Structure, Morphology and Optical Properties of Chiral N-(4-X-phenyl)-N-[1(S)-1-phenylethyl]thiourea, X= Cl, Br, and NO2

Abstract
:
1. Introduction

2. Results
2.1. Synthesis and basic properties
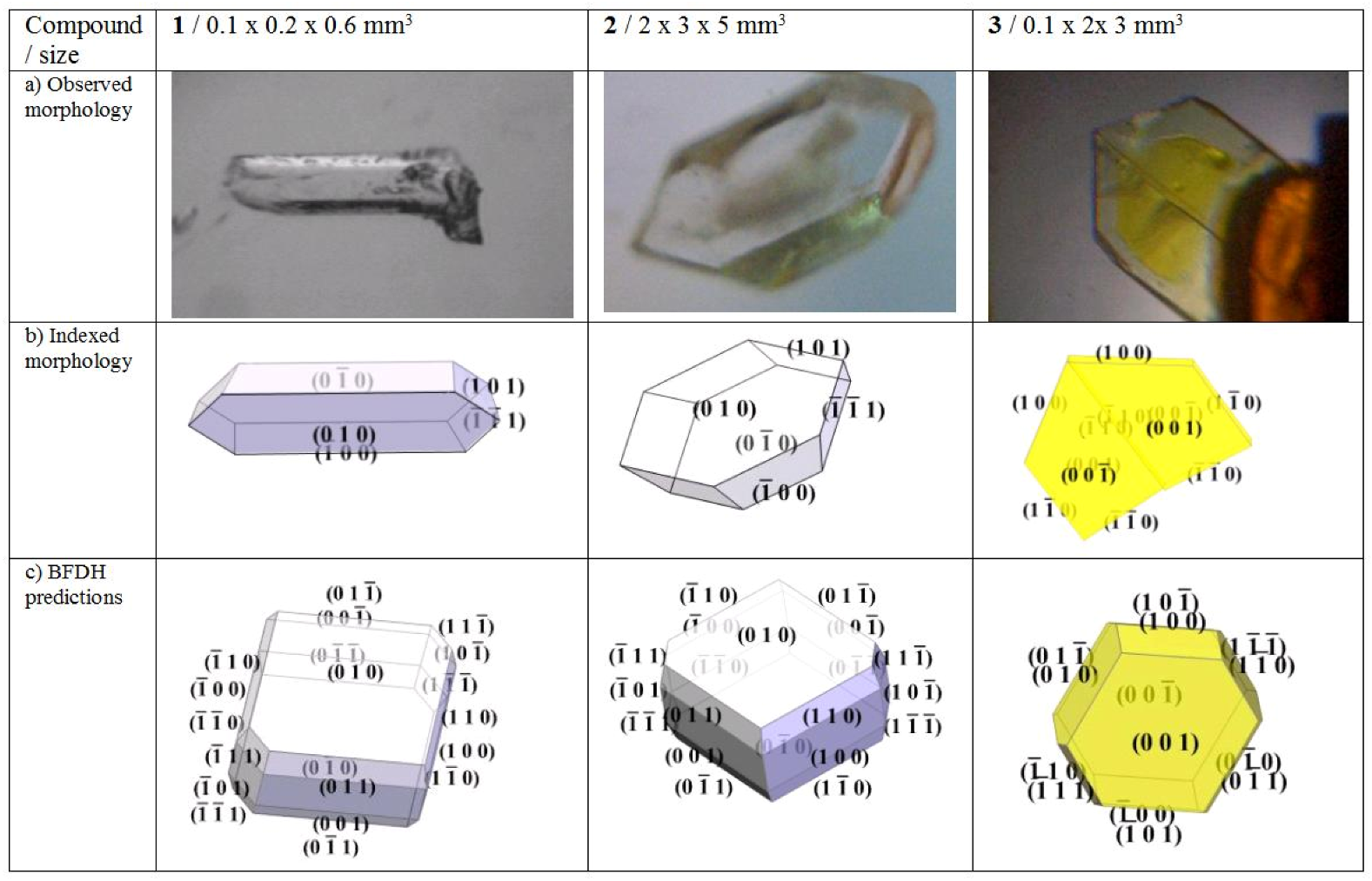
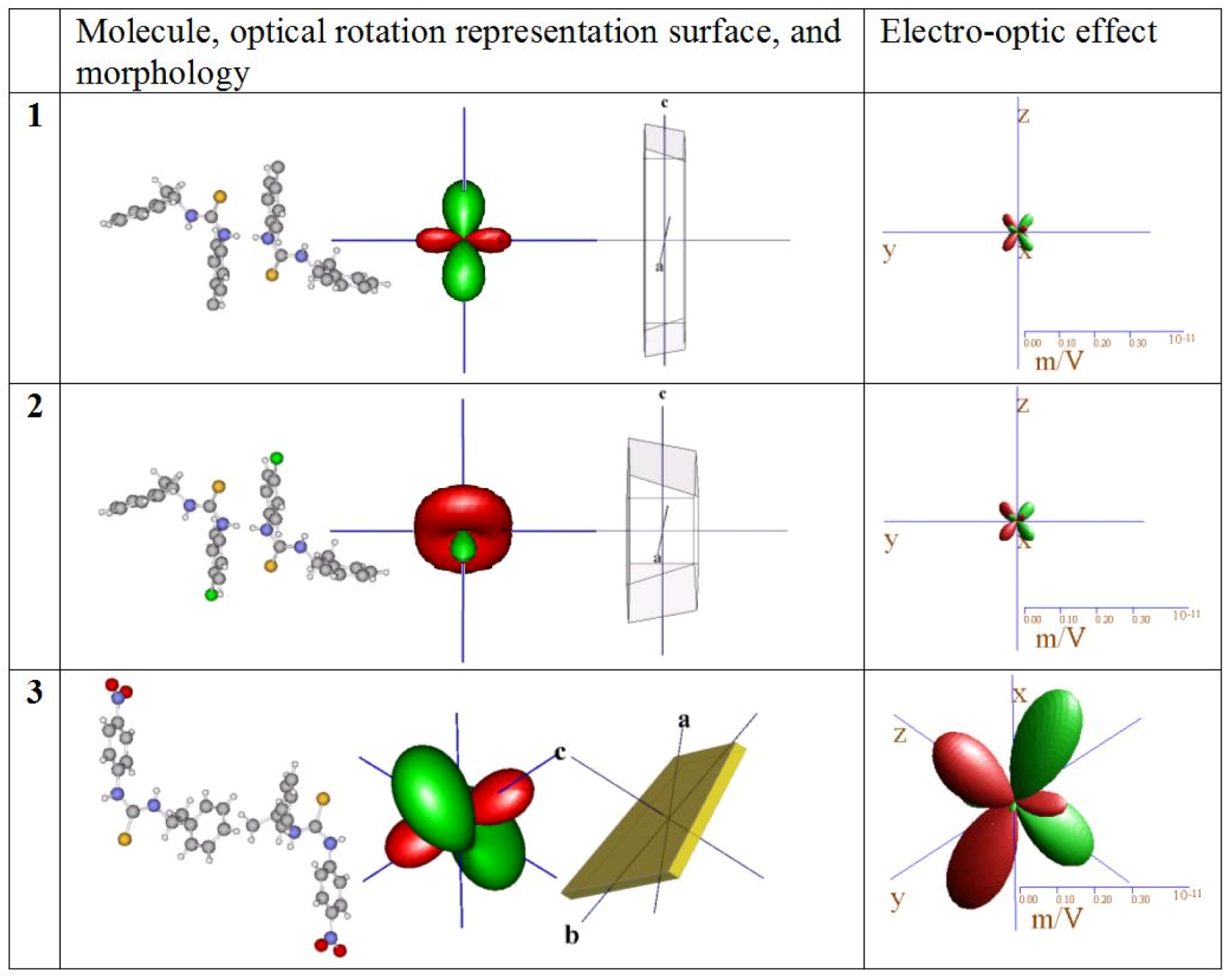
 ). Crystals of 1 cleave on (010).
). Crystals of 1 cleave on (010).2.2. Crystal morphology and twinning
 ) faces to be dominant in 3 and (010) as well as (
) faces to be dominant in 3 and (010) as well as (  ) largest in 1 and 2. If strong polarity is added to the calculation for 3 by which the central distances of (h k l) are extended and those of (
) largest in 1 and 2. If strong polarity is added to the calculation for 3 by which the central distances of (h k l) are extended and those of (  ) are shortened as a simple approach to simulate the symmetry independence of Friedel pairs, the model can be made to exhibit exclusively the observed faces.
) are shortened as a simple approach to simulate the symmetry independence of Friedel pairs, the model can be made to exhibit exclusively the observed faces. 
2.3. X-ray diffraction studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Formula | C15H15N2ClS | C15H15N2BrS | C15H15N3O2S |
| Formula weight | 290.80 | 335.26 | 301.36 |
| Crystal system | monoclinic | monoclinic | triclinic |
| Space group | P21 | P21 | P1 |
| T/K | 130 | 130 | 293* |
| Color / description | colorless needle | colorless plate | yellow plate |
| Size (mmm3) | 0.59x0.22x0.12 | 0.50x0.20x0.14 | 0.50x0.48x0.05 |
| a/Å | 7.4610(3) | 7.6500(3) | 7.737(3) |
| b/ Å | 24.627(1) | 24.645(1) | 8.267(3) |
| c/ Å | 7.8620(3) | 7.7780(3) | 13.616(7) |
| α/º | 90 | 90 | 91.43(2) |
| β/º | 100.478(2) | 101.113(2) | 102.06(2) |
| γ/º | 90 | 90 | 112.99(2) |
| V/Å3 | 1420.5(1) | 1438.9(1) | 778.5(3) |
| Z | 4 | 4 | 2 |
| Dc/gcm_3 | 1.360 | 1.548 | 1.286 |
| µ(Mo Kα)/mm-1 | 0.403 | 2.989 | 0.215 |
| Measured / unique data | 4842 / 4842 | 5864 / 5864 | 4641 / 4641 |
| Flack enantiopole | -0.2(1) | -0.02(2) | -0.1(2) |
| Rlin, No. refined parameters | 0.061, 345 | 0.096, 345 | 0.088, 381 |
| Observed data, I>2σ(I) | 3835 | 4686 | 2984 |
| R, obs.; Rw all data | 0.0775; 0.2210 | 0.0672; 0.1730 | 0.0764; 0.2174 |
| GOOF; compl. (ϑ=25º) | 1.111; 99.6 | 1.065; 99.4 | 1.026; 96.2 |
2.4. Refractive indices
3. Discussion
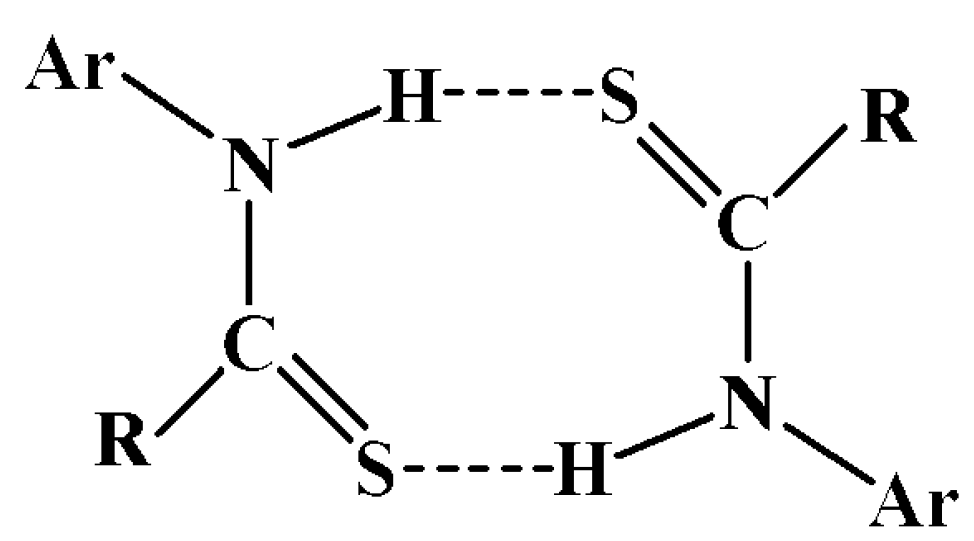
3.1. Thioamide R22(8) {…H-N-C=S}2 dimer


| 1: H…A, D…A, <(DHA) | 2: H…A, D…A, <(DHA) | 3: H…A, D…A, <(DHA) | |
|---|---|---|---|
| N1-H1N…S2 | 2.51 Å, 3.345(6) Å, 158.4o | 2.50 Å, 3.338(9) Å, 159.1o | 2.59 Å, 3.395(7) Å, 155.2o |
| N3-H3N…S1 | 2.48 Å, 3.327(6) Å, 162.0o | 2.50 Å, 3.342(9) Å, 161.6o | 2.62 Å, 3.400(6) Å, 151.8o |
| N5-H5N…O2 | N/A | N/A | 2.52 Å, 3.36(1) Å, 166.1o |
3.2. Optical features
3.2.1. Model calculations





3.2.2. Estimation of optical features
| 1 | 2 | 3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nα, eo | 1.513, (0.99,0,0.12) | 1.552, (0.98,0,0.21) | 1.504, (0.18,-0.88,-0.45) | |||||||||
| nβ, e o | 1.606, (0, 1, 0) | 1.634, (0, 1, 0) | 1.573, (-0.97,-0.07,-0.24) | |||||||||
| nγ, e o | 1.661, (0.12,0,-0.99) | 1.710, (0.21,0,-0.98) | 1.767, (0.18,0.45,-0.86) | |||||||||
| Opt. rotation | 2.9 0 -5.8 | -4.3 0 -9.7 | 13.7 -10.7 4.8 | |||||||||
| 0 -8.4 0 | 0 -8.0 0 | -10.7 -20.0 -21.3 | ||||||||||
| ρ(ij) (o/mm) | -5.8 0 9.1 | -9.7 0 -0.1 | 4.8 -21.3 32.4 | |||||||||
| normalized | r112 | -0.01 | r231 | -0.38 | r112 | -0.12 | r231 | -0.45 | r111 | -0.43 | r221 | 2.40 |
| r121 | 0.07 | r233 | -0.48 | r121 | -0.18 | r233 | -0.35 | r112 | -0.15 | r222 | -1.82 | |
| El. Opt. effect | r123 | -0.44 | r312 | -0.44 | r123 | -0.54 | r312 | -0.44 | r113 | 0.11 | r223 | 1.82 |
| r’(ij)k/ε (pm/V) | r222 | 0.19 | r332 | -0.19 | r222 | 0.16 | r332 | -0.16 | r121 | -0.04 | r231 | -1.00 |
| r122 | 0.22 | r232 | 2.08 | |||||||||
| r123 | 0.10 | r233 | 0.94 | |||||||||
| r131 | -0.24 | r331 | 1.27 | |||||||||
| r132 | -0.08 | r332 | 0.03 | |||||||||
| r133 | 0.04 | r333 | -3.07 | |||||||||

Supplementary Data
Acknowledgements
- Sample Availability: Samples of the compounds 1-3 are available from MDPI.
Appendix 1. Dipole-Dipole interaction model






References
- Kaminsky, W. Experimental and phenomenological aspects of circular birefringence and related properties in transparent crystals. Rep. Prog. Phys. 2000, 63, 1575–1640, The following review is only one of many examples. [Google Scholar] [CrossRef]
- Rohl, A.; Moret, M.; Kaminsky, W.; Claborn, K.; Kahr, B. Hirshfeld Surfaces Identify Errors in Computations of Intermolecular Interactions in Crystals: Pentamorphic 1,8-Dihydroxyanthraquinone. Cryst. Growth Design 2008, 8, 4517–4525, Dihydroxyanthraquinone, for example, is pentamorphic. [Google Scholar]
- Ho, S.Y.; Bettens, R.P.A.; Dakternieks, D.; Duthie, A.; Tiekink, E.R.T. Prevalence of the thioamide {…H–N–CLS}2 synthon—solid-state (X-ray crystallography), solution (NMR) and gas-phase (theoretical) structures of O-methyl-N-aryl-thiocarbamides. Cryst. Eng. Comm. 2005, 7, 682–689. [Google Scholar] [CrossRef]
- Humeres, E.; Zucco, C.; Nunes, M.; Debacher, N.; Nunes, R. Hydrolysis of N-aryl thioncarbamate esters. Modified Marcus equation for reactions with asymmetric intrinsic barriers. J. Phys. Org. Chem. 2002, 15, 570–575. [Google Scholar] [CrossRef]
- Fowke, J.; Chung, F.; Jin, F.; Qi, D.; Cai, Q.; Conaway, C.; Cheng, J.; Shu, X.; Gao, Y.; Zheng, W. Urinary isothiocyanate levels, brassica, and human breast cancer. Cancer Res. 2003, 63, 3980–3986. [Google Scholar]
- These thiocarbamates are typical of the structures often used (or found) in high-throughput screens and often have multiple activities. Something this small is often a fragment or part of the overall pharmacophore. It may have low (micromolar to millimolar) affinity for part of the receptor binding epitope and will eventually be complemented by a second fragment of approximately the same size that occupies the other part of the binding epitope. As far as for specific activity, these compounds are not part of a predictable class of biologically active compounds. Activity studies would require unbiased high throughput screens and fragment approaches.
- Taylor, R.; Tiekink, E.R.T. Crystal and molecular-structure of O-ethyl-N-phenylthiocarbamate. Z. Kristallogr. 1994, 209, 64–67. [Google Scholar] [CrossRef]
- Benson, R.E.; Broker, G.A.; Daniels, L.M.; Tiekink, E.R.T.; Wardell, J.L. (E)-O-Ethyl N-(4-nitrophenyl)thiocarbamate. Acta Cryst. 2006, E62, 4106–4108. [Google Scholar]
- Ellis, C.A.; Miller, M.A.; Spencer, J.; Zukerman-Schpector, J.; Tiekink, E.R.T. Co-crystallization experiments of thiocarbamides with bipyridine-type molecules. Cryst. Eng. Comm. 2009, 11, 1352–1361. [Google Scholar] [CrossRef]
- Jian, F.F.; Yu, H.Q.; Qiao, Y.B.; Liang, T.L. O-Isobutyl N-phenylthiocarbamate. Acta Cryst. 2006, E62, 3416–3417. [Google Scholar]
- Ho, S.Y.; Lai, C.S.; Tiekink, E. R. T. O-Methyl N-phenylthiocarbamate. Acta Cryst. 2003, E59, 1155–1156. [Google Scholar]
- Kuan, F.S.; Mohr, F.; Tadbuppa, P.P.; Tiekink, E.R.T. Principles of crystal packing in O-isopropyl-N-aryl-thiocarbamides: iPrOC(LS)N(H)C6H4-4-Y: Y 5 H, Cl, and Me. Cryst. Eng. Comm. 2007, 9, 574–581. [Google Scholar] [CrossRef]
- Kaminsky, W.; Goldberg, K.I.; West, D.X. Synthesis and structures of two N,N’-bis(2-pyridinyl)thioureas and N-(2-pyridinyl)-N’-(benzoyl)thiourea. J. Mol. Struct. 2002, 605, 9–15. [Google Scholar] [CrossRef]
- Hermetet, A.K.; Ackerman, L.J.; Eilts, K.K.; Johnson, T.K.; Swearingen, J.K.; Giesen, J.M.; Goldberg, K.I.; Kaminsky, W.; West, D.X. Structural, spectral and thermal studies of N-2-(4,6-lutidyl)-N’-chlorophenylthioureas. J. Mol. Struct. 2002, 605, 214–247. [Google Scholar]
- Valdes-Martinez, J.; Henandez-Ortega, S.; Espinosa-Perez, G.; Presto, C.A.; Hermetet, A.K.; Haslow, K.D.; Ackerman, L.J.; Szczepura, L.F.; Goldberg, K.I.; Kaminsky, W.; West, D.X. Structural, spectral and thermal studies of substituted N-(2-pyridyl)-N’-phenylthioureas. J. Mol. Struct. 2002, 608, 77–87. [Google Scholar] [CrossRef]
- Szczepura, L.F.; Kelman, D.R.; Hermetet, A.K.; Ackerman, L.J.; Goldberg, K.I.; Claborn, K.A.; Kaminsky, W.; West, D.X. Structural, spectral and thermal studies of N-2-(picolyl)-N’-4-chlorophenylthioureas. J. Mol. Struct. 2002, 608, 245–251. [Google Scholar] [CrossRef]
- Giesen, J.M.; Claborn, K.A.; Goldberg, K.I.; Kaminsky, W.; West, D.X. Structural, thermal and spectral studies of N-2-pyridyl-, N-2-picolyl- and N-2-(4,6-lutidyl)-N’-(3-methoxy-phenyl)thioureas. J. Mol. Struct. 2003, 613, 223–233. [Google Scholar]
- Kaminsky, W.; Kelman, D.R.; Giesen, J.M.; Goldberg, K.I.; Claborn, K.A.; Szczepura, L.F.; West, D.X. Structural and spectral studies of N-2-(pyridyl)-, N-2-(3-, 4-, 5-, and 6-picolyl)- and N-2-(4,6-lutidyl)-N’-2-thiomethoxyphenylthioureas. J. Mol. Struct. 2002, 616, 79–89. [Google Scholar] [CrossRef]
- Kelman, D.R.; Claborn, K.A.; Kaminsky, W.; Goldberg, K. I.; West, D. X. Structural, spectral and thermal studies of N-2-(pyridyl)- and N-2-(picolyl)-N’-(3-chlorophenyl)thioureas. J. Mol. Struct. 2002, 642, 119–127. [Google Scholar] [CrossRef]
- Kelman, D.R.; Claborn, K.A.; Kaminsky, W.; Goldberg, K.I.; Li, D.T.; West, D.X. Structural studies of N-2-(6-aminopyridine)-N’-arylthioureas. J. Mol. Struct. 2003, 654, 145–152. [Google Scholar] [CrossRef]
- Valdes-Martınez, J.; Hernandez-Ortega, S.; Rubio, S.M.; Li, D.T.; Swearingen, J.K.; Kaminsky, W.; Kelman, D.R.; West, D.X. Study of the sulfur atom as hydrogen bond acceptorin N(2)-pyridylmethyl-N’-arylthioureas. J. Chem. Crystallogr. 2004, 34, 533–540. [Google Scholar] [CrossRef]
- Valdes-Martınez, J.; Hernandez-Ortega, S.; Hermetet, A.K.; Ackerman, L.J.; Presto, C.A.; Swearingen, J.K.; Kelman, D.R.; Goldberg, K.I.; Kaminsky, W.; Douglas, X.; West, D.X. Structural studies of N-2-(3-picolyl)- and N-2-(4-picolyl)-N’-tolylthioureas. J. Chem. Crystallogr. 2002, 32, 431–438. [Google Scholar] [CrossRef]
- Hermetet, A.K.; Ackerman, L.J.; Swearingen, J.K.; Presto, C.A.; Kelman, D.R.; Giesen, J.M.; Goldberg, K.I.; Kaminsky, W.; West, D.X. Structural studies of N-2-(6-picolyl)-N’-tolylthioureas. J. Chem. Crystallogr. 2002, 32, 17–25. [Google Scholar] [CrossRef]
- Allen, F.H.; Motherwell, W.D.S.; Raithby, P.R.; Shields, G.P.; Taylor, R. Systematic analysis of the probabilities of formation of bimolecular hydrogen-bonded ring motifs in organic crystal structures. New J. Chem. 1999, 23, 25–34. [Google Scholar] [CrossRef]
- Avalos, M.; Babiano, R.; Cabanillas, A.; Cintas, P.; Higes, F.J.; Jimenez, J.L.; Palacios, J.C. Cycloaddition Chemistry of 1,3-Thiazolium-4-olate Systems. Reaction with Nitroalkenes and Interpretation of Results Using PM3 Calculations. J. Org. Chem. 1996, 61, 3738–3748. [Google Scholar]
- Kaminsky, W. From *.cif to virtual morphology: new aspects of predicting crystal shapes as part of the WinXMorph program. J. Appl. Cryst. 2007, 40, 382–385. [Google Scholar] [CrossRef]
- Kaminsky, W. WinXMorph: a computer program to draw crystal morphology, growth sectors and cross-sections with export files in VRML V2.0 utf8-virtual reality format. J. Appl. Crystallogr. 2005, 38, 566–567. [Google Scholar] [CrossRef]
- Bravais, A. du Cristal Considere Comme un Simple Assemblage de Points. In Etude Cristallographiques; Gauthier-Villars: Paris, France, 1866. [Google Scholar]
- Friedel, G. Studies on the law of Bravais. Bull. Soc. Franc. Mineral 1907, 22, 326–455. [Google Scholar]
- Donnay, J.D.H.; Harker, D. A new law of crystal morphology extending the law of Bravais. Am. Mineral. 1937, 22, 446–467. [Google Scholar]
- Otwinowsky, Z.; Minor, W. Methods in Enzymology; Carter, C.W., Jr., Sweet, R.M., Eds.; Academic Press: New York, NY, USA, 1997; Volume 276, pp. 307–326. [Google Scholar]
- Altomare, A.; Burla, C.; Camalli, M.; Cascarano, L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: a new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. J. Completion and refinement of crystal structures with SIR92. Appl. Cryst. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97: Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Mackay, S.; Edwards, C.; Henderson, A.; Gilmore, C.; Stewart, N.; Shankland, K.; Donald, A. MaXus: A Computer Program for the Solution and Refinement of Crystal Structures from Diffraction Data; University of Glasgow: Glasgow, UK, 1997. [Google Scholar]
- Waasmaier, D.; Kirfel, A. New analytical scattering-factor functions for free atoms and ions. Acta Crystallogr. A 1995, 51, 416–431. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Claborn, K.A.; Kahr, B.; Kaminsky, W. Calculation of optical properties of the tetraphenyl-X family of isomorphous crystals (X=C, Si, Ge, Sn, Pb). Cryst. Eng. Comm. 2002, 4, 252–256, Our approach is described in detail here. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Mitchell, A.S; Spackman, M.A. Hirshfeld Surfaces: A New Tool for Visualising and Exploring Molecular Crystals. Chem. Eur. J. 1998, 11, 2136–2141. [Google Scholar]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. B 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Spackman, M.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Comm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. B 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Beurskens-Kerssen, G.; Kroon, J.; Endemann, H.J.; van Laar, J.; Bijvoet, J.M. Crystallography and Crystal Perfection; Ramachandran, G., Ed.; Academic Press: London, UK, 1963; p. 225. [Google Scholar]
- Bijvoet, J.M.; Peerdeman, A.F.; Van Bomel, A.J. Determination of the absolute configuration of optically active compounds by means of X-rays. Nature 1951, 168, 271–272. [Google Scholar] [CrossRef]
- Born, M.; Goeppert-Mayer, M. Dynamische Gittertheorie der Kristalle. In Handbuch der Physik; Geiger, H., Scheel, K., Eds.; Springer-Verlag: Berlin, Germany, 1933; Vol. 24/2, p. 623. [Google Scholar]
- Bruhat, G.; Grivet, P. Rotatoire du quartz pour des rayons perpendiculaires a l'axe optique et sa dispersion dans l'ultra-violet. J. Phys. Radium 1935, 6, 12–26. [Google Scholar] [CrossRef]
- van Laar, J.; Endemann, H.J.; Bijvoet, J.M. Remarks on the relation between microscopic and macroscopic crystal optics. Acta Crystallogr. A 1968, 24, 52–56. [Google Scholar] [CrossRef]
- Reijnhart, R. Classical Calculations Concerning the Double Refraction, Optical Rotation and Absolute Configuration of Te, Se, Cinnabar (HgS) α- and β-Quartz, α-Cristobalite, NaNO2, NaClO3 and NaBrO3. PhD thesis, University of Delft, Delft, The Netherlands, 1970. [Google Scholar]
- Devarajan, V.; Glazer, A.M. Theory and computation of optical-rotatory power in inorganic crystals. Acta Crystallogr. A 1986, 42, 560. [Google Scholar] [CrossRef]
- Tessman, J.R.; Kahn, A.H.; Shokley, W. Electron polarizabilities of ions in crystals. Phys. Rev. 1953, 92, 891–895. [Google Scholar]
- Kondru, R.K.; Wipf, P.; Beratan, D.N. Atomic contributions to the optical rotation angle as a quantitative probe of molecular chirality. Science 1998, 282, 2247–2250. [Google Scholar] [CrossRef]
- Claborn, K.; Kaminsky, W.; Herreros-Cedres, J.; Weckert, E.; Kahr, B. Optical rotation of Achiral Pentaerythritol. J. Am. Chem. Soc. 2006, 128, 14746–14747. [Google Scholar]
- Kaminsky, W.; Glazer, A.M. Crystal optics of Mannitol, C6H14O6: Crystal growth, structure, basic physical properties, birefringence, optical activity, Faraday effect, electro-optic related effects and model calculations. Z. Kristallogr. 1997, 212, 283–296. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 5th ed; John Wiley & Sons Inc.: New York, NY, USA, 1976; p. 399. [Google Scholar]
- Ashcroft, N.W.; Mermin, N.D. Solid State Physics; Holt, Rinehart and Winston: London, UK, 1976; p. 546. [Google Scholar]
- Bohatý, L. Crystallographic aspects of the linear electrooptic effect. Z. Kristallogr. 1984, 166, 97–119. [Google Scholar] [CrossRef]
- One might be astonished by the large polarizability assigned to H, which only reflects the delocalized character of the C-H bonds.
- Haussuehl, S. Kristallphysik; Physik-Verlag: Verlag Chemie:: Weinheim, Germany, 1983. [Google Scholar]
- Jackson, J.D. Classical Electrodynamics, 2nd ed; John Wiley & Sons: New York, NY, USA, 1975; p. 220. [Google Scholar]
- Ewald, P.P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 64, 253–287. [Google Scholar] [CrossRef]
- Kaminsky, W.; Glazer, A.M. Comparison of experimental optical properties of TGS with calculations using the DES model. Phase Transit. 1997, 66, 1–21. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kaminsky, W.; Responte, D.; Daranciang, D.; Gallegos, J.B.; Ngoc Tran, B.-C.; Pham, T.-A. Structure, Morphology and Optical Properties of Chiral N-(4-X-phenyl)-N-[1(S)-1-phenylethyl]thiourea, X= Cl, Br, and NO2. Molecules 2010, 15, 554-569. https://doi.org/10.3390/molecules15010554
Kaminsky W, Responte D, Daranciang D, Gallegos JB, Ngoc Tran B-C, Pham T-A. Structure, Morphology and Optical Properties of Chiral N-(4-X-phenyl)-N-[1(S)-1-phenylethyl]thiourea, X= Cl, Br, and NO2. Molecules. 2010; 15(1):554-569. https://doi.org/10.3390/molecules15010554
Chicago/Turabian StyleKaminsky, Werner, Donald Responte, Dan Daranciang, Jose B. Gallegos, Bao-Chau Ngoc Tran, and Tram-Anh Pham. 2010. "Structure, Morphology and Optical Properties of Chiral N-(4-X-phenyl)-N-[1(S)-1-phenylethyl]thiourea, X= Cl, Br, and NO2" Molecules 15, no. 1: 554-569. https://doi.org/10.3390/molecules15010554