Structural Necessity of Indole C5-O-Substitution of seco-Duocarmycin Analogs for Their Cytotoxic Activity

Abstract
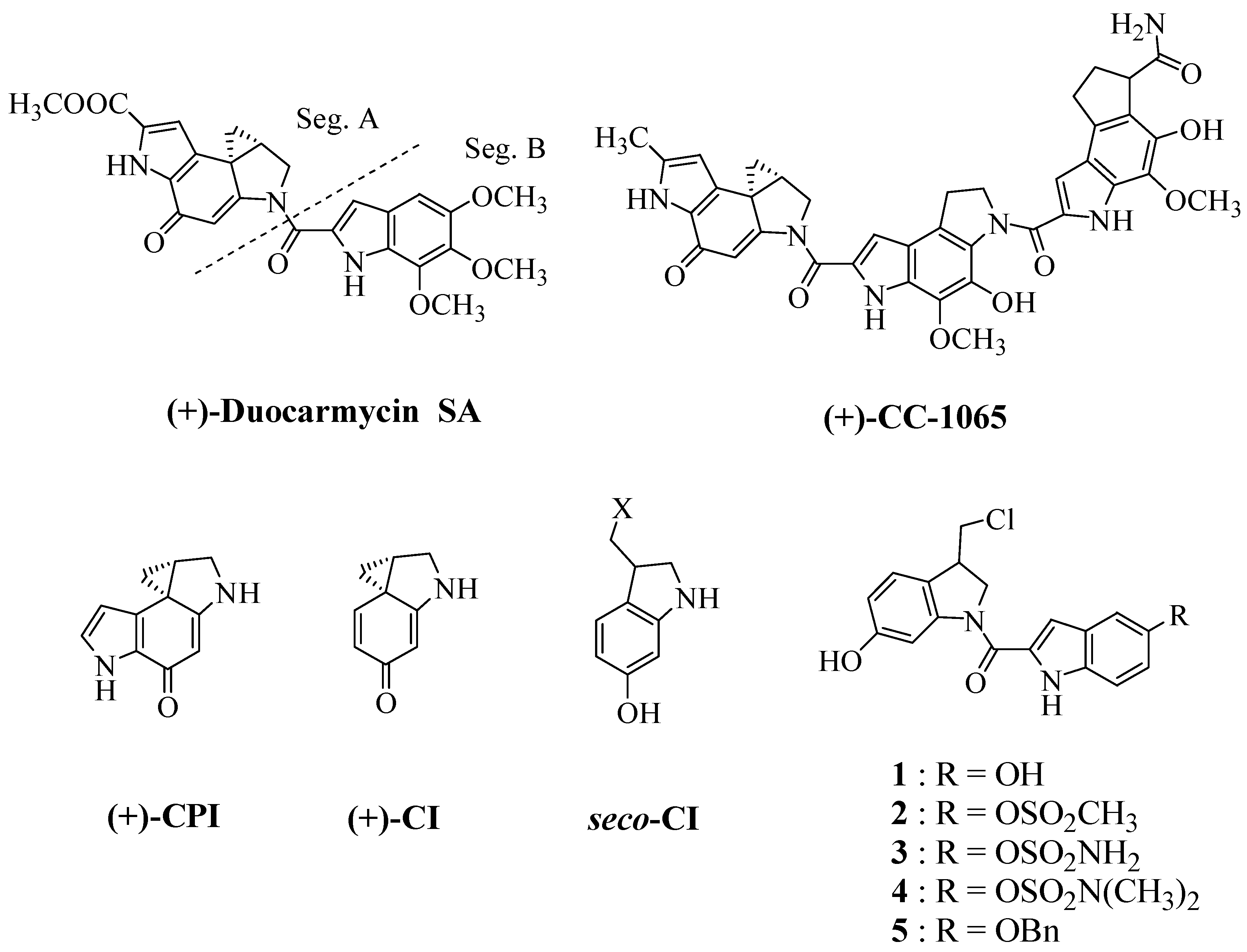
:1. Introduction


2. Results and Discussion
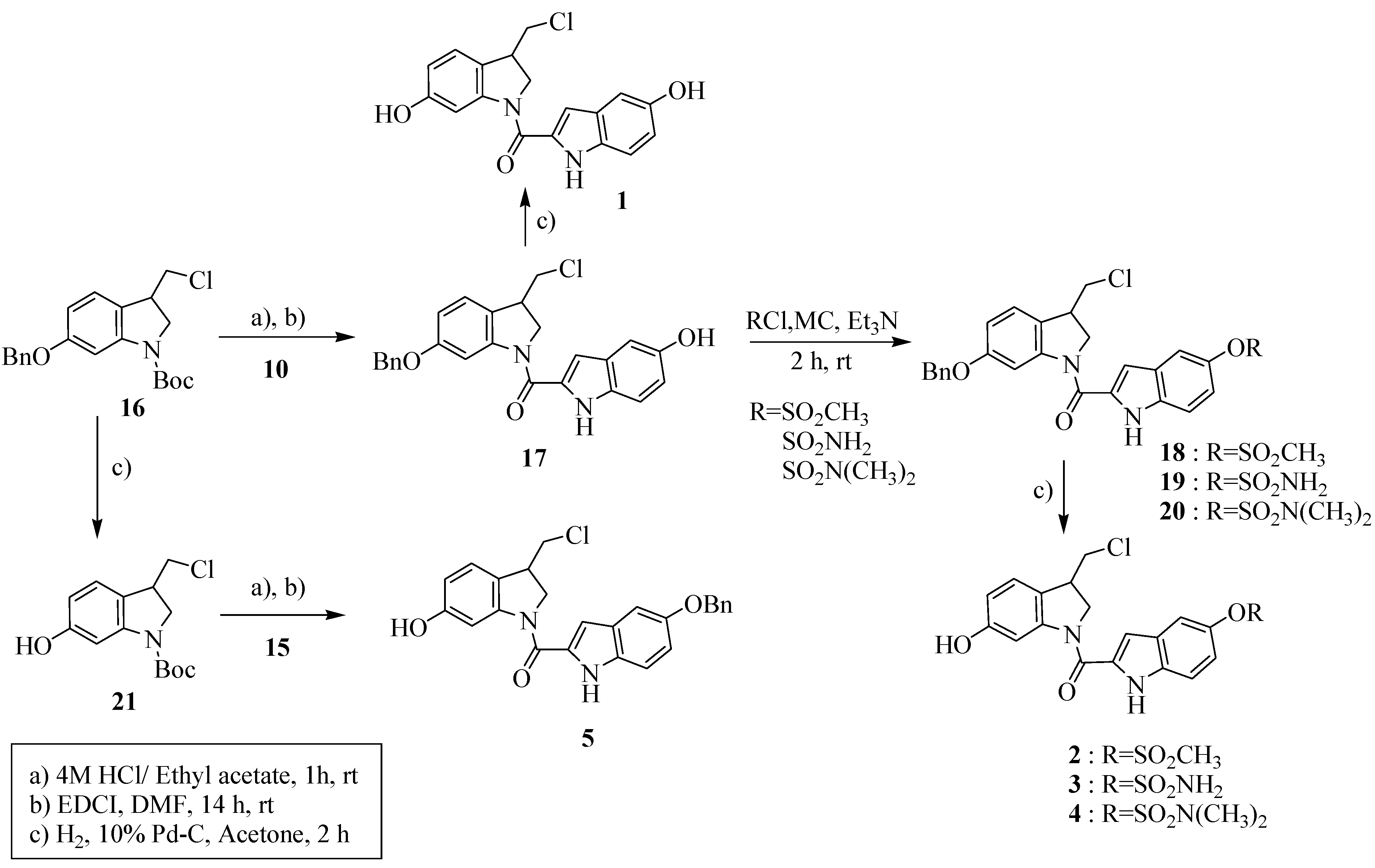
2.1. Synthesis
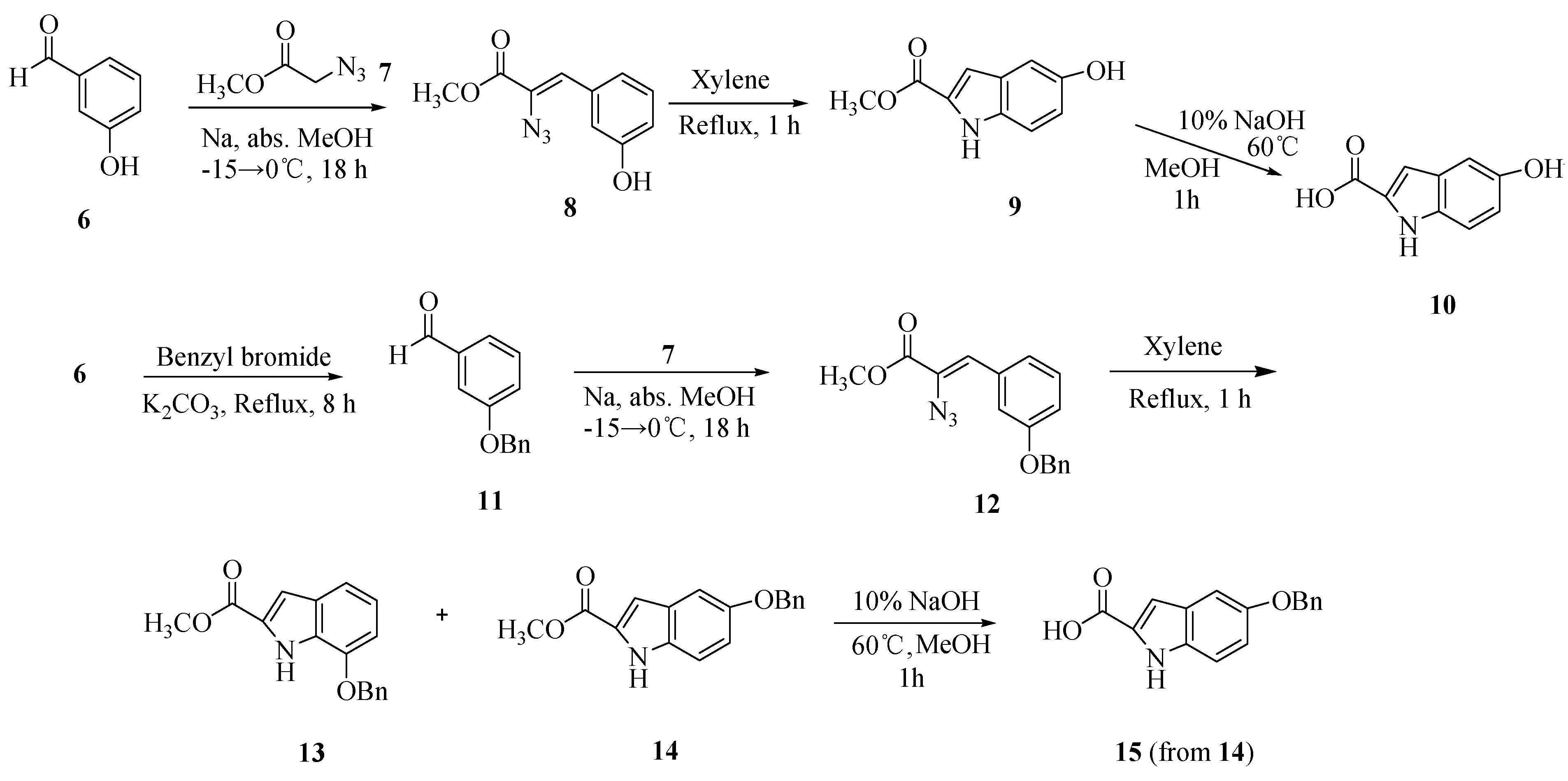
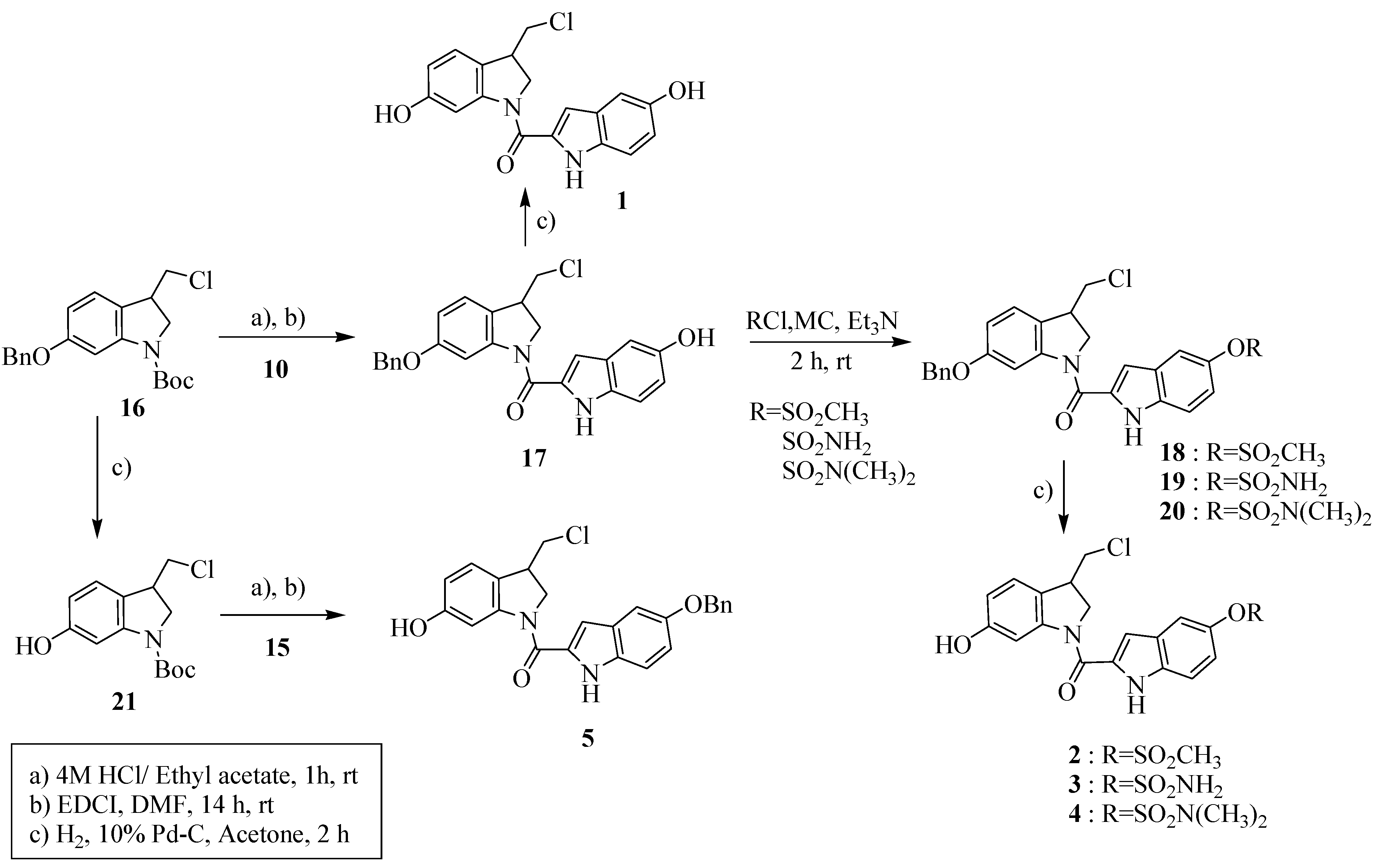
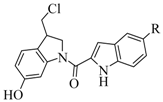
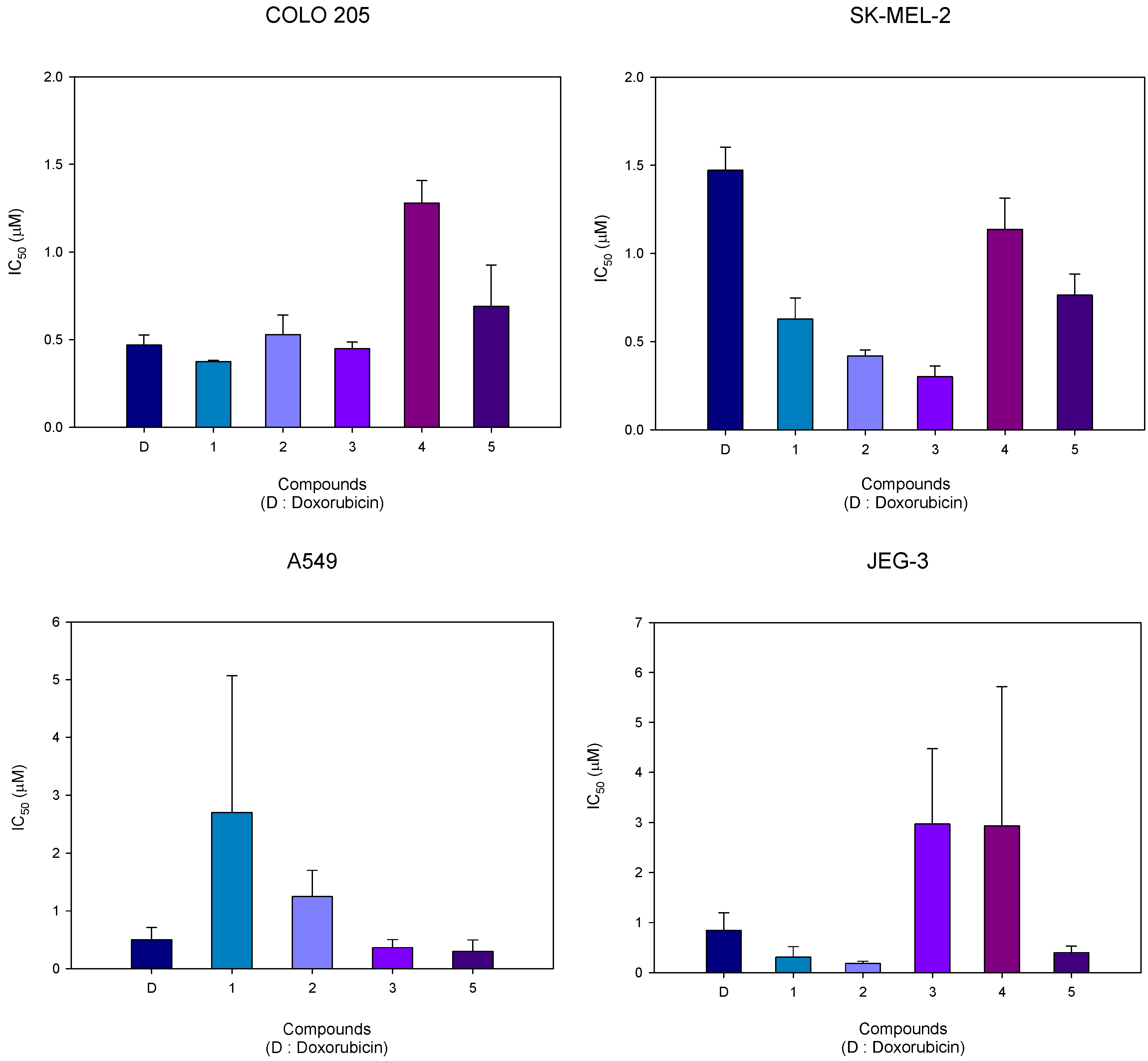
2.2. Cytotoxic activity

| Compound | R | IC50 (µM)a) | |||
|---|---|---|---|---|---|
| COLO 205 | SK-MEL-2 | A549 | JEG-3 | ||
| Doxorubicin | – | 0.469 | 1.472 | 0.499 | 0.847 |
| 1 | OH | 0.374 | 0.629 | 2.709 | 0.310 |
| 2 | OSO2CH3 | 0.528 | 0.419 | 1.250 | 0.182 |
| 3 | OSO2NH2 | 0.448 | 0.301 | 0.365 | 2.974 |
| 4 | OSO2N(CH3)2 | 1.279 | 1.137 | >5 | 2.936 |
| 5 | OBn | 0.690 | 0.764 | 0.298 | 0.397 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

3. Experimental
3.1. General
3.2. Cytotoxicity
4. Conclusions
References
- Boger, D.L.; Ishizaki, T.; Zarrinmayeh, H.; Kitos, P.A.; Suntornwat, O. Synthesis and preliminary evaluation of agents incorporating the pharmacophore of the duocarmycin/pyrindamycin alkylation subunit: Identification of the CC-1065/duocarmycin common pharmacophore. J. Org. Chem. 1990, 55, 4499–4502. [Google Scholar] [CrossRef]
- Sugiyama, H.; Ohmori, K.; Chan, K.L.; Hosoda, M.; Asai, A.; Saito, H.; Saito, I. A novel guanine N3 alkylation by antitumor antibiotic duocarmycin A. Tetrahedron Lett. 1993, 34, 2179–2182. [Google Scholar] [CrossRef]
- Fukuda, Y.; Nakatani, K.; Terashima, S. Synthetic studies on duocarmycin. 2. Synthesis and cytotoxicity of natural (+)-duocarmycin A and its three possible stereoisomers. Tetrahedron 1994, 50, 2809–2820. [Google Scholar] [CrossRef]
- Boger, D.L.; Johnson, S. Prodrugs of the cytostatic CC-1065 that can be activated in a tumor-selective manner. Angew. Chem. Int. Ed. Engl. 1996, 108, 1542–1580. [Google Scholar] [CrossRef]
- McGovren, J.P.; Clarke, G.L.; Pratt, E.A.; Dekoning, T.F. Preliminary toxicity studies with the DNA-binding antibiotics, CC-1065. J. Antibiot. 1984, 37, 63–70. [Google Scholar] [CrossRef]
- Boger, D.L.; Johnson, S. CC-1065 and the duocarmycins: Understanding their biological function through mechanistic studies. Angew. Chem. Int. Ed. Engl. 1996, 35, 1438–1474. [Google Scholar] [CrossRef]
- Boger, D.L. The Duocarmycins: Synthetic and mechanistic studies. Acc. Chem. Res. 1995, 28, 20–29. [Google Scholar] [CrossRef]
- Boger, D.L.; McKie, J.A.; Han, N.; Tarbie, C.M.; Riggs, H.W.; Kitos, P.A. A Hammett correlation for CC-1065 and duocarmycin analogs: Magnitude of substituent electronic effects on functional reactivity. Bioorg. Med. Chem. Lett. 1996, 6, 659–664. [Google Scholar] [CrossRef]
- Yasuzawa, T.; Muroi, K.; Ichimura, M.; Takahashi, I.; Ogawa, T.; Takahashi, K.; Sano, H.; Saotoh, Y. Duocarmycins, potent antitmor antibiotics produced by Streotomyces sp. Structure and chemistry. Chem. Pharm. Bull. 1995, 43, 378–391. [Google Scholar] [CrossRef]
- Boger, D.L.; Munk, S.A.; Zarrinmayeh, H.; Ishizaki, T.; Haught, J.; Bina, M. An alternative and convenient strategy for generation of substantial quantities of singly 5′-32p-end-labeled double-stranded DNA for binding studies: Development of a protocol for examination of functional features of (+)-CC-1065 and the duocarmycins that contribute to their sequence-selective DNA alkylation properties. Tetrahedron 1991, 47, 2661–2681. [Google Scholar] [CrossRef]
- Boger, D.L.; Ishizaki, T.; Zarrinmayeh, H.; Munk, S.A.; Kitos, P.A.; Suntorwart, O. Duocarmycin-pyrindamycin DNA alkylation properties and identification, synthesis, and evaluation of agents incorporating the pharmacophore of the duocarmycin-pyrindamycin alkylation subunit. Identification of the CC-1065 duocarmycin common pharmacophore. J. Am. Chem. Soc. 1990, 112, 8961–8971. [Google Scholar]
- Boger, D.L.; Ishizaki, T.; Zarrinmayeh, H.; Kitos, P.A.; Suntornwat, O. Synthesis and preliminary evaluation of agents incorporating the pharmacophore of the duocarmycin/pyrindamycin alkylation subunit: identification of the CC-1065/duocarmycin common pharmacophore. J. Org. Chem. 1990, 55, 4499–4502. [Google Scholar] [CrossRef]
- Boger, D.L.; Johnson, D.S.; Yun, W. (+)- and ent-(-)-Duocarmycin SA and (+)- and ent-(-)-N-BOC-DSA DNA alkylation properties. Alkylation site models that accommodate the offset AT-rich adenine N3 alkylation selectivity of the enantiomeric agents. J. Am. Chem. Soc. 1994, 116, 1635–1656. [Google Scholar] [CrossRef]
- Baird, R.; Winstein, S. Neighboring carbon and hydrogen. Li. Dienones from Ar1[UNK]-3 participation. Isolation and behavior of spiro[2,5]octa-1,4-diene-3-one. J. Am. Chem. Soc. 1963, 85, 567–578. [Google Scholar] [CrossRef]
- Borger, D.L.; Yun, W. CBI-TMI: Synthesis and evaluation of a key analog of the duocarmycins. Validation of a direct relationship between chemical solvolytic stability and cytotoxic potency and communication of the structural feature responsible for the distinguishing behavior of enantiomeric pairs of agents. J. Am. Chem. Soc. 1994, 116, 7996–8006. [Google Scholar]
- Asai, A.; Nagamura, S.; Saito, H. A novel properties of duocarmycin and its analogs for covalent reaction. J. Am. Chem. Soc. 1994, 116, 4171–4177. [Google Scholar]
- Boger, D.L.; Colman, R.S.; Invergo, B.J.; Sakya, S.M.; Ishizaki, T.; Munk, S.A.; Zarrinmayeh, H.; Kitos, P.A.; Thompson, S.C. Synthesis and evaluation of aborted and extended CC-1065 functional analogs: (+)- and (-)-CPI-PDE-I1, (+)- and (-)-CPI-CDPI1, and (±)-, (+)-, and (-)-CPI-CDPI3. Preparation of key partial structures and definition of an additional functional role of the CC-1065 central and right-hand subunits. J. Am. Chem. Soc. 1990, 112, 4623–4632. [Google Scholar]
- Warpehoski, M.A.; Harper, D.E.; Mitchel, M.A.; Monroe, T.J. Reversibility of the covalent reaction of CC-1065 and analogs with DNA. Biochemistry 1992, 31, 2502–2508. [Google Scholar] [CrossRef]
- Boger, D.L.; Yun, W. Reversibility of the duocarmycin A and SA DNA alkylation reaction. J. Am. Chem. Soc. 1993, 115, 9872–9873. [Google Scholar] [CrossRef]
- Parrish, J.P.; Kastrinsky, D.B.; Stauffer, F.; Hedrick, M.P.; Hwang, I.; Boger, D.L. Establishment of substituent effects in the DNA binding subunit of CBI analogs of the duocarmycins and CC-1065. Bioorg. Med. Chem. 2003, 11, 3815–3838. [Google Scholar] [CrossRef]
- Howard, T.T.; Lingerfelt, B.M.; Purnell, B.L.; Scott, A.E.; Price, C.A.; Townes, H.M.; McNulty, L.; Handl, H.L.; Summerville, K.; Hudson, S.J.; Bowen, J.P.; Kiakos, K.; Hartley, J.A.; Lee, M. Novel furano analogues of duocarmycin C1 and C2: Design, synthesis, and biological evaluation of seco-iso-cyclopropylfurano[2,3-e]indoline (seco-iso-CFI) and seco-cyclopropyltetrahydrofurano [2,3-f]quinoline (seco-CFQ) analogues. Bioorg. Med. Chem. 2002, 10, 2941–2952. [Google Scholar]
- Rosa, F.A.F.; Rebelo, R.A.; Nascimento, M.G. Synthesis of new indolecarboxylic acids related to the plant hormone indoleacetic acid IAA. J. Braz. Chem. Soc. 2003, 14, 11–15. [Google Scholar]
- Fukuda, Y.; Itoh, Y.; Nakatani, K.; Terashima, S. Synthetic studies on duocarmycin. 1. Total synthesis of dl-duocarmycin A and its 2-epimer. Tetrahedron 1994, 50, 2793–2808. [Google Scholar] [CrossRef]
- Coowar, D.; Bouissac, J.; Hanbali, M.; Paschaki, M.; Mohier, E.; Luu, B. Effects of indole fatty alcohols on the differentiation of neural stem cell derived neurospheres. J. Med. Chem. 2004, 47, 6270–6282. [Google Scholar] [CrossRef]
- Choi, T.; Ma, E. Efficient synthesis of achiral seco-CI subunit of duocarmycin pharmacophore. Bull. Korean Chem. Soc. 2009, 30, 2815–2818. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Brossi, A. Synthesis of (-)-1-hydroxy-N-methyl morphinan-6-one and its O-methyl ether from 4-hydroxy-N-formyl morphinan-6-one. J. Org. Chem. 1983, 48, 1469–1471. [Google Scholar] [CrossRef]
- Lin, C.-F.; Yang, J.-S.; Chan, C.-Y.; Kuo, S.-C.; Lee, M.-R.; Huang, L.-J. Synthesis and anticancer activity of benzyloxybenzaldehyde derivatives against HL-60 cells. Bioorg. Med. Chem. Lett. 2005, 13, 1537–1544. [Google Scholar] [CrossRef]
- Loge, O.; Heindl, J.; Albrecht, R. β2-Agonists containing metabolically labile groups. II. The influence of ester groups in the aryl system. Eur. J. Med. Chem. Chim Ther. 1985, 20, 57–60. [Google Scholar]
- Okada, M.; Iwashita, S.; Koizumi, N. Efficient general method for sulfamoylation of a hydroxyl group. Tetrahedron Lett. 2000, 41, 7047–7051. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Choi, T.; Ma, E. Structural Necessity of Indole C5-O-Substitution of seco-Duocarmycin Analogs for Their Cytotoxic Activity. Molecules 2010, 15, 7971-7984. https://doi.org/10.3390/molecules15117971
Choi T, Ma E. Structural Necessity of Indole C5-O-Substitution of seco-Duocarmycin Analogs for Their Cytotoxic Activity. Molecules. 2010; 15(11):7971-7984. https://doi.org/10.3390/molecules15117971
Chicago/Turabian StyleChoi, Taeyoung, and Eunsook Ma. 2010. "Structural Necessity of Indole C5-O-Substitution of seco-Duocarmycin Analogs for Their Cytotoxic Activity" Molecules 15, no. 11: 7971-7984. https://doi.org/10.3390/molecules15117971
APA StyleChoi, T., & Ma, E. (2010). Structural Necessity of Indole C5-O-Substitution of seco-Duocarmycin Analogs for Their Cytotoxic Activity. Molecules, 15(11), 7971-7984. https://doi.org/10.3390/molecules15117971