Isolation and Identification of Two New Polyynes from a North American Ethnic Medicinal Plant--Oplopanax horridus (Smith) Miq.

Abstract
:
1. Introduction

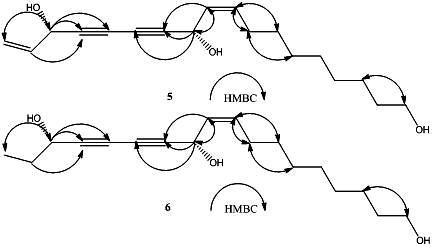
2. Results and Discussion


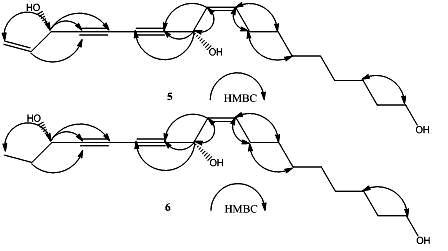{kind=link}
{kind=link}
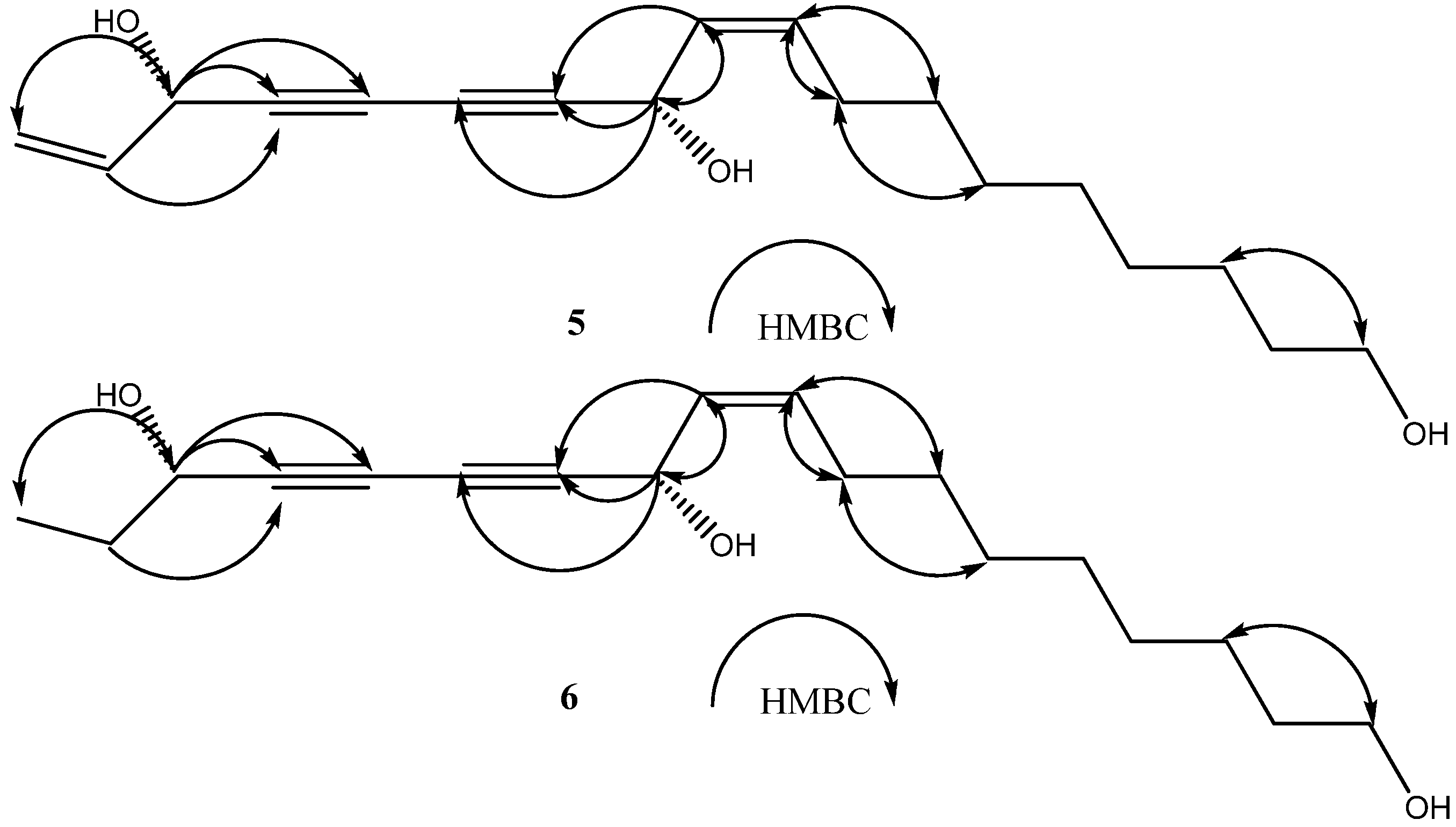{kind=link}
| Carbon position | compound 5 | compound 6 | |||
|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | ||
| 1 | 3.64, t (2H, 6.5 ) | 63.0 | 3.64, t (2H, 6.5 ) | 63.0 | |
| 2 | 1.56, m (2H ) | 32.6 | 1.57, m ( 2H ) | 32.6 | |
| 3 | 1.31, m (2H) | 25.6 | 1.31, m (2H) | 25.6 | |
| 4 | 1.31, m (2H) | 29.1 a | 1.31, m (2H) | 29.1 b | |
| 5 | 1.31, m (2H) | 29.2 | 1.31, m (2H) | 29.2 | |
| 6 | 1.31, m (2H) | 29.0 a | 1.31, m (2H) | 29.0 b | |
| 7 | 1.39, m (2H) | 28.8 | 1.38, m (2H) | 28.8 | |
| 8 | 2.11, dq (2H, 7.1, 1.5 ) | 27.5 | 2.11, dq (2H, 7.1, 1.5 ) | 27.5 | |
| 9 | 5.51, ddt (1H, 10.6, 8.2, 1.5 ) | 127.9 | 5.52, ddt (1H, 10.6, 8.2, 1.5 ) | 128.0 | |
| 10 | 5.58, ddt (1H, 10.6, 7.3, 1.5 ) | 134.2 | 5.58, ddt (1H, 10.6, 7.3, 1.5 ) | 134.1 | |
| 11 | 5.19, d (1H, 8.0) | 58.5 | 5.19, br.d (1H, 8.0) | 58.5 | |
| 12 | - | 79.8 | - | 79.1 | |
| 13 | - | 68.7 | - | 68.8 c | |
| 14 | - | 70.1 | - | 68.8 c | |
| 15 | - | 78.5 | - | 80.9 | |
| 16 | 4.93, br.d ( 1H, 5.5) | 63.3 | 4.37, t ( 1H, 6.6) | 63.8 | |
| 17 | 5.93, ddd ( 1H, 17.4, 10.0, 5.5 ) | 136.0 | 1.74, m (2H) | 30.6 | |
| 18 | 5.22, dt ( 1H, 10.0, 1.0 );5.46, dt ( 1H, 17.4, 1.0 ) | 117.1 | 1.00, t ( 3H, 7.5) | 9.3 | |
 + 189.3° (c = 0.23, CHCl3)} and 4a { + 230.6° (c = 0.11, CHCl3)} were identical with those of the new polyynes 5 { +194.4° (c = 0.16, CHCl3)}and 6{ + 233.0° (c = 0.3, CHCl3)}, respectively. The above evidence indicated that 5 and 6 should have the same absolute configurations with the known compounds 3 and4. Thus, the complete structures of the new polyynes, oplopantriol A (5) and oplopantriol B (6), were elucidated to be (11S,16S,9Z)-9,17-octadecadien-12,14-diyne-1,11,16-triol and (11S,16S,9Z)-9-octadecaen-12,14- diyne-1,11,16-triol, which were named as oplopantriol A (5) and oplopantriol B (6), respectively.
+ 189.3° (c = 0.23, CHCl3)} and 4a { + 230.6° (c = 0.11, CHCl3)} were identical with those of the new polyynes 5 { +194.4° (c = 0.16, CHCl3)}and 6{ + 233.0° (c = 0.3, CHCl3)}, respectively. The above evidence indicated that 5 and 6 should have the same absolute configurations with the known compounds 3 and4. Thus, the complete structures of the new polyynes, oplopantriol A (5) and oplopantriol B (6), were elucidated to be (11S,16S,9Z)-9,17-octadecadien-12,14-diyne-1,11,16-triol and (11S,16S,9Z)-9-octadecaen-12,14- diyne-1,11,16-triol, which were named as oplopantriol A (5) and oplopantriol B (6), respectively.3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Alkaline Hydrolysis of Compounds 3 and 4
+194.4° (c = 0.16, CHCl3); UV (CHCl3) λmax (log ξ): 215 (0.63), 226 (1.10), 255 (4.09) 261 (3.95), 273 (4.13) and 287 (4.07) nm; IR (KBr) νmax : 3357, 3022, 2929, 2855, 2251, 2150, 1675, 1464, 1405, 1303, 1021, 933 and 880 cm-1; 1H and 13C NMR data (Table 1); Positive mode ESI-MS m/z: 313 [M+Na]+ (100); Negative mode HR-ESI-MS m/z: 289.1867 [M-H]¯, Calcd for C18H25O3: 289.1804). + 233.0° (c = 0.3, CHCl3); UV(CHCl3) λmax (log ξ): 207 (1.07), 226 (1.19), 232 (1.17), 253 (4.02), 263 (3.98), 265 (3.95), 272 (4.01) and 288 (3.84) nm; IR (KBr) νmax : 3355, 3021, 2930, 2856, 2232, 2143, 1656, 1463, 1305, 1095, 1017, 970 and 866 cm-1; 1H and 13C NMR data (Table 1); Positive mode ESI-MS m/z: 315 [M+Na]+ (100); Negative mode HR-ESI-MS m/z: 291.1966 [M-H]¯, Calcd for C18H27O3: 291.1960).4. Conclusions
Acknowledgements
References and Notes
- Wu, C.Y. Flora of China; Science Press: Beijing, China, 1988; Volume 54, p. 16. [Google Scholar]
- Takeda, K.; Minato, H.; Ishikawa, M. Studies on sesquiterpenoids—XII Structure and absolute configuration of oplopanone, a new sesquiterpene from oplopanax japonicus (NAKAI) NAKAI. Tetrahedron 1966, 22 Suppl. 7, 219–225. [Google Scholar] [CrossRef]
- McCutcheon, A.R.; Roberts, T.E.; Gibbons, E.; Ellis, S.M.; Babiuk, L.A.; Hancock, R.E.W.; Towers, G. H.N. Antiviral screening of British Columbian medicinal plants. J. Ethnopharmacol. 1995, 49, 101–110. [Google Scholar] [CrossRef]
- Inui, T.; Wang, Y.H.; Deng, S.X.; Smith, D.C.; Franzblau, S.G.; Pauli, G.F. Counter-current chromatography based analysis of synergy in an anti-tuberculosis ethnobotanical. J. Chromatogr. A 2007, 1151, 211–215. [Google Scholar]
- McCutcheon, A.R.; Ellis, S.M.; Hancock, R.E.W.; Towers, G.H.N. Antibiotic screening of medicinal plants of the British Columbian native peoples. J. Ethnopharmacol. 1992, 37, 213–223. [Google Scholar] [CrossRef]
- Noda, K.; Fujikawa, T.; Ecredia, K.K. Skin care preparation for improving skin lineae atrophicae such as striae gravidarum/Extract containing triterpenoid saponins prepared preferably from plants belonging Araliaceae family and Cucurbitaceae family for relieving striae gravidarum by external application. Jpn. Kokai Tokkyo Koho 2003. JP Pat. 2003128570, A. [Google Scholar]
- McCutcheon, A.R.; Ellis, S.M.; Hancock, R.E.W.; Towers, G.H.N. Antifungal screening of medicinal plants of British Columbian native peoples. J. Ethnopharmacol. 1994, 44, 157–169. [Google Scholar] [CrossRef]
- Dou, D.Q.; Hu, X.Y.; Zhao, Y.R.; Kang, T.G.; Liu, F.Y.; Kuang, H.X.; Smith, D.C. Studies on the anti-psoriasis constituents of Oplopanax elatus Nakai. Nat. Prod. Res. 2009, 23, 334–342. [Google Scholar] [CrossRef]
- Wang, C.Z.; Aung, H.H.; Mehendale, S.R.; Shoyama, Y.; Yuan, C.S. High performance liquid chromatographic analysis and anticancer potential of Oplopanax horridus: Comparison of stem and berry extracts. Fitoterapia 2010, 81, 132–139. [Google Scholar] [CrossRef]
- Turner, N.J. Traditional Use of Devil's-Club (Oplopanax horridus; Araliaceae) by Native Peoples in Western North America. J. Ethnobiol. 1982, 2, 17–38. [Google Scholar]
- Kobaisy, M.; Abramowski, Z.; Lermer, L.; Saxena, G.; Hancock, R.E.W.; Towers, G.H.N.; Doxsee, D.; Stokes, R.W. Antimycobacterial polyynes of Devil's Club (Oplopanax horridus), a North American native medicinal plant. J. Nat. Prod. 1997, 60, 1210–1213. [Google Scholar] [CrossRef]
- Papajewski, S.A.; Guse, J.H.; Klaiber, I.; Roos, G.; Suessmuth, R.; Vogler, B.; Walter, C.U.; Kraus, W. Bioassay Guided Isolation of a New C18-Polyacetylene, (+)-9(Z),17-Octadecadiene- 12,14-diyne-1,11,16-triol, from Cussonia barteri. Planta Med. 1998, 64, 479–481. [Google Scholar] [CrossRef]
- Crosby, D.G.; Aharonson, N. The structure of carotatoxin, a natural toxicant from carrot. Tetrahedron 1967, 23, 465–472. [Google Scholar] [CrossRef]
- Lemmich, E. The absolute configuration of the acetylenic compound falcarindiol. Phytochemistry 1981, 20, 1419–1420. [Google Scholar] [CrossRef]
- Ratnayake, A.S.; Hemscheidt, T. Olefin cross-metathesis as a tool in natural product degradation. the stereochemistry of (+)-falcarindiol. Org. Lett. 2002, 4, 4667–4669. [Google Scholar] [CrossRef]
- Zheng, G.R.; Lu, W.; Cai, J.C. Stereoselective total synthesis of (3R,8S)-falcarindiol, a common polyacetylenic compound from Umbellifers. J. Nat. Prod. 1999, 62, 626–628. [Google Scholar] [CrossRef]
- Sabitha, G.; Bhaskar, V.; Reddy, C.S.; Yadav, J.S. Stereoselective approaches for the total synthesis of polyacetylenic (3R,8S)-falcarindiol. Synthesis 2008, 1, 115–121. [Google Scholar]
- Bernart, M.W.; Cardellina, J.H.; Balaschak, M.; Alexander, M.R.; Shoemaker, R.H.; Boyd, M.R. Cytotoxic falcarinol oxylipins from dendropanax arboreus. J. Nat. Prod. 1996, 59, 748–753. [Google Scholar] [CrossRef]
- Schmiech, L.; Alayrac, C.; Witulski, B.; Hofmann, T. Structure determination of bisacetylenic oxylipins in carrots (Daucus carota L.) and enantioselective synthesis of falcarindiol. J. Agric. Food Chem. 2009, 57, 11030–11040. [Google Scholar] [CrossRef]
- Seger, C.; Godejohann, M.; Spraul, M.; Stuppner, H.; Hadacek, F. J. J. Chromatogr. A 2006, 1136, 82–88. [CrossRef]
- Lechner, D.; Stavri, M.; Oluwatuyi, M.; Pereda-Miranda, R.; Gibbons, S. The anti-staphylococcal activity of Angelica dahurica (Bai Zhi). Phytochemistry 2004, 65, 331–335. [Google Scholar]
- Stavri, M.; Ford, C.H.J.; Bucar, F.; Streit, B.; Hall, M.L.; Williamson, R.T.; Mathew, K.T.; Gibbons, S. Bioactive constituents of Artemisia monosperma. Phytochemistry 2005, 66, 233–239. [Google Scholar]
- Sample Availability: Samples of all the compounds are available from the authors.
© 2010 by the authors;
Share and Cite
Huang, W.-H.; Zhang, Q.-W.; Wang, C.-Z.; Yuan, C.-S.; Li, S.-P. Isolation and Identification of Two New Polyynes from a North American Ethnic Medicinal Plant--Oplopanax horridus (Smith) Miq. Molecules 2010, 15, 1089-1096. https://doi.org/10.3390/molecules15021089
Huang W-H, Zhang Q-W, Wang C-Z, Yuan C-S, Li S-P. Isolation and Identification of Two New Polyynes from a North American Ethnic Medicinal Plant--Oplopanax horridus (Smith) Miq. Molecules. 2010; 15(2):1089-1096. https://doi.org/10.3390/molecules15021089
Chicago/Turabian StyleHuang, Wei-Hua, Qing-Wen Zhang, Chong-Zhi Wang, Chun-Su Yuan, and Shao-Ping Li. 2010. "Isolation and Identification of Two New Polyynes from a North American Ethnic Medicinal Plant--Oplopanax horridus (Smith) Miq." Molecules 15, no. 2: 1089-1096. https://doi.org/10.3390/molecules15021089