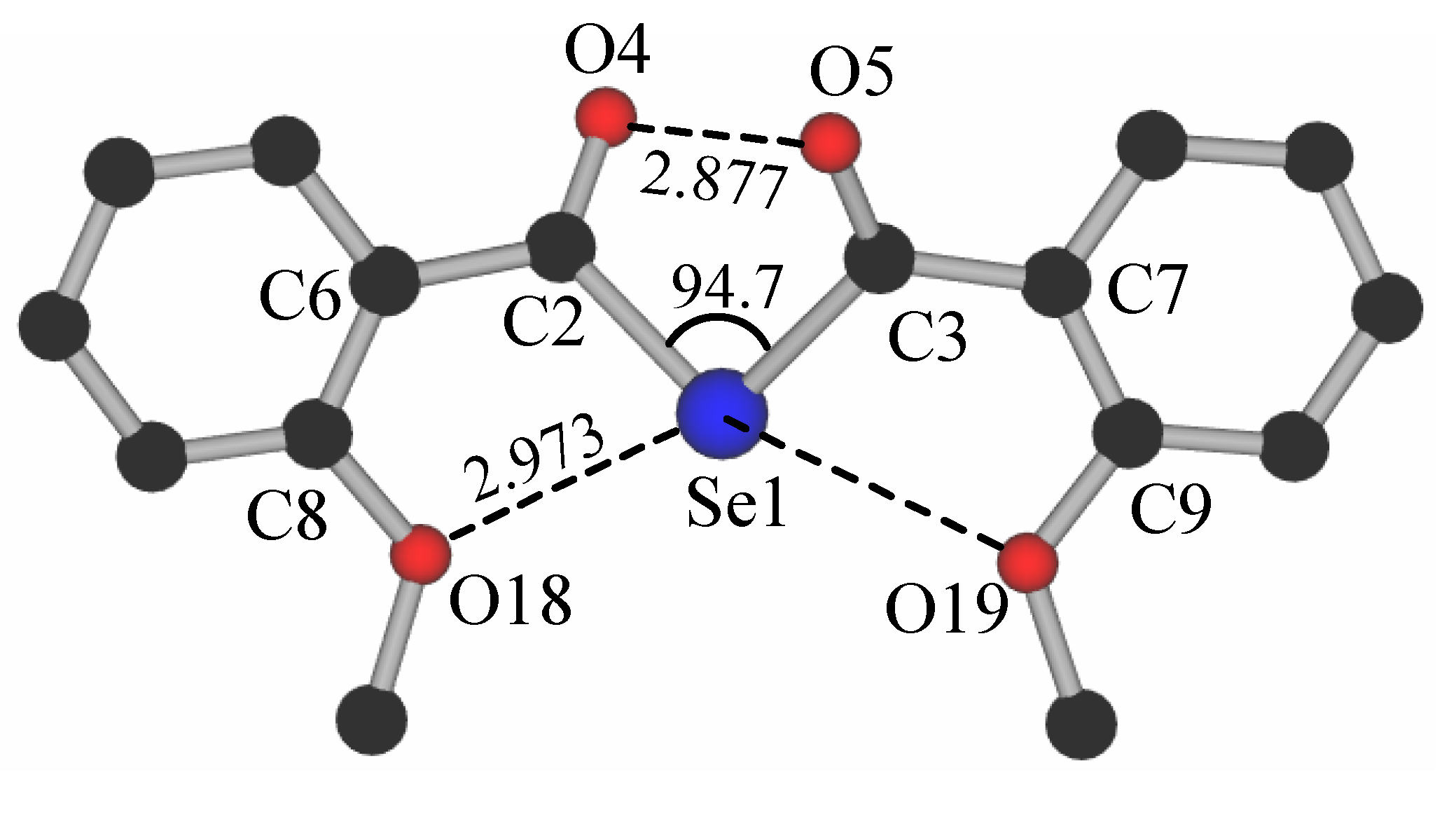
Packing
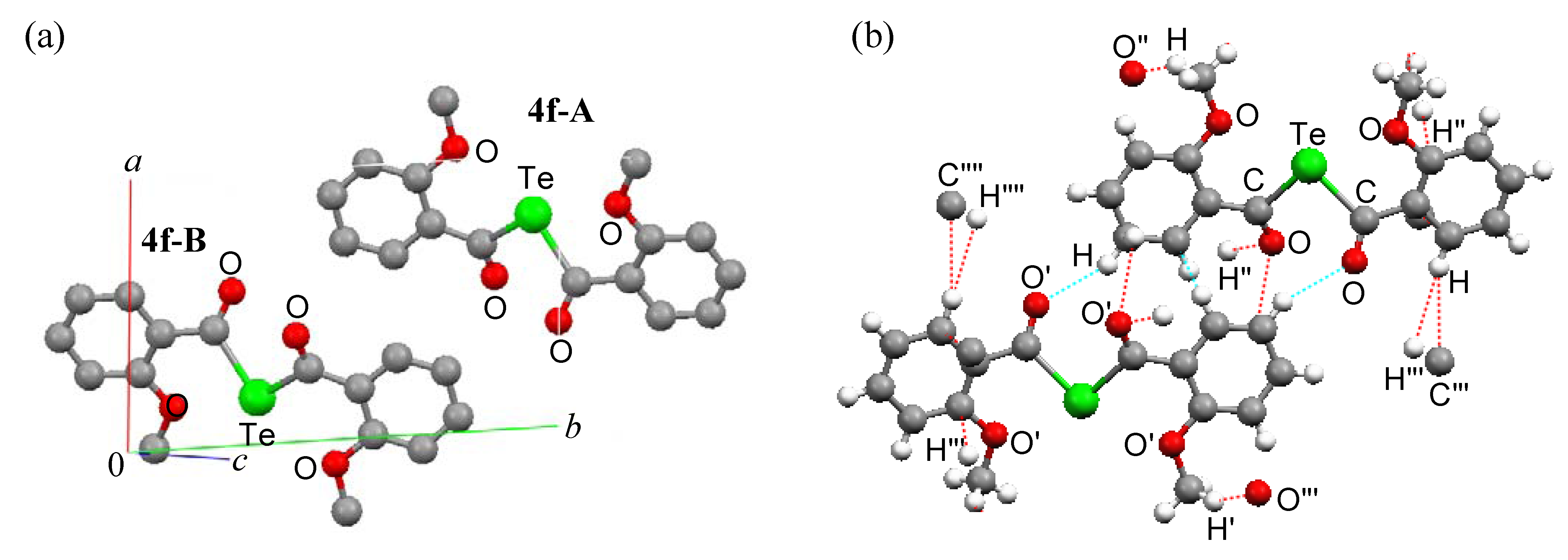
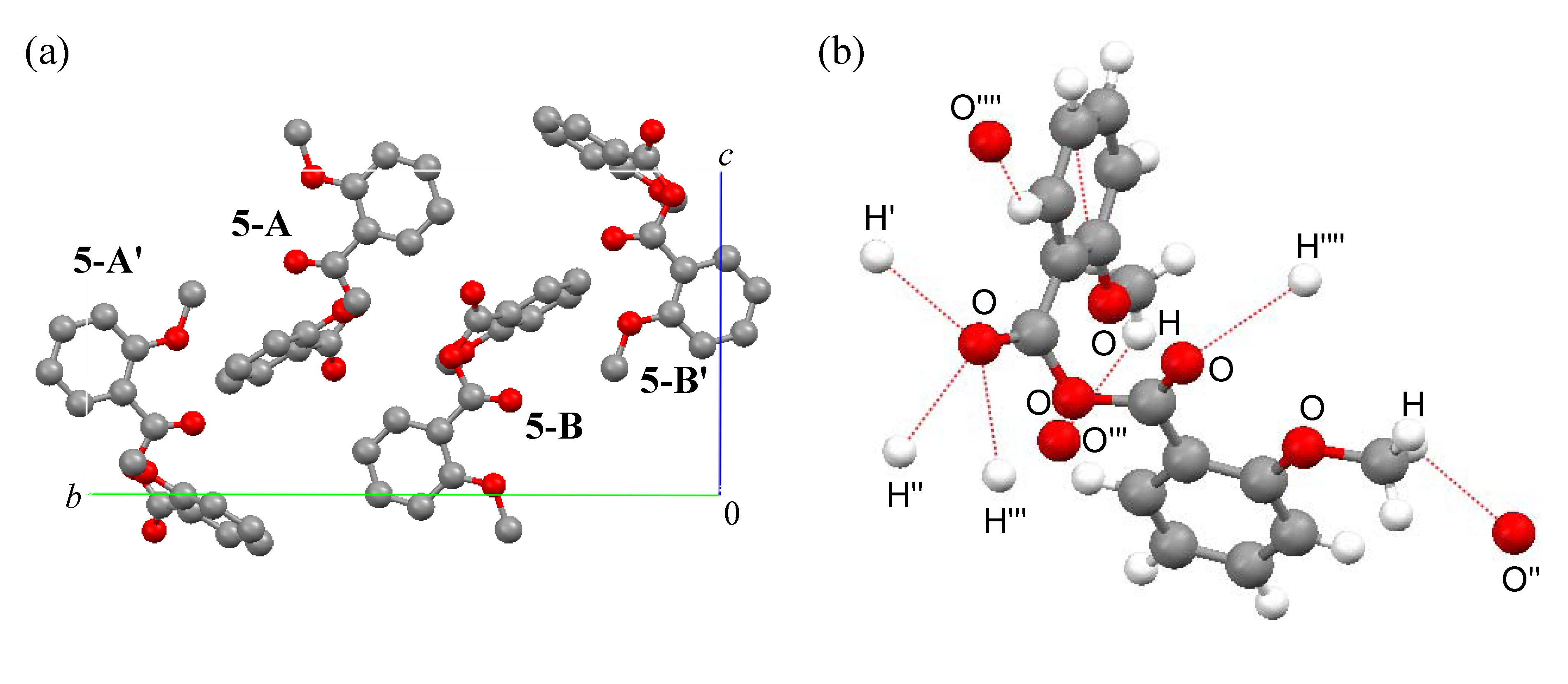
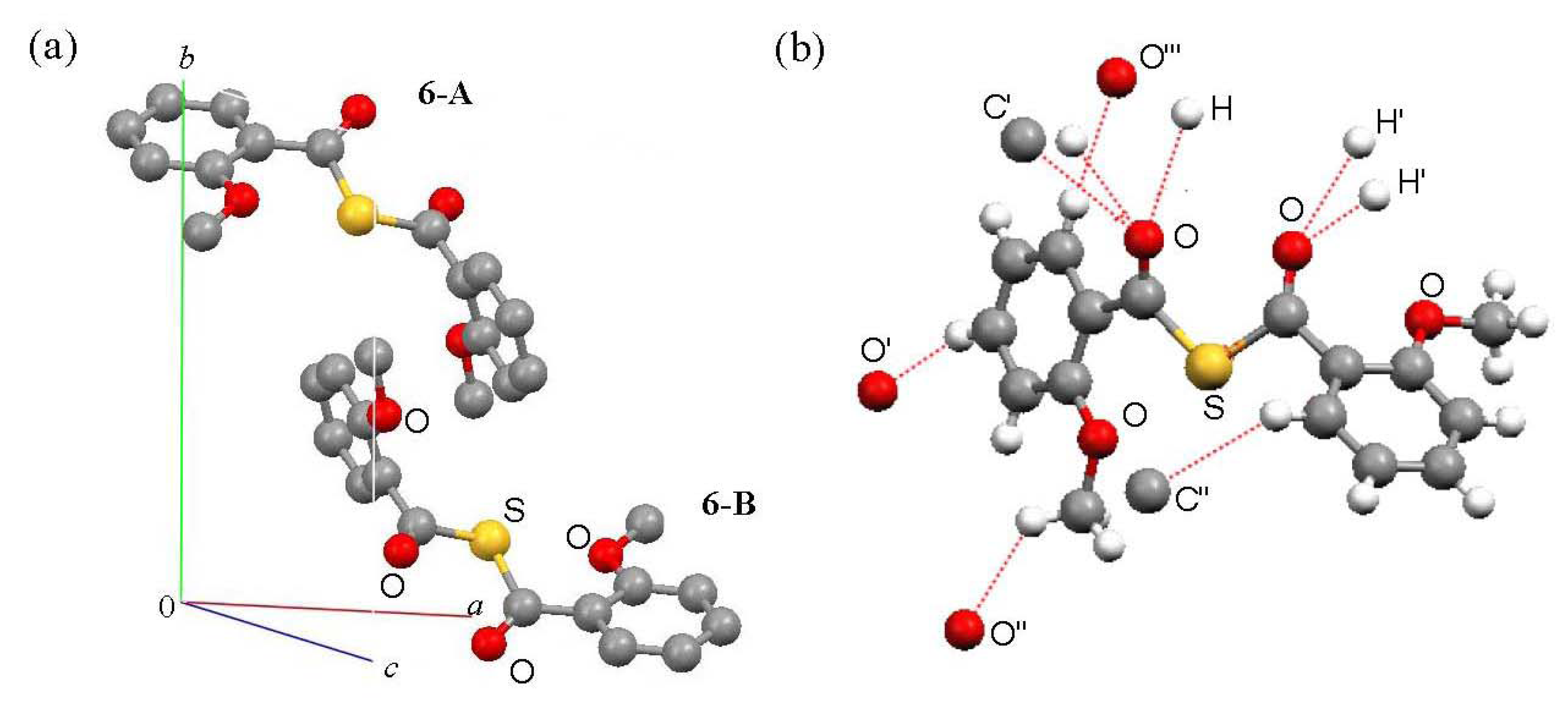
Ab initio calculations
 |
Reactions diacyl tellurides with Grignard reagents
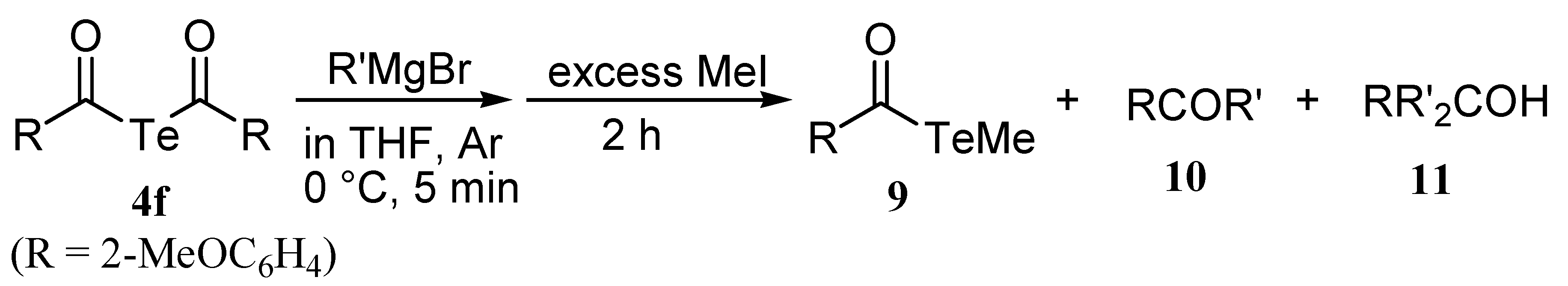
 |
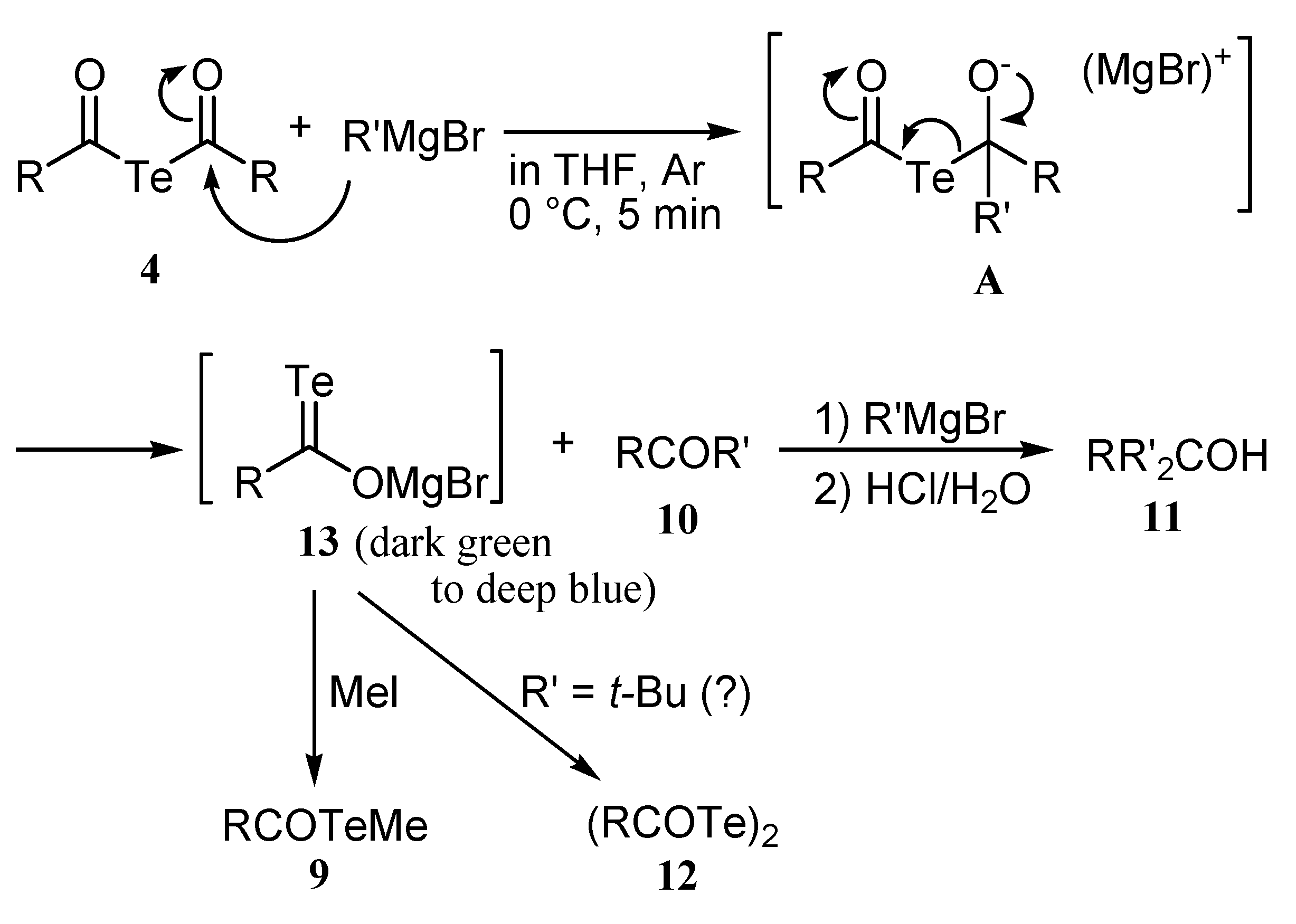
Conclusions
Experimental
General
Materials
X-ray Measurements [20,21,22, 23 and 24]
Preparation of (2-MeOC6H4CO)2S (5) [26] and (2-MeOC6H4CO)2O (6) [27]
Syntheses of di(acyl) tellurides 4a-k
Reactions of compounds 4 with Grignard reagents
Acknowledgements
References and Notes
- Bergmann, J.; Engman, L. Preparation of Selena- and Tellura Phthalic Anhydride. Org. Prep. Proc. Int. 1978, 10, 289–290. [Google Scholar]
- Severengiz, T.; du Mont, W.W.; Renoir, D.; Voss, H. Novel Reactions of Acyl Halides with Bis(trimethylsilyl) Telluride: C, Te and C, C Bond Formation. Angew. Chem. Int. Ed. Engl. 1985, 24, 1041–1042. [Google Scholar] Severengiz, T.; du Mont, W.-W. Evidence for the 1,3-Silyl Shift Equilibrium between the C=O- and C=Te-Bonded Isomers of a Stable Trimethylsilyl Tellurocarboxylate. J. Chem. Soc., Chem. Commun. 1987, 820–821. [Google Scholar]
- Sewing, D.; du Mont, W.W.; Pohl, S.; Saak, W. Diacyltelluride: Synthesen durch Reaktionen von Acylchloriden mit Bis(trialkylsilyl)telluriden; Strukturbestimmunge an Di(1-adamantoyl)tellurid und Adamantancarbonsäureanhydrid. Z. Anorg. Allg. Chem. 1998, 624, 1363–1368. [Google Scholar]
- Kato, S.; Kakigano, T.; Ishida, M. Tellurium Isologues of Acid Anhydrides: The first Acyclic Bis(acyl) Tellurides. Z. Chem. 1986, 26, 179–180. [Google Scholar] Kakigano, T.; Kanda, T.; Ishida, M.; Kato, S. Synthesis and Characterization of Tellurocarboxylic Acid Salts and Bis(acyl) Ditellurides. Chem. Lett. 1987, 475–478. [Google Scholar]
- The reactions appear to produce the telluride quantitatively and the low yield of 4d is due to the loss during purification
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Minyaev, R.S.; Minkin, V.I. Theoretical study of O→X (S, Se, Te) coordination in organic compounds. Can. J. Chem. 1998, 76, 776–788. [Google Scholar] [CrossRef]
- CCDC 719922 (4f), CCDC 719923 (5) and CCDC 719924 (6) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- Kucsman, A.; Kapovits, I. Organic Sulfur Chemistry: Theoretical and Experimental Advances 1985, 191–245. [CrossRef]
- Niyomura, O.; Kato, S.; Inagaki, S. An Unusual Planar Diacyl Ditelluride (2-MeOC6H4CO)2Te: The Origin of its Planarity. J. Am. Chem. Soc. 2000, 122, 3132–2133. [Google Scholar]
- Nishio, M. The CH/π Interaction. Evidence, Nature, and Consequences; Wiley-VCH: New York, NY, USA, 1998. [Google Scholar] Nishio, M. CH/π hydrogen bonds in crystals. Cryst. Eng. Comm. 2004, 6, 138–158. [Google Scholar]
- Huzinaga, S.; Andzelm, J.; Klobukowski, M.; Radzio-Andzelm, Y.; Sakai, Y.; Tatewaki, H. Gaussian Basis Sets for Molecular Calculations; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A. Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 2003; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Kulkarni, Y.D.; Srivastava, S.; Athar, M. Synthesis, Characterization & Antibacterial Actvity of Dibenzoyl Telluride & Heterocyclic Systems Derived from Them. Indian J. Chem. 1986, 25A, 57–59. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 56–58. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the elrectron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- The effective core potentials (ECPs) LANL2DZ were used for heavy atoms S, Se and Te. Except for H, the d polarization function for the ECP basis set of all atoms are taken from the following literature: Duning, T.H., Jr.; Hay, P.J. Methods of Electronic Structure Theory, 3rd ed.; Schaefer, H.F., Ed.; Plenum Press: New York, NY, USA, 1977; Vol. 2. [Google Scholar] Hay, P.J.; Waldt, W.R. Ab initio effective core potentials for molecular calculation. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] Hay, P.J.; Waldt, W.R. Ab initio effective core potentials for molecular calculation. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] Hay, P.J.; Waldt, W.R. Ab initio effective core potentials for molecular calculation. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar]
- Ph3COH: mp 160–161 °C; Acrey, S.F. Ber. Deutsch. Chem. Ges.; Über einige Synthesen mit Hülfe von Phenylnatrium und Alkylmagnesiumbromiden; 1904; Volume 37, pp. 2753–2768. [Google Scholar] Peters, F.N., Jr.; Grifith, S.; Griggs, D.R., Jr.; French, H.E. Action of Phenylmagnesium Bromide on Organic Acids. J. Am. Chem. Soc. 1925, 47, 449–454. [Google Scholar]
- Bachelor Thesis of Gifu University, 1972.
- Kawahara, Y.; Kato, S.; Kanda, T.; Murai, T.; Ebihara, M. Synthesis of Rubidium and Cesium Tellurocarboxyaltes and an X-Ray Structural Analysis of Heavy Alkali Metal Monochalcogeno carbxoylates. Bull. Chem. Soc. Jpn. 1995, 68, 3507–3517. [Google Scholar] Kato, S.; Kawahara, Y.; Kageyama, H.; Yamada, R.; Niyomura, O.; Murai, T.; Kanda, T. Thion (RCSOH), Selenon (RCSeOH), and Telluron (RCTeOH) Acids as Predominant Species. J. Am. Chem. Soc. 1996, 118, 1262–1267. [Google Scholar] Kanda, T.; Nakaiida, S.; Murai, T.; Kato, S. Te-Alkyl Tellurocarboxylates: Isolation and Charcterization. Tetrahedron Lett. 1989, 30, 1829–1832. [Google Scholar] Kato, S.; Niyomura, O.; Nakaiida, S.; Kawahara, Y.; Kanda, T.; Yamada, R.; Hori, H. First Isolation and Characterization of Sodium and Potasium Tellurocarboxylates. Inorg. Chem. 1996, 38, 519–530. [Google Scholar]
- North, A.C.T.; Philips, D.C.; Mathews, F.S. A semi-Empirical Method of Absorption Correction. Acta Crystallogr. Sect. A 1968, 24, 351–359. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL 97 Program for the Refinement of Crystal Structure; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Cromer, D.T.; Waber, J.T. International Tables for X-ray Crystallography; The Kynoch Press: Birmingham, England, 1974; Vol. IV, Table 2.2.A. [Google Scholar]
- Creagh, D.C.; McAuley, W. J. International Tables for X-ray Crystallography; Wilson, A.J.C., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 1992; Vol. C, Table 4.2.6.8; pp. 219–266. [Google Scholar]
- Masumoto, H.; Tsutsumi, H.; Kanda, T.; Komada, M.; Murai, T.; Kato, S. A Convenient Synthesis of Diacyl Sulfides. Sulfur Lett. 1989, 10, 103–115. [Google Scholar]
- Rambacher, P.; Mäke, S. Verfahren zur Herstellung von Anhydriden aromatischer Carbonsäuren. Angew. Chem. 1968, 80, 487. [Google Scholar] [CrossRef]
- Data Availability: the electronic supplementary information: ESI-Table 1 is available from the corresponding author.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Niyomura, O.; Nakaiida, S.; Yamada, R.; Kato, S.; Ishida, M.; Ebihara, M.; Ando, F.; Koketsu, J. Diaroyl Tellurides: Synthesis, Structure and NBO Analysis of (2-MeOC6H4CO)2Te – Comparison with Its Sulfur and Selenium Isologues. The First Observation of [MgBr][R(C=Te)O] Salts. Molecules 2009, 14, 2555-2572. https://doi.org/10.3390/molecules14072555
Niyomura O, Nakaiida S, Yamada R, Kato S, Ishida M, Ebihara M, Ando F, Koketsu J. Diaroyl Tellurides: Synthesis, Structure and NBO Analysis of (2-MeOC6H4CO)2Te – Comparison with Its Sulfur and Selenium Isologues. The First Observation of [MgBr][R(C=Te)O] Salts. Molecules. 2009; 14(7):2555-2572. https://doi.org/10.3390/molecules14072555
Chicago/Turabian StyleNiyomura, Osamu, Shoho Nakaiida, Ryo Yamada, Shinzi Kato, Masaru Ishida, Masahiro Ebihara, Fumio Ando, and Jugo Koketsu. 2009. "Diaroyl Tellurides: Synthesis, Structure and NBO Analysis of (2-MeOC6H4CO)2Te – Comparison with Its Sulfur and Selenium Isologues. The First Observation of [MgBr][R(C=Te)O] Salts" Molecules 14, no. 7: 2555-2572. https://doi.org/10.3390/molecules14072555
APA StyleNiyomura, O., Nakaiida, S., Yamada, R., Kato, S., Ishida, M., Ebihara, M., Ando, F., & Koketsu, J. (2009). Diaroyl Tellurides: Synthesis, Structure and NBO Analysis of (2-MeOC6H4CO)2Te – Comparison with Its Sulfur and Selenium Isologues. The First Observation of [MgBr][R(C=Te)O] Salts. Molecules, 14(7), 2555-2572. https://doi.org/10.3390/molecules14072555