3. Experimental
3.1. General
Reactions were monitored by gas chromatography (GC) on a Varian CP-3800 gas chromatograph fitted with a methyl silicone (CP-Sil 8 CB) capillary column (30 m × 0.25 mm × 0.25 μm); carrier gas: He; flow rate: 1 mL/min; oven temperature program: 50−290 °C at a rate of 8 °C/min; injector temperature: 250 °C; flame ionization detector temperature: 300 °C; retention times (tR) are expressed in minutes. The reaction products were purified by conventional column chromatography (Merck silica gel 60, 70−230 mesh) or by flash chromatography (Scharlab silica gel 60, 230−400 mesh), in both cases using appropriate mixtures of hexane and Et2O. 1H-NMR spectra were recorded on a Bruker DPX 300 spectrometer (300 MHz, CDCl3, TMS) and a Bruker DPX 400 (400 MHz) spectrometer. Chemical shift values are reported in parts per million (ppm, δ scale) and coupling constants (J) are in hertz (Hz). All described coupling constants refer to a three-bond coupling distance (3J). 13C-NMR spectra were recorded on the same instruments (75 or 100 MHz, CDCl3, TMS). Chemical shifts are also reported in ppm and carbon substitution degrees were established by DEPT multipulse sequence. 2D NMR experiments (DQF-COSY, HSQC, HMBC, NOESY) were carried out for all compounds of dihydronopol series (17, 18, 27, 32, 37) and for the isochromene 23, on the same instrument. Infrared (IR) spectra were recorder on a FT-IR Perkin-Elmer 1760X spectrometer using a thin film between KBr plates (neat). Mass spectra (MS) were obtained in all cases by GC− MS analysis carried out on a Hewlett-Packard 5990 A II gas chromatograph coupled to a Hewlett-Packard 5989B mass spectrometer using the electron impact (EI) ionization method (70 eV); the parameters for the GC unit were the same as those described previously for the GC analyses. High-resolution mass spectra (HRMS) were obtained on a trisector EBE Waters Micromass AutoSpect NT spectrometer using EI (70 eV).
3.2. Starting materials
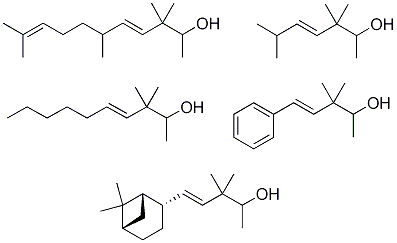
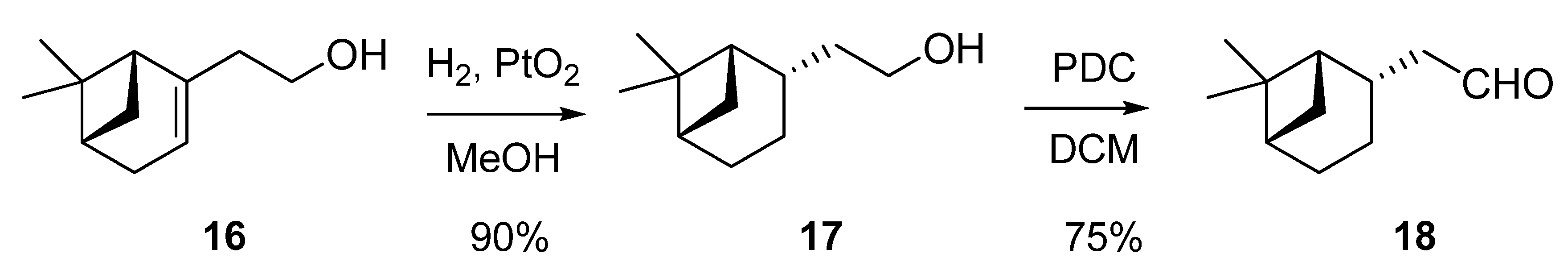
Isovaleraldehyde (3-methylbutanal, 12): Aldrich, 97% (GC); tR 2.26. Heptanal (13): Aldrich, 95% (GC); tR 6.32. (±)-Citronellal (3,7-dimethyl-6-octenal, 14): Fluka, 90% (GC); tR 16.20. 2-Phenyl-acetaldehyde (15): Fluka, 50% solution in diethylphthalate. To separate 2-phenylacetaldehyde (195 °C, 1 atm), tR 11.40, from the non-volatile diethylphthalate (295 °C, 1 atm) a vacuum distillation was performed. (1S,2S,5S)-Dihydronopal (18) was obtained, as described below, from (1R)-(–)-nopol [(1R)-2-(6,6-dimethylbicyclo[3.1.1]-2-hepten-2-yl)ethanol, 16): Aldrich, 98% (GC); [α]25D = –31.8 (c 1.15, MeOH); tR 21.60.
(1S,2S,5S)-2-(6,6-Dimethylbicyclo[3.1.1]hept-2-yl)ethanol (17): a solution of the alkene 16 (250 mg, 1.5 mmol) in absolute MeOH (12.5 mL) was hydrogenated over PtO2 (23 mg) under the low-pressure of a H2 gas filled balloon for 90 min. At this time the GC analysis indicated the hydrogenation was complete and the catalyst being filtered off and washed with MeOH. The solvent was evaporated under reduced pressure to afford 17 (227 mg, 90%) as a colourless oil; [α]25D = –22.1(c 1.25, MeOH); tR 24.65; IR (ν, cm-1): 3332 (OH), 2907 (cyclohexane), 1383 and 1366 (C(CH3)2); MS (m/z, %): 168 (M+, 1), 150 (M+−H2O, 1), 135 (M+−H2O−Me, 5), 123 (M+−C2H4OH, 14), 107 (C8H11+, 33); 1H-NMR (400 MHz): δ 3.65 (dt, J1A-1B=10.1, J1A-2A-2B=7.0, 1H, H-1A), 3.62 (dt, J1A-1B=10.1, J1A-2A-2B=7.0, 1H, H-1B), 1.68 (q, J2-2’-1=7.0, 2H, H-2), 1.87−1.82 (m, 1H, CH-1’), 2.12 (ddq, J2’-3’a-2-=7.1, J2’-3’s=11.0, J2’-1’=2.0, 1H, H-2’), 1.47 (ddt, J3’a-3’s=14.2, J3’a-2’-4’s=5.8, J3’a-4’a=11.1, 1H, H-3’a), 2.01−1.92 (m, 1H, H-3’s), 1.89−1.82 (m, 1H, H-4’a), 1.97−1.90 (m, 1H, H-4’s), 1.91−1.87 (m, 1H, H-5’), 0.90 (d, J7’s-7’a=9.5, 1H, H-7’s), 2.33 (ddt, J7’a-7’s=9.3, J7’a-4’a=2.0, J7’a-1’-5’=6.2, 1H, H-7’a), 1.19 (s, 3H, Me-6’), 1.01 (s, 3H, Me’-6’); 13C-NMR (100 MHz): δ 61.68 (C-1), 40.76 (C-2), 46.40 (C-1’), 37.48 (C-2’), 22.34 (C-3’), 26.43 (C-4’), 41.42 (C-5’), 38.69 (C-6’), 33.60 (C-7’), 28.16 (Me-6’), 23.22 (Me’-6’).
(1S,2S,5S)-6,6-Dimethylbicyclo[3.1.1]hept-2-yl-ethanal (18): a solution of dihydronopol (17, 907 mg, 5.4 mmol) in dry CH2Cl2 (8 mL) was added to a solution of PDC (3.05 g, 8.1 mmol) in CH2Cl2 (22 mL) at 25 °C and stirred for 20 h under argon. Then, the mixture was diluted with diethylether–hexane, filtered and the solvent evaporated under reduced pressure to afford 18 (672 mg, 75%) as a yellow-pale oil, [α]25D = –19.3 (c 1.10, MeOH). The GC analysis (tR 21.44) indicated the conversion was complete. IR (ν, cm-1): 2907 and 2713 (cyclohexane), 1725 (C=O), 1384 and 1367 (C(CH3)2); MS (m/z, %): 166 (M+, 1), 151 (M+−Me, 9), 148 (M+−H2O, 4), 133 (M+−H2O−Me, 12), 123 (M+−CH2CHO, 27), 107 (C8H11+, 33), 79 (C6H7+, 82); 1H-NMR (400 MHz): δ 0.90 (d, J7’s-7’a=9.8, 1H, H-7’s), 0.95 (s, 3H, Me-6’), 1.12 (s, 3H, Me’-6’), 1.37 (m, 1H, H-3’a), 1.99 (ddt, J3’s-3’a=14.6, J3’s-4’a=3.0, J3’s-4’s-2’=10.4, 1H, H-3’s), 1.74−1.79 (m, 1H, H-1’), 1.73−1.92 (m, 2H, H-4’), 1.91−1.89 (m, 1H, H-5’), 2.28 (ddt, J7’a-7’s=9.3, J7’a-4’a=2.0, J7’a-1’-5’=6.0, 1H, H-7’a), 2.44 (ddd, J2A-1=2.0, J2A-2B=16.3, J2A-2’=7.3, 1H, H-2A), 2.47 (ddd, J2B-1=2.0, J2A-2B=16.3, J2B-2’=7.3, 1H, H-2B), 2.58 (ddt, J2’-2=7.3, J2’-3’s=17.1, J2’-1’=2.3, 1H, H2-2’); 13C-NMR (100 MHz): δ 202.54 (C-1), 51.81 (C-2), 46.11 (C-1’), 34.83 (C-2’), 21.83 (C-3’), 25.95 (C-4’), 40.89 (C-5’), 38.46 (C-6’), 33.20 (C-7’), 27.73 (Me-6’), 22.96 (Me’-6’).
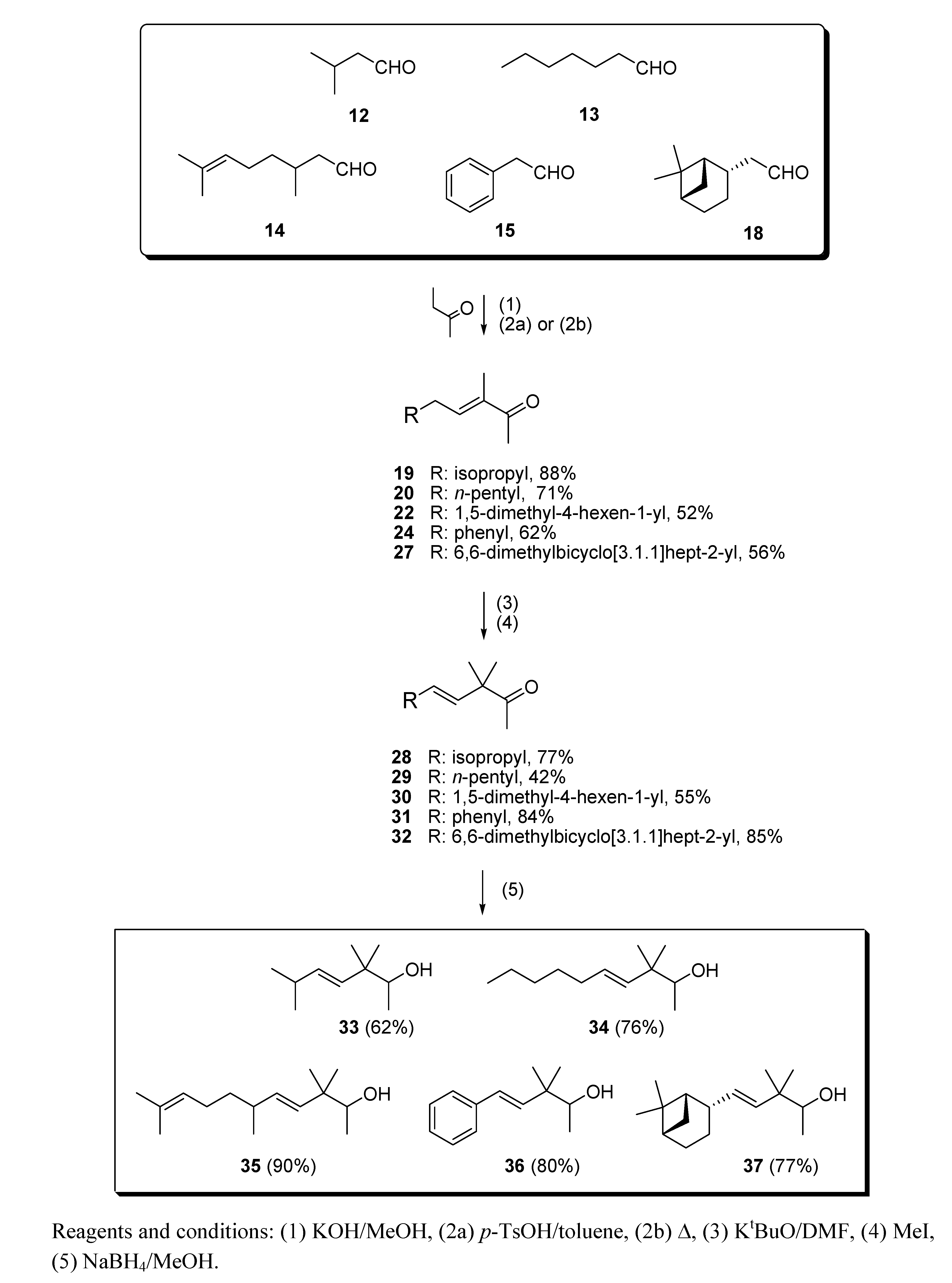
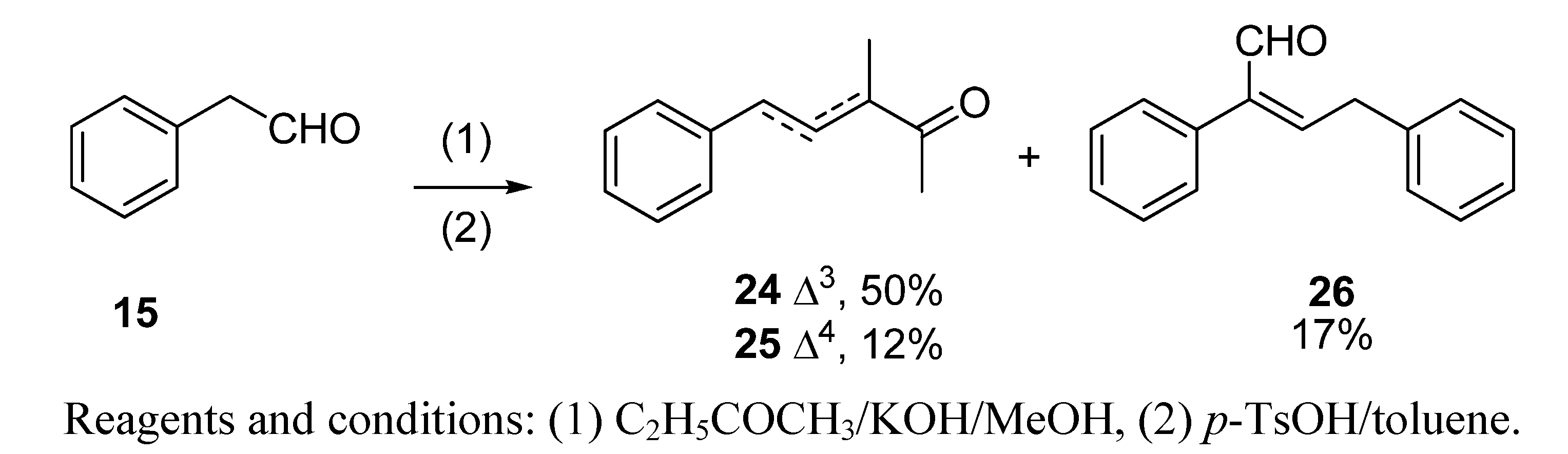
3.3. Aldol condensation of 12−15 and 18 with butanone to give 19, 20, 22, 24 and 27
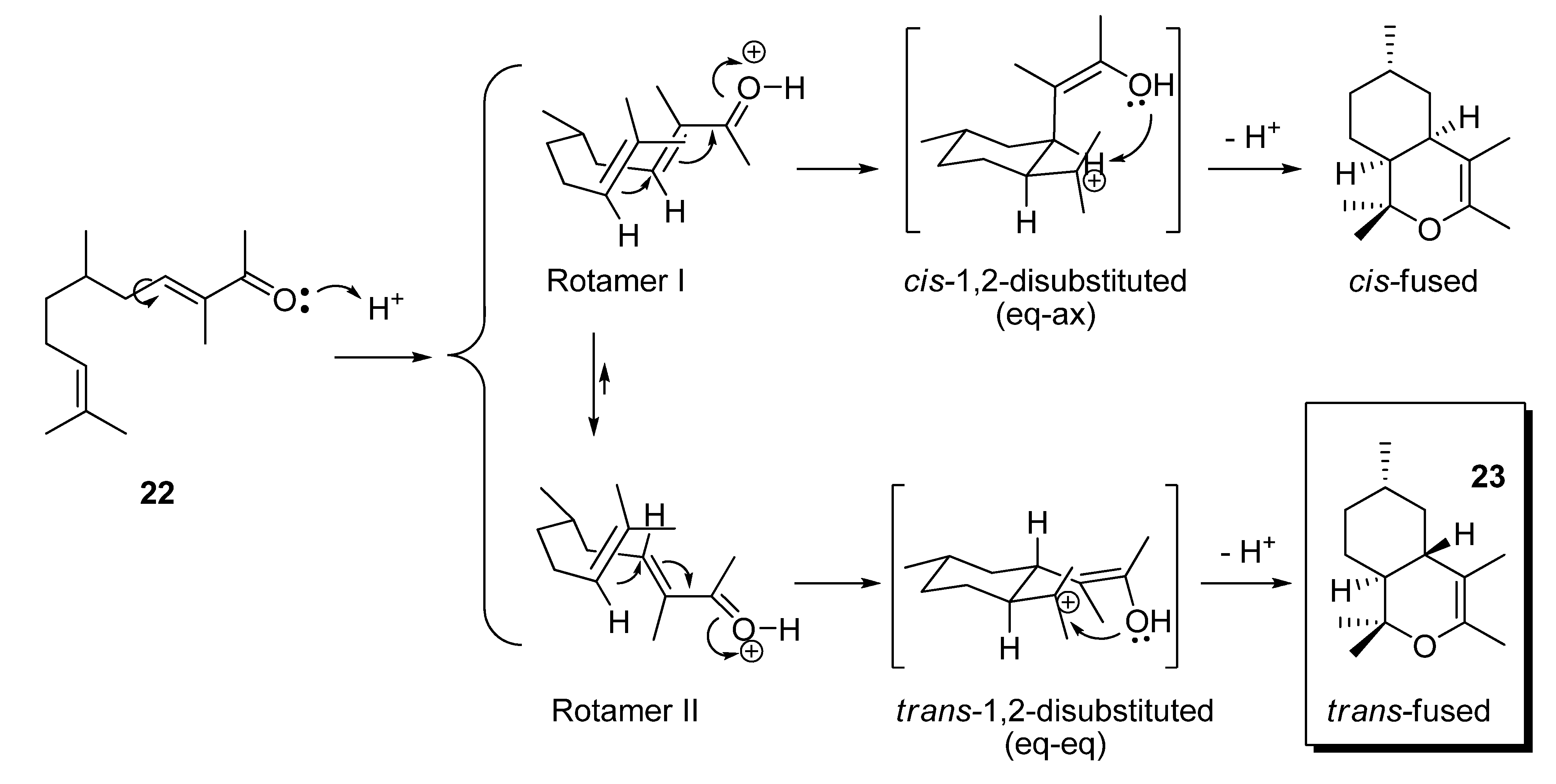
(a) With subsequent acidic catalyzed dehydration: a 6.0 M solution of starting aldehydes (12−15 and 18) in MeOH (1.0 mL, 6.0 mmol) was added dropwise to a stirred solution of butanone (1.73 g, 24.0 mmol) and KOH (15 mg, 0.25 mmol) in MeOH (1.5 mL) at 0 °C for 1 h. Then, the mixture was allowed to warm to room temperature and stirring was continued for a further 8 h. The reaction was quenched with a 1N aqueous solution of AcOH (100 mL), the solvent was then partially evaporated in vacuo and the resulting crude diluted with Et2O (25 mL) and washed with 1 N AcOH solution (25 mL) and brine (3×25 mL). The crude was dried over anhyd Na2SO4 and evaporated to yield a yellow residue, which was used in the next reaction without further purification. Then, a Dean−Stark apparatus was fitted to a flask containing a solution of the above aldol crude reaction and TsOH·H2O (40 mg, 0.2 mmol) in dry toluene (10 mL), and the mixture was refluxed for 90 min. The solution was allowed to cool down and washed with an aqueous saturated NaHCO3 solution (3×25 mL), 1N AcOH solution (25 mL) and brine (3×25 mL), dried over anhyd Na2SO4 and evaporated in vacuo. The aldol condensations of butanone (a) with 12 afforded (E)-3,6-dimethylhept-3-en-2-one (19) in a 88% yield, (b) with 13 afforded (E)-3-methyldec-3-en-2-one (20) and (Z)-2-pentylnon-2-enal (21) in 71% and 24% yields, respectively, (c) with 14 afforded 1,1,3,4,6-pentamethyl-4a,5,6,7,8,8a-hexahydro-1H-isochromene (23) in a 91% yield, (d) with 15 afforded (E)-3-methyl-5-phenylpent-3-en-2-one (24) and its isomer (E)-3-methyl-5-phenylpent-4-en-2-one (25) in a 62% yield (24:25, 4:1) and (E)-2,4-diphenylbut-2-enal (26) in a 17% yield and (e) with 18 afforded (E)-3-methyl-5-((1S,2S,5S)-6,6-dimethylbicyclo[3.1.1]hept-2-yl)pent-3-en-2-one (27) in a 56% yield.
(b) With subsequent basic catalyzed dehydration: a solution of 14 (2.0 g, 12.9 mmol) in MeOH (4.0 mL) was added dropwise to a stirred solution of butanone (3.74 g, 51.9 mmol) and KOH (30 mg, 0.5 mmol) in MeOH (2.0 mL) at 0 °C for 1 h. Then, the mixture was allowed to warm to room temperature and stirring was continued for a further 8 h. A condenser was then fitted to the flask and the mixture heated at ca. 50 °C for 2 h. The solution was allowed to reach room temperature and quenched with 1N AcOH solution (25 mL). The mixture was extracted with Et2O (3×25 mL), and the combined organic extracts were neutralized by washing with brine (3×25 mL). The crude was dried over anhyd Na2SO4 and evaporated in vacuo to afford (E)-3,6,10-trimethylundeca-3,9-dien-2-one (22) in a 52% yield.
(E)-3,6-Dimethylhept-3-en-2-one (19): colourless oil; tR 12.42; IR (ν, cm-1): 1671 and 1642 (α,β-unsaturated C=O), 1466 (-CH2-C=C-) and 1433 (CH3-C=C-); MS (m/z, %) 140 (M+, 14), 125 (M+−Me, 22), 97 (M+−[Me-C=O], 12), 83 (CH3-CO-CH-CH3+, 62), 69 (20), 55 (65), 43 (Me-C=O+, 100); 1H-NMR (300 MHz): δ 0.96 (d, J=6.6, 6H, H-7 and Me-6), 1.76 (br s, 3H, Me-3), 1.79 (n, J=6.7, 1H, H-6), 2.14 (t, J=7.1, 2H, H-5), 2.31 (s, 3H, H-1), 6.66 (tq, J1=7.4, J2=1.4, 1H, H-4); 13C NMR (75 Hz): δ 11.03 (Me-3), 22.28 (C-7, Me-6), 25.20 (C-1), 28.20 (C-6), 37.97 (C-5), 137.98 (C-3), 142.47 (C-4), 199.54 (C-2).
(E)-3-Methyldec-3-en-2-one (20): the crude reaction mixture (20+21) was purified by flash chromatography (eluent: n-hexane/Et2O 95:5) to yield 20 as a yellow-pale oil; tR 22.98. IR (ν, cm-1): 1671 and 1642 (α,β-unsaturated C=O), 1460 (-CH2-C=C-); MS (m/z, %): 168 (M+, 2), 153 (M+−Me, 8), 125 ( M+−[Me-C=O], 7), 111 (M+−[CH3-(CH2)3-], 4), 85 (M+−[Me-CO-C(Me)=CH], 14), 83 (M+−[CH3-(CH2)5-], 16), 69 (33), 55 (40), 43 (Me-C=O+, 100); 1H-NMR (300 MHz): δ 0.90 (t, J=6.7, 3H, H-10), 1.25-1.36 (m, 4H, H-7, H-8), 1.42-1.50 (m, 4H, H-6, H-9), 1.76 (br s, 3H, Me-3), 2.24 (q, J=7.3, 2H, H-5), 2.31 (s, 3H, H-1), 6.64 (tq, J1=7.3, J2=1.3, 1H, H-4); 13C-NMR (75 MHz): δ 11.02 (Me-3), 13.98 (C-10), 22.50 (C-9), 25.34 (C-1), 28.54 (C-5), 29.03 (C-7), 29.09 (C-6), 31.57 (C-8), 137.48 (C-3), 143.97 (C-4), 199.91 (C-2).
1,1,3,4,6-Pentamethyl-4a,5,6,7,8,8a-hexahydro-1H-isochromene (23): the crude reaction product was purified by flash chromatography (eluent: n-hexane) to yield 23 as a colourless oil; tR 26.00; IR (ν, cm‑1): 1738, 1715, 1677, 1456; MS (m/z, %): 208 (M+, 22), 193 (M+−Me, 8), 190 (M+−H2O, 1), 175 (M+−Me−H2O, 2), 165 (M+−[O-C(Me)], 27), 150 (M+−[O-C(Me)]−Me, 7), 137 ((M++1)−[O-C(Me)=C(Me)], 12), 123 (24), 109 (38), 95 (18), 83 (C6H11+, 10), 81 (C6H9+, 20), 69 (C5H9+, 28), 55 ([C-O-C(Me)]+, 37), 43 ([O-C(Me)]+, 100), 41 (74); 1H-NMR (300 MHz): δ 0.59 (q, J5ax-5eq-4a-6=12, 1H, H-5ax), 0.80−1.00 (m, 1H, H-7ax), 0.91−1.00 (m, 1H, H-8ax),0.92 (d, J=6.6, 3H, Me-6), 1.00 (s, 3H, Meax-1), 1.12−1.22 (m, 1H, H-8a), 1.22 (s, 3H, Meeq-1), 1.33−1.46 (m, 1H, H-6), 1.53 (br s, 3H, Me-4), 1.55−1.68 (m, 1H, H-4a), 1.63−1.72 (m, 1H, H-8eq), 1.63−1.72 (m, 1H, H-7eq), 1.71 (br s, 3H, Me-3), 1.98−2.05 (m, 1H, H-5eq); 13C-NMR (75 MHz): δ 14.05 (Me-4), 17.22 (Me-3), 19.20 (Meax-1), 22.64 (Me-6), 27.68 (Meeq-1), 27.74 (C-8), 32.77 (C-6), 35.32 (C-7), 38.37 (C-4a), 38.75 (C-5), 48.47 (C-8a), 75.40 (C-1), 103.15 (C-4), 141.75 (C-3).
(E)-3,6,10-Trimethylundeca-3,9-dien-2-one (22): the crude reaction product was purified by flash chromatography (eluent: n-hexane/Et2O 95:5) to yield 22 as a yellow-pale oil; tR 28.69; IR (ν, cm-1): 1672 and 1642 (α,β-unsaturated C=O), 1455−1439 (CH3-C=C-); MS (m/z, %): 208 (M+, 1), 193 (M+−Me, 3), 165 (M+−[Me-C=O], 9), 150 (3), 136 (5), 125 ([(Me)2C=CH-(CH2)2-CH(Me)-CH2-]+, 9), 123 (14), 109 (26), 97 ([-CH2-CH=C(Me)-CO-Me]+, 3), 95 (10), 83 ([(Me)2C=CH-(CH2)2-]+, [CH=C(Me)-CO-Me]+, 11), 81 (12), 69 ([(Me)2C=C-CH2-]+, 52), 55 ([(Me)2C=CH-]+, 30), 43 (Me-C=O+, 94), 41 (100); 1H-NMR (300 MHz): δ 0.93 (d, J=6.6, 3H, Me-6), 1.18–1,42 (m, 2H, H-7), 1.34–1.64 (m, 1H, H-6), 1.61 (br s, 3H, Me-10), 1.69 (br s, 3H, H-11), 1.77 (br s, 3H, Me-3), 1.90–2.28 (m, 4H, H-5, H-8), 2.31 (s, 3H, H-1), 5.09 (br t, J=7.0, 1H, H-9), 6.66 (br t, J=7.4, 1H, H-4); 13C-NMR (75 MHz): δ 11.20 (Me-3), 17.54 (Me-10), 19.59 (Me-6), 25.35 (C-1), 25.47 (C-8), 25.61 (C-11), 32.66 (C-6), 36.29 (C-7), 36.82 (C-5), 124.31 (C-9), 131.37 (C-10), 138.15 (C-3), 142.66 C-4), 199.70 (C-2).
(E)-3-Methyl-5-phenylpent-3-en-2-one (24) and (E)-3-methyl-5-phenylpent-4-en-2-one (25): the crude reaction product was purified by vacuum distillation (60 °C, 0.08 Torr) to yield a 4:1 mixture of 24 and 25 as a brown oil; tR 29.80 (24) and 28.76 (25); IR (ν, cm-1): 3084, 3060, 3027 (C=C, Ar), 1694 (CH3COCH(CH3)C), 1694 and 1667 (C=C(CH3)COCH3), 1452 (CH2C=C); MS (m/z, %) of 24: 175 (M++1, 11), 174 (M+, 9), 159 (M+–Me, 29), 131 (M+–MeCO, 100), 91 (C7H7+, 99), 77 (C6H5+, 12). MS (m/z, %) of 25: 174 (M+, 9), 131 (M+–MeCO, 100), 116 (M+–MeCO−Me, 15), 91 (C7H7+, 46), 77 (C6H5+, 8); 1H-NMR (400 MHz): δ 1.27 (d, JMe-3-3=7, 3H, Me-C-3, 25), 1.89 (d, JMe-3-4=1.2, 3H, Me-C-3, 24), 2.19 (s, 3H, Me-1, 25), 2.30 (s, 3H, Me-1, 24), 3.35 (q, J3-4-Me-3=7.5, 1H, H-3, 25), 3.59 (d, J4-5=7.5, 2H, H-5, 24), 3.59 (d, J4-5=7.5, 2H, H-4, 25), 6.52 (d, J5-4=15.9, 1H, H-5, 25), 6.76 (tq, J4-5=7.2, J4-Me-3=1.2, 1H, H-4, 24), 7.15−7.50 (m, 5H, Ph, 24 and25); 13C-NMR (100 MHz): δ 11.34 (Me-C3, 24), 25.51 (C-1, 24), 35.36 (C-5, 24), 127.64 (C-4’,24), 128.48 (C-5’ and -3’, 24), 128.76 (C-2’ and -6’, 24), 138.07 (C-3, 24), 138.92 (C-1’, 24), 141.49 (C-4, 24), 199.79 (C-2, 24); 16.15 (Me-3, 25), 28.14 (C-1, 25), 51.34 (C-3, 25), 126.24 (C-5’ and -3’, 25), 127.46 (C-4’,25), 128.48 (C-2’ and -6’, 25), 128.74 (C-4, 25), 132.15 (C-5, 25), 135.81 (C-1’, 25), 208.08 (C-2, 25).
(E)-3-Methyl-5-((1S,2S,5S)-6,6-dimethylbicyclo[3.1.1]hept-2-yl)pent-3-en-2-one (27): the crude reaction product was purified by flash chromatography (eluent: n-hexane/Et2O 4:1) to yield 27 as a colourless oil; [α]25D = –16.3 (c 1.35, MeOH); tR 38.06; IR (ν, cm-1): 2982 and 2907 (cyclohexane), 1669 and 1641 (CH=C(CH3)COCH3), 1468 and 1431 (CH=CCH3), 1365 and 1387 (C(CH3)2); MS (m/z, %): 220 (M+, 1), 205 (M+−Me, 9), 177 (M+−Me−CO, 13), 137 (M+−CH=C(CH3)COCH3, 7), 123 (M+ −CH2CH=C(CH3)COCH3, 27), 83 (CH=C(CH3)COCH3+, 20), 43 (CH3CO+, 86); 1H-NMR (400 MHz): δ 0.89 (d, J7’s-7’a=9.0, 1H, H-7’s), 1.07 (s, 3H, CH3-9’), 1.19 (s, 3H, CH3-8), 1.50 (ddt, J3’a-3’s=14.5, J3’a-4’a=11.5, J3’a-4’s-2’=5.9, 1H, H-3’a), 1.76 (q, JMe-3-4=1.1, 3H, CH3-3), 1.88–1.82 (m, 1H, H-1’), 1.95–1.89(m, 1H, H-2’), 1.91–1.85 (m, 1H, H-4’a), 2.00–1.91 (m, 1H, H-4’s), 2.04–1.93 (m, 1H, H-3’s), 2.19 (ddt, J5’-1’=2.0, J5’-7’a=2.6, J5’-4’s-4’a=7.4, 1H, H-5’), 2.29 (s, 3H, CH3-1), 2.39–2.32 (m, 1H, H-7’a), 2.35–2.30 (m, 2H, H-5), 6.62 (tq, J4-5=7.3 and J4-Me–3=1.3, 1H, H-4); 13C-NMR (100 MHz): δ 11.36 (Me-C-3), 22.28 (C-3’), 23.18 (Me-9’), 25.43 (C-1), 26.36 (C-4’), 28.12 (Me-8’), 33.81 (C-7’), 36.78 (C-5), 38.71 (C-6’), 41.13 (C-5’), 41.33 (C-2’), 45.79 (C-1’), 137.83 (C-3), 143.41 (C-4), 199.92 (C-2).
3.4. Deconjugative α-methylation of 19, 20, 22,24 and 27 to give 28−32
A solution of the appropriate α,β-unsaturated ketone [19, 20, 22,24 (+25) or 27] (60.5 mmol) in dry DMF (4 mL) was added dropwise to a stirred solution of KtBuO (6.98 g, 61.0 mmol) in dry DMF (30 mL) at room temperature for 30 min. After the addition was completed, the reaction was stirred for 10 min and then cooled to 0 °C. Pre-cooled MeI ( 22.81 g, 160.7 mmol) was added quickly and the reaction mixture stirred at that temperature for 10 min and then allowed to warm to room temperature. Brine (10 mL) and 1N AcOH solution (10 mL) were added and the crude was extracted with hexane/Et2O 1:1 (75 mL). The resulting organic solution was washed with 1N AcOH solution (2×30 mL) and brine (3×30 mL), dried over anhyd Na2SO4 and evaporated under reduced pressure to afford crude β,γ-unsaturated ketones 28–32, which were all purified by flash chromatography (eluent: hexane/Et2O). The deconjugative α-methylation reaction (a) of 19 afforded (E)-3,3,6-trimethylhept-4-en-2-one (28) in a 77% yield, (b) of 20 afforded (E)-3,3-dimethyldec-4-en-2-one (29) in a 42% yield, (c) of 22 afforded (E)-3,3,6,10-tetramethylundeca-4,9-dien-2-one (30) in a 55% yield, (d) of a 4:1 mixture of 24 and 25 afforded (E)-3,3-dimethyl-5-phenylpent-4-en-2-one (31) in a 84% yield, and (e) of 27 afforded (E)-3,3-dimethyl-5-((1S,2S,5S)-6,6-dimethylbicyclo[3.1.1]hept-2-yl)pent-4-en-2-one (32) in a 85% yield.
(E)-3,3,6-Trimethylhept-4-en-2-one (28): yellow-pale oil; IR (ν, cm-1): 1712 (C=O), 1672 (H-C=C-H) and 1467 (-CH-C=C-C-); MS (m/z, %): 154 (M+, 1), 111(M+−C(CH3)2, 51), 69 ((CH3)2CH-CH=CH-+, 100), 55 ((CH3)2CH-C-+, 46), 43 (CH3-C=O+, 51); 1H-NMR (300 MHz): δ 0.98 (d, J=6.9, 6H, Me-7 and Me-C6), 1.20 (s, 6H, 2 Me-C3), 2.09 (s, 3H, Me-1), 2.29 (o, J=6.3, 1H, H-6), 5.42 (d, J4-5=15.7, 1H, H-4), 5.50 (dd, J5-4=15.8, J5-6=5.6, 1H, H-5); 13C-NMR (75 MHz): δ 22.35 (2 Me-C3), 24.01 (C-7 and Me-C6), 25.21 (C-1), 31.16 (C-6), 49.85 (C-3), 131.33 (C-5), 137.48 (C-4), 211.92 (C-2).
(E)-3,3-Dimethyldec-4-en-2-one (29): yellow-pale oil; IR (ν, cm-1): 1713 (C=O) and 1467 (-CH-CH=CH-C-); MS (m/z, %): 182 (M+, 1), 167 (M+−Me, 1), 139 (M+−Me-C=O, 17), 97 (CH3-(CH2)4-CH-+, 8), 85 (Me-CO-C(Me)2-+, 1), 83 (33), 69 (100), 57 (CH3-(CH2)-+, 4), 55 (28), 43 (Me-C=O+); 1H-NMR (300 MHz): δ 0.89 (t, J=6.9, 3H, Me-10), 1.20 (s, 6H, 2 Me-C3), 1.22–1.41 (m, 6H, H-7, H-8, H-9), 2.03 (q, J=6.5, 2H, H-6), 2.10 (s, 3H, Me-1), 5.46 (d, J=15.6, 1H, H-4), 5.55 (dd, J5-4=6.0, J5-6=15.6, 1H, H-5); 13C-NMR (75 MHz):δ 13.88 (C-10), 22.42 (C-9), 23.99 (2 Me-C-3), 25.27 (C-1), 28.94 (C-7), 31.28 (C-8), 32.62 (C-6), 50.06 (C-3), 130.53 (C-5), 134.16 (C-4), 211.92 (C-2).
(E)-3,3,6,10-Tetramethylundeca-4,9-dien-2-one (30): yellow-pale oil; IR (ν, cm-1): 1712 (C=O), 1677 and 1628 (C=C), 1455 (-CH-C=C-C-); MS (m/z, %): 222 (M+, 1), 207 (M+−Me, 1), 179 (M+−Me-C=O, 5), 137 (M+−(Me)2C-CO-Me, 2), 124 ((Me)2-C=C-(CH2)2-CH(Me)-CH+, 3), 123 (13), 109 ((Me)2-C=CH-(CH2)2-CH(Me)-CH+−Me, 23), 95 (12), 83 ((Me)2-C=CH-(CH2)2+, 32), 69 ((Me)2-C=CH-CH2+,100), 55 ((Me)2-C=CH+, 21), 43 (Me-C=O+, 54); 1H-NMR (300 MHz): δ 0.90 (d, J=6.6, 3H, Me-C-6), 1.13 (s, 6H, 2 Me-C-3), 1.17–1.29 (m, 2H, H-7), 1.51 (br s, 3H, Me-11), 1.61 (br s, 3H, Me-C-10), 1.83 (q, 2H, H-8), 2.03 (s, 3H, Me-1), 2.40 (q, J=7.4, 1H, H-6), 5.01 (br t, J=7.1, 1H, H-9), 5.30 (dd, J5-4=15.7, J5-6=6.8, 1H, H-5), 5.38 (d, J=15.7, 1H, H-4);13C-NMR (75 MHz):δ 17.62 (C-11), 20.63 (Me-C-6), 24.05 and 24.12 (2 Me-C-3), 25.30 (C-1), 25.57 (Me-C-10), 25.80 (C-8), 36.56 (C-6), 37.06 (C-7), 50.02 (C-3), 124.47 (C-9), 131.31 (C-10), 132.77 (C-5), 136.27 (C-4), 211.86 (C-2).
(E)-3,3-Dimethyl-5-phenylpent-4-en-2-one (31): brown oil; IR (ν, cm-1): 3082, 3059 and 3026 (C=C, Ar), 1690 (CH3COC(CH3)2), 1679 and 971 (HC=CH), 1363 and 1363 (C(CH3)2), 749 and 694 (Ph); MS (m/z, %): 188 (M+, 1), 173 (M+-CH3, 1), 145 (M+−CH3CO, 30), 131 (M+−Me−CH3CO, 5), 91 (C7H7+, 99), 77 (C6H5+, 4), 43 (CH3CO+, 12), 28 (CO+, 100); 1H-NMR (400 MHz): δ 1.32 (s, 6H, 2CH3-C-3), 2.13 (s, 3H, Me-1), 6.26 (d, J4-5=16.2, 1H, H-4), 6.45 (d, J5-4=16.3, 1H, H-5), 7.21 (tt, J4’-3’-5’=7.2, J4’-2’-6’=1.5, 1H, H-4’), 7.29 (t, J3’-2’-4’=7.3, 2H, H-3’ and 5’), 7.35 (dd, J2’-3’/6’-5’=7.1, J2’-4’/6’-4’=1.3, 2H, H-2’ and 6’); 13C-NMR (100 MHz): δ 23.91 (2CH3-C-3), 25.46 (C-1), 50.36 (C-3), 126.18 (C-2’ and C-6’), 127.47 (C-4), 128.48 (C-3’ and C-5’), 129.29 (C-4’), 133.99 (C-5), 136.85 (C-1’), 210.60 (C-2).
(E)-3,3-Dimethyl-5-((1S,2S,5S)-6,6-dimethylbicyclo[3.1.1]hept-2-yl)pent-4-en-2-one (32): yellow-pale oil; [α]25D = –22.3 (c 1.15, MeOH); Rt 35.85; IR (ν, cm-1): 2939 and 2909 (cyclohexane), 1710 (CH3COC(CH3)2), 1672 and 972 (HC=CH), 1383 and 1364 (C(CH3)2). MS (m/z, %): 234 (M+, 1), 219 (M+–Me, 1), 191 (M+−CH3CO, 23), 149 (M+−CH3COC(CH3)2, 15), 93 (C7H9+, 3), 69 (CH3COCHCH3+, 100), 43 (CH3CO+, 77); 1H-NMR (400 MHz): δ 2.08 (s, 3H, Me-1), 5.37 (dd, J4-5=15.8, J4-2´=1.6, 1H, H-4), 5.67 (dd, J5-4=15.8, J5-2’=6.8, 1H, H-5), 1.91–1.99 (m, 1H, CH-1’), 2.73 (ddddd, J2’-3’s=10.5, J2’-5 =6.8, J2’-1’=6.0, J2’-3’a =2.5, J2’-4=1.7, 1H, H-2’), 1.60 (dddd, J3’a-3’s=15.3, J3’a-4’a=10.5, J3’a-2’=6.0, J3’a-4’s=4.5, 1H, H-3’a), 1.93–2.02 (m, 1H, H-3’s), 1.81–1.92 (m, 1H, H-4’a), 1.92–2.00 (m, 1H, H-4’s), 1.86–1.96 (m, 1H, H-5’), 0.99 (d, J7’s-7’a=9.7, 1H, H-7’s), 2.32 (dddd, J7’s-7’a=9.7, J7’s-1’=6.6, J7’s-5’=5.7, J7’s-4’a=1.6, 1H, H-7’a), 1.19 (s, 3H, Me-8), 0.95 (s, 3H, Me-9), 1.19 (s, 6H, 2CH3-C-3); 13C-NMR (100 MHz): δ 21.42 (C-3’), 23.53 (Me-9’), 24.03 (2CH3-C-3), 25.35 (C-1), 26.00 (C-4’), 27.83 (Me-8’), 32.31 (C-7’), 38.53 (C-6’),41.08 (C-5’), 43.34 (C-2’), 46.99 (C-1’), 49.96 (C-3), 132.09 (C-4), 137.15 (C-5), 211.80 (C-2).
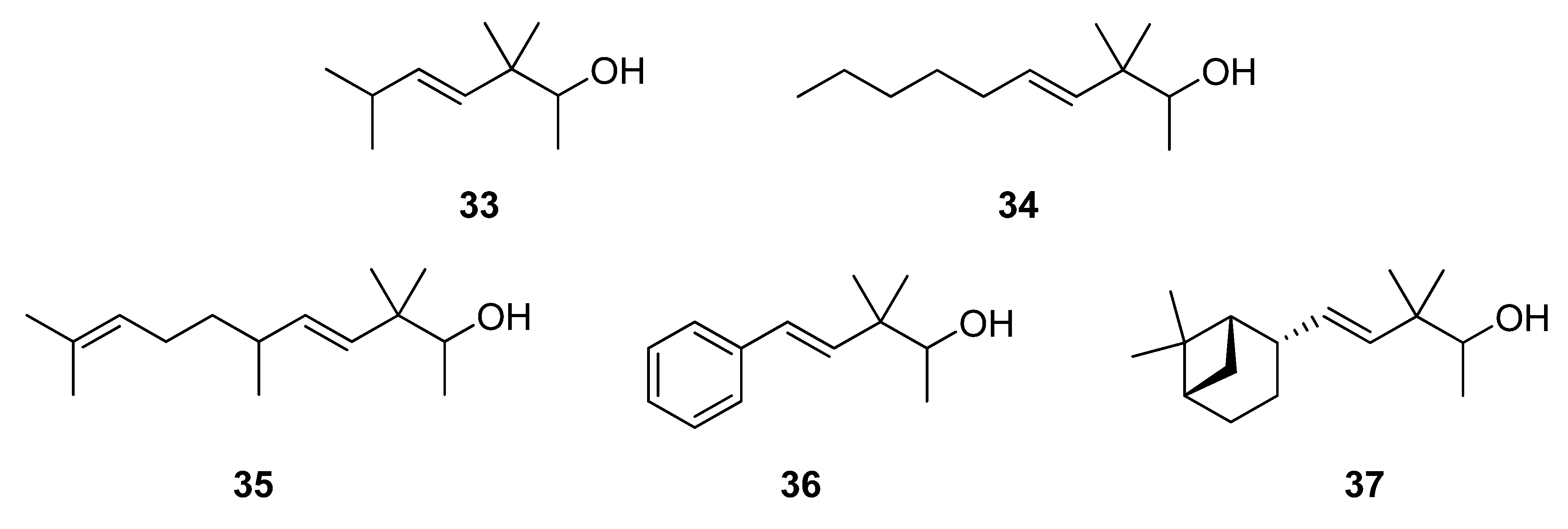
3.5. Reduction of 28–32 with NaBH4 to give 33–37
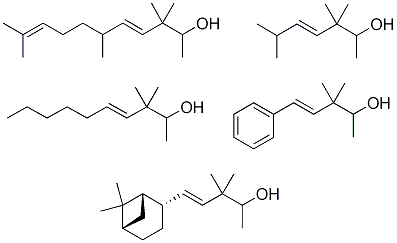
Solid NaBH4 (2.77 g, 71.8 mmol) was added portionwise to a stirred solution of starting β,γ-unsaturated ketone 28–32 (54.7 mmol) in MeOH (50 mL) at 0 °C. After 15 min the reaction was allowed to warm to rt and left to react for 45 min. Then, the solvent was partially evaporated under reduced pressure and the resulting suspension was diluted with hexane/Et2O 1:2 (75 mL), cooled again to 0 °C and neutralized with 1N AcOH solution. The organic layer was washed again with 1N AcOH solution (50 mL) and brine (3 × 50 mL), then dried over anhyd Na2SO4 and the solvent evaporated in vacuo to yield crude alcohols 33–37, which were purified by flash chromatography (eluent: hexane/Et2O). The reduction with NaBH4 (a) of 28 afforded (E)-3,3,6-trimethylhept-4-en-2-ol (33) in a 62% yield, (b) of 29 afforded (E)-3,3-dimethyldec-4-en-2-ol (34) in a 76% yield, (c) of 30 afforded (E)-3,3,6,10-tetramethylundeca-4,9-dien-2-ol (35) in a 90% yield, (d) of 31 afforded (E)-3,3-dimethyl-5-phenylpent-4-en-2-ol (36) in a 80% yield, and (e) of 32 afforded (E)-3,3-dimethyl-5-((1S,2S,5S)-6,6-dimethylbicyclo[3.1.1]hept-2-yl)pent-4-en-2-ol (37) in a 77% yield.
(E)-3,3,6-Trimethylhept-4-en-2-ol (33): yellow-pale oil; tR 11.62; IR (ν, cm-1): 3684 (OH), 1466 (CH-C=C-C); MS (m/z, %): 142 (M+−14, 1), 127 (1), 123 (M+−Me–H2O, 1), 113 (M+−(CH3)2CH, 2), 112 (25), 69 ((CH3)2CH-CH=CH+, 100), 56 ((CH3)2CH-CH=C+, 22), 55 (34), 43 ((CH3)2CH+, 38); 1H- NMR (300 MHz): δ 0.97 (s, 6H, 2CH3-C-3), 0.99 (d, J=6.6, 6H, Me-7 and Me-C-6), 1.09 (d, J=6.3, 3H, Me-1), 2.28 (o, J=6.7, 1H, H-6), 3.46 (q, J=6.4, 1H, H-2), 5.34 (d, J4-5=15.7, 1H, H-4), 5.43 (dd, J5-4=15.7, J5-6=6.2, 1H, H-5); 13C-NMR (75 MHz): δ 17.24 (C-1), 21.78*a (Me-C-3), 22.79*b (Me-C-6), 22.81*b (Me-7), 24.03*a (Me’-C-3), 31.35 (C-6), 40.44 (C-3), 74.13 (C-2), 133.55 (C-5), 136.98 (C-4) (*these signals may be interchanged); HRMS m/z, calcd. for C10H20O, 156.1514 (M+), found 156.1467.
(E)-3,3-Dimethyldec-4-en-2-ol (34): yellow-pale oil; tR 22.25; IR (ν, cm-1): 3841−3400 (OH), 1467 (CH2-CH=CH); MS (m/z, %): 169 (M+−Me, 1), 167 (M+−OH, 1), 140 (13), 139 (Me-CH-OH+, 3), 125 (4), 112 (5), 97 (CH3-(CH2)4-CH=CH+, 11), 83 (31), 69 (100), 55 (34), 43 (27), 41 (50); 1H-NMR (300 MHz): δ 0.89 (t, J=6.5, 3H, Me-10), 0.97 (s, 6H, 2Me-C-3), 1.09 (d, J=6.3, 3H, Me-1), 1.25–1.44 (m, 6H, H-7, H-8, H-9), 2.02 (q, J=6.6, 2H, H-6), 3.46 (q, J=6.2, 1H, CH-2), 5.38 (d, J4-5=15.9, 1H, H-4), 5.47 (dd, J5-6=6.1, J5-4=15.7, 1H, H-5); 13C-NMR (75 MHz): δ 14.02 (C-10), 17.24 (C-1), 21.73*a (Me-C-3), 22.47 (C-9), 24.03*a (Me-C-3), 29.30 (C-7), 31.36*b (C-8), 32.83*b (C-6), 40.68 (C-3), 74.11 (C-2), 129.83 (C-5), 136.57 (C-4) (*these signals may be interchanged); HRMS m/z, calcd. for C12H20O, 184.1827 (M+), found 184.1183.
(E)-3,3,6,10-Tetramethylundeca-4,9-dien-2-ol (35): yellow-pale oil; tR 31.25; IR (ν, cm-1): 3430 (OH) and 1455 (CH2-CH=CH); MS (m/z, %): 180 ((M++1)−[Me-CH-OH], 1), 137 (M+−[(Me)2C-CHOH-Me], 8), 123 (19), 109 ([(Me)2-C=CH-(CH2)2-CH(Me)-CH]+−Me, 50), 95 (21), 83 ([(Me)2-C=CH-(CH2)2]+, 29), 69 ([(Me)2-C=CH-CH2]+,100), 55 ([(Me)2-C=CH]+, 30), 45 (54); 1H-NMR (300 MHz): δ 0.96–0.98 (m, 3H, Me-C-6), 0.98 (s, 6H, 2 Me-3), 1.09 (d, J=6.5, 3H, Me-1), 1.29 (q, J=7.4, 2H, CH2-7), 1.58 (br s, 3H, Me-C-10), 1.68 (br s, 3H, Me-11), 1.93 (q, J= 7.6, 2H, CH2-8), 2.11 (m, J=6.8, 1H, CH-6), 3.46 (q, J=6.2, 1H, CH-2), 5.09 (br t, J=7.2, 1H, CH-9), 5.29 (dd, J1=16.0, J2= 6.6, 1H, CH-5), 5.36 (d, J=15.6, 1H, CH-4); 13C-NMR (75 MHz): δ 17.28 (C-1), 17.62 (C-11), 21.04 (Me-C6), 21.97 (Me-C3), 23.98 (Me’-C3), 25.67 (Me-C10), 25.89 (C-8), 36.72 (C-6), 37.26 (C-7), 40.59 (C-3), 74.13 (C-2), 124.59 (C-9), 131.19 (C-10), 135.00 (C-5), 135.58 (C-4); HRMS m/z, calcd. for C15H28O, 224.2140 (M+), found 224.2157.
(E)-3,3-Dimethyl-5-phenylpent-4-en-2-ol (36): yellow oil; tR 32.30; IR (ν, cm-1): 3406, 1093, 1071 (OH); 1383 (C(CH3)2); 1646, 972 (CH=CH). 1945, 1875, 1802, 911, 748, 693 (Ph); 1H-NMR (400 MHz): 1.10 (s, 6H, 2Me-C-3), 1.13 (d, J=6.4, 3H, Me-1), 3.58 (q, J=6.3, 1H, H-2), 6.22 (d, J4-5=16.3, 1H, H-4), 6.39 (d, J5-4=16.3, 1H, H-5), 7.19 (tt, J4’-3’-5’=7.2 and J4’-2’-6’=0.9, 1H, H-4’), 7.28 (dt, J3’-2’-4’=7.4 and J3’-5’=1.5, 2H, H-3’ and H-5’), 7.36 (br d, J2’-3’=7.4H, 2H, H-2’ and H-6’); 13C-NMR (100 MHz): 17.75 (C-1), 22.27 and 23.56 (2 M-C-3), 41.23 (C-3), 74.44 (C-2), 126.08 (C-2’ and C-6’), 127.08 (C-4), 128.41 (C-4’), 128.45 (C-3’ and C-5’), 136.94 (C-5), 137.48 (C-1’); HRMS m/z, calcd. for C13H18O, 190.1358 (M+), found 190.1345.
(E)-3,3-Dimethyl-5-((1S,2S,5S)-6,6-dimethylbicyclo[3.1.1]hept-2-yl)pent-4-en-2-ol (37): yellow-pale oil; [α]25D = −20.9 (c 0.85, MeOH); tR 36.70; IR (ν, cm-1): 3475–3300 (OH), 1090, 1069 (CH-OH), 1384, 1366 (C(CH3)2); 1654, 975 (CH=CH); 1H-NMR (400 MHz): δ 1.08 (d, J=6.4, 3H, H-1), 3.46 (q, J=6.4, 1H, H-2), 5.29 (dd, J4-5=15.8 and J4-2´=1.5, 1H, H-4), 5.62 (dd, J5-4=15.8, J5-2’=7.0, 1H, H-5), 1.89–1.99 (m, 1H, H-1’), 2.73 (dtt, J2’-1’-4=1.1, J2’-3’a-5=6.0, J2’-3’s=11.8 1H, H-2’), 1.61 (ddt, J3’a-4’a=5.6, J3’a-3’s=10.2, J3’a-2’-4’s=10.0, 1H, H-3’a), 1.93–2.03 (m, 1H, H-3’s), 1.81–1.92 (m, 1H, H-4’a), 1.92–1.99 (m, 1H, H-4’s), 1.86–1.97 (m, 1H, H-5’), 0.99 (d, J7’s-7’a=9.9, 1H, H-7’s), 2.32 (dt, J7’s-7’a=8.8, J7’a-1’-5’=6.1, 1H, H-7’a), 1.19 (s, 3H, Mes-C-6), 0.97 (s, 3H, Mes-C-6), 0.97 (s, 3H, Me-C-3), 0.96 (s, 3H, Me’-C-3); 13C-NMR (100 MHz): δ 17.25 (C-1), 21.69 and 21.74 (Me’-C-3), 21.82 and 21.77 (C-3’), 23.59 (Me-9), 24.03 (Me-C-3), 26.09 (C-4’), 27.90 (Me-8), 32.43 (C-7’), 38.58 (C-6’), 40.54 (C-3), 41.13 (C-5’), 43.60 (C-2’), 47.48 and 47.35 (C-1’), 74.21 and 74.18 (C-2), 134.46 (C-4), 136.74 (C-5); HRMS m/z, calcd. for C16H28O, 236.2140 (M+), found 236.1993.
3.6. Sensory evaluation
Direct smelling analysis. Blotting paper strips were impregnated with compounds 33–37, previously diluted with Et2O (25 mg/200 μL), and smelt by perfumers at that moment (after solvent evaporation), 3 h and 24 h later. The olfactory description in each session therefore corresponded to the top, heart and base notes, respectively.
GC sniffing analysis. Odour assessment of compounds 33–37 was achieved by a group of perfumers using a Hewlett-Packard Model 5890 Series II gas chromatograph equipped with a thermal conductivity detector (TCD) and handmade sniffing port. Separation was done with a 10% Carbowax 20M over Chromosorb W/AW 80−100 mesh packed column (1.8 m×6 mm OD×2.2 mm ID); injector temperature: 250 °C; detector temperature: 250 °C, oven temperature program: 60 °C (0 min) to 240 °C (20 min) at 4 °C/min. Sample size for each injection was approximately 1 μL in a 1:10 split mode.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









