Catalytic Performance of Ceria Nanorods in Liquid-Phase Oxidations of Hydrocarbons with tert-Butyl Hydroperoxide

Abstract
:
1. Introduction
2. Results and Discussion

2.1. Catalytic Oxidation of Ethylbenzene
2.1.1. General


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2/g) |
|---|---|
| CeNR | 78 |
| CeNR-BuOOH | 55 |
| CeNR- H2O2 | 90 |


2.1.2. Stability of CeNR in the Liquid-Phase Oxidation Reaction
| Catalyst | Temperature (°C) | Conversion 6/120 h | PhEtOOT Selectivity 6/120 h | PhEt=O Selectivity 6/120 h |
|---|---|---|---|---|
| CeNR | 55 | 6/51 | 100/100 | 0/0 |
| CeNR (run 1) | 70 | 15/64 a | 100/82 a | 0/18 a |
| CeNR (run 2) | 70 | 11/64 | 100/82 | 0/18 |
| CeNR-BuOOH | 70 | 16/97 a | 100/34 a | 0/64 a |
| CeNR-H2O2 | 70 | 10/78 | 100/71 | 0/29 |
| CeNR | 90 | 31/96 | 100/43 | 0/57 |
| CeNR | 105 | 22/88 | 100/35 | 0/65 |
| Ce(SO4)2.9H2O | 70 | 0/15 | -/66 | -/34 |
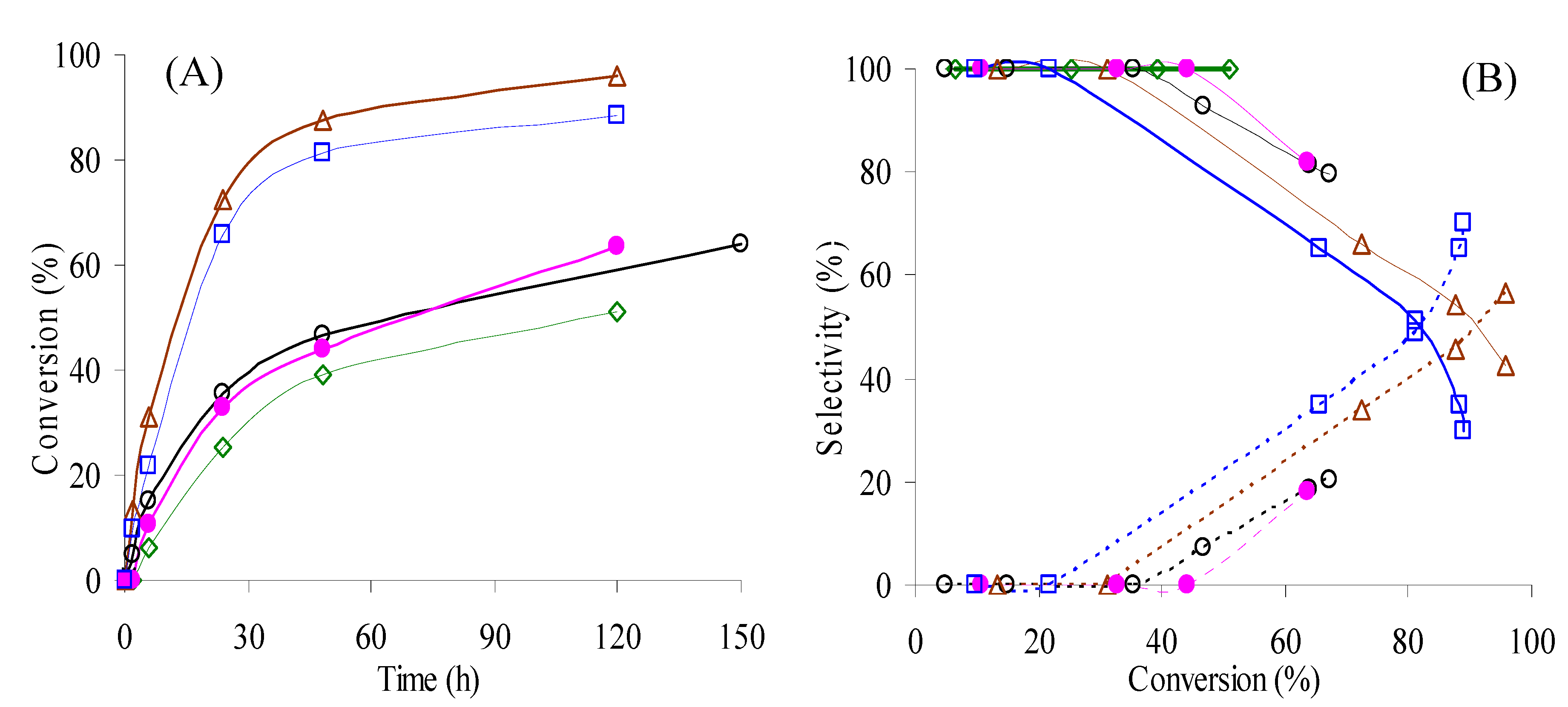
 ), 70 °C (run 1 - (O); run 2 - (
), 70 °C (run 1 - (O); run 2 - (  )), 90 °C (
)), 90 °C (  ), 105 °C (
), 105 °C (  ), using acetonitrile as solvent. (B) - Dependence of selectivity to PhEtOOT (solid lines) and acetophenone (dashed lines) on conversion for the reactions conditions indicated in (A) - the same symbols were used for each reaction.
), 70 °C (run 1 - (O); run 2 - ( )), 90 °C ( ), 105 °C ( ), using acetonitrile as solvent. (B) - Dependence of selectivity to PhEtOOT (solid lines) and acetophenone (dashed lines) on conversion for the reactions conditions indicated in (A) - the same symbols were used for each reaction.
), using acetonitrile as solvent. (B) - Dependence of selectivity to PhEtOOT (solid lines) and acetophenone (dashed lines) on conversion for the reactions conditions indicated in (A) - the same symbols were used for each reaction.
), 70 °C (run 1 - (O); run 2 - ( )), 90 °C ( ), 105 °C ( ), using acetonitrile as solvent. (B) - Dependence of selectivity to PhEtOOT (solid lines) and acetophenone (dashed lines) on conversion for the reactions conditions indicated in (A) - the same symbols were used for each reaction.
2.1.3. Oxidant Effect
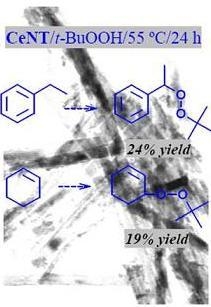
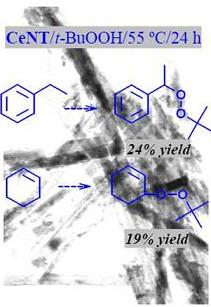
2.2. Oxidation of Different Substrates
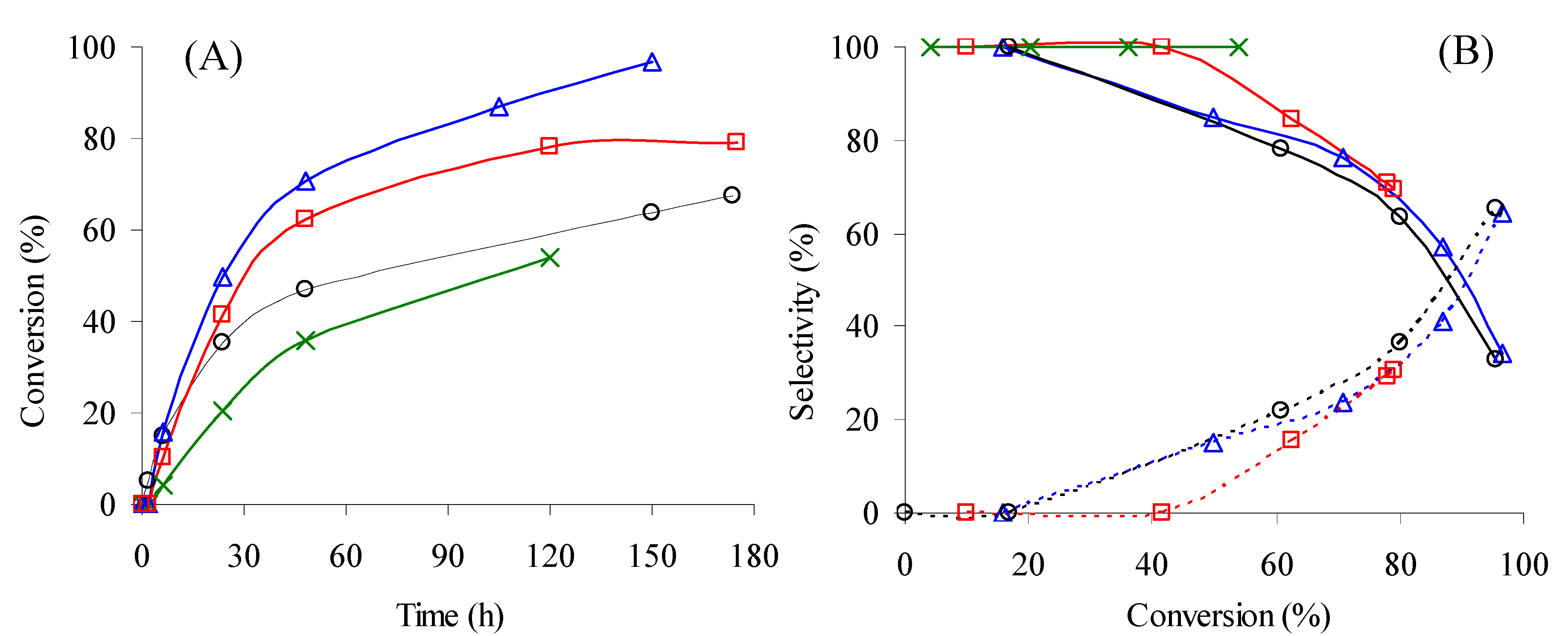
 ),CeNR pre-treated with H2O2 (
),CeNR pre-treated with H2O2 (  ) or t-BuOOH (
) or t-BuOOH (  ). (B) - Dependence of selectivity to PhEtOOT (solid lines) and acetophenone (dashed lines) on conversion for the reactions conditions indicated in (A) - the same symbols were used for each reaction.
),CeNR pre-treated with H2O2 ( ) or t-BuOOH ( ). (B) - Dependence of selectivity to PhEtOOT (solid lines) and acetophenone (dashed lines) on conversion for the reactions conditions indicated in (A) - the same symbols were used for each reaction.
). (B) - Dependence of selectivity to PhEtOOT (solid lines) and acetophenone (dashed lines) on conversion for the reactions conditions indicated in (A) - the same symbols were used for each reaction.
),CeNR pre-treated with H2O2 ( ) or t-BuOOH ( ). (B) - Dependence of selectivity to PhEtOOT (solid lines) and acetophenone (dashed lines) on conversion for the reactions conditions indicated in (A) - the same symbols were used for each reaction.
| Substrate | Conversion (%) | Product | Selectivity (%) |
|---|---|---|---|
 | 25 |  | 100 |
 | 22 |  | 86a |
 | 33 |  | 100 |
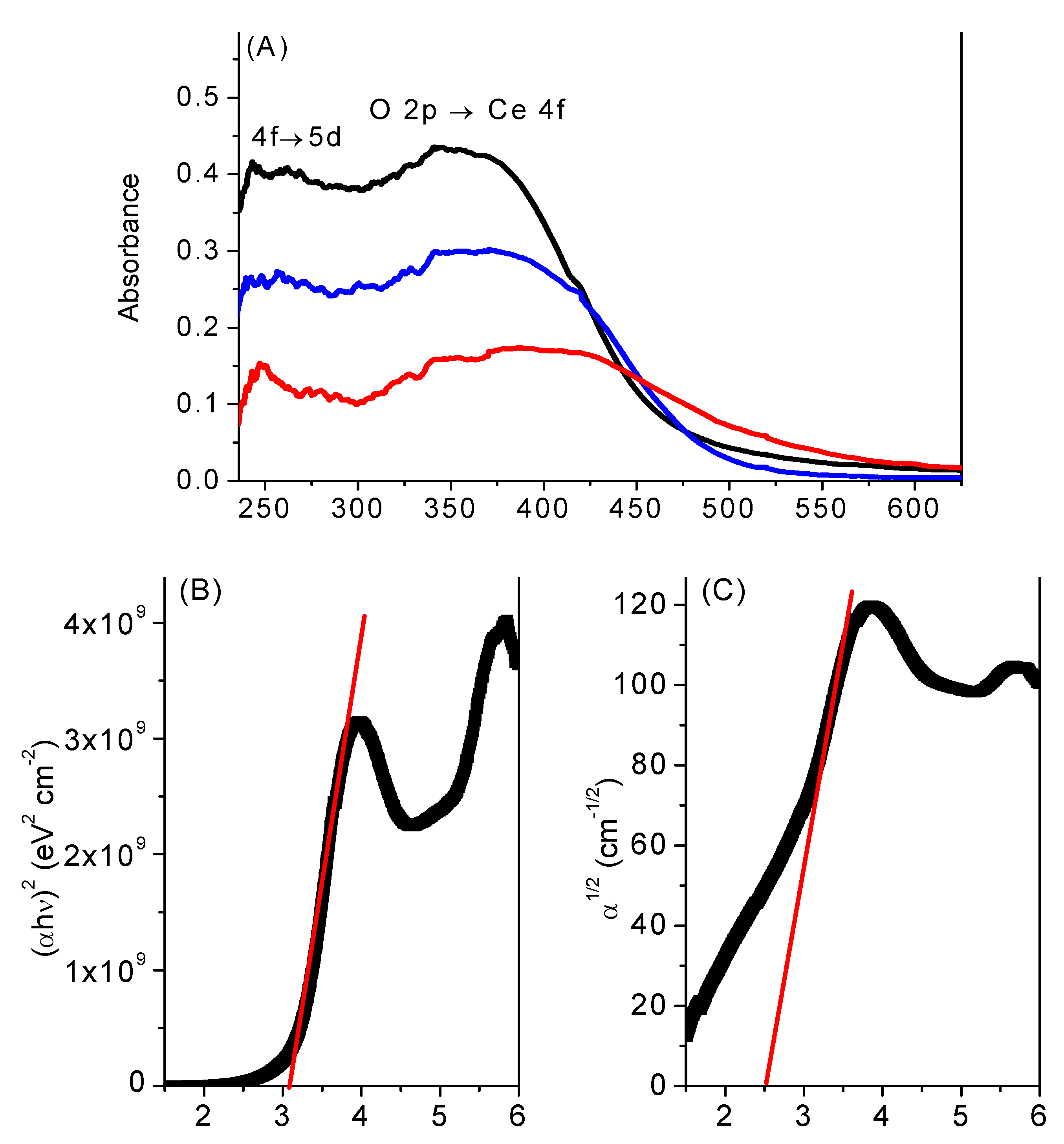
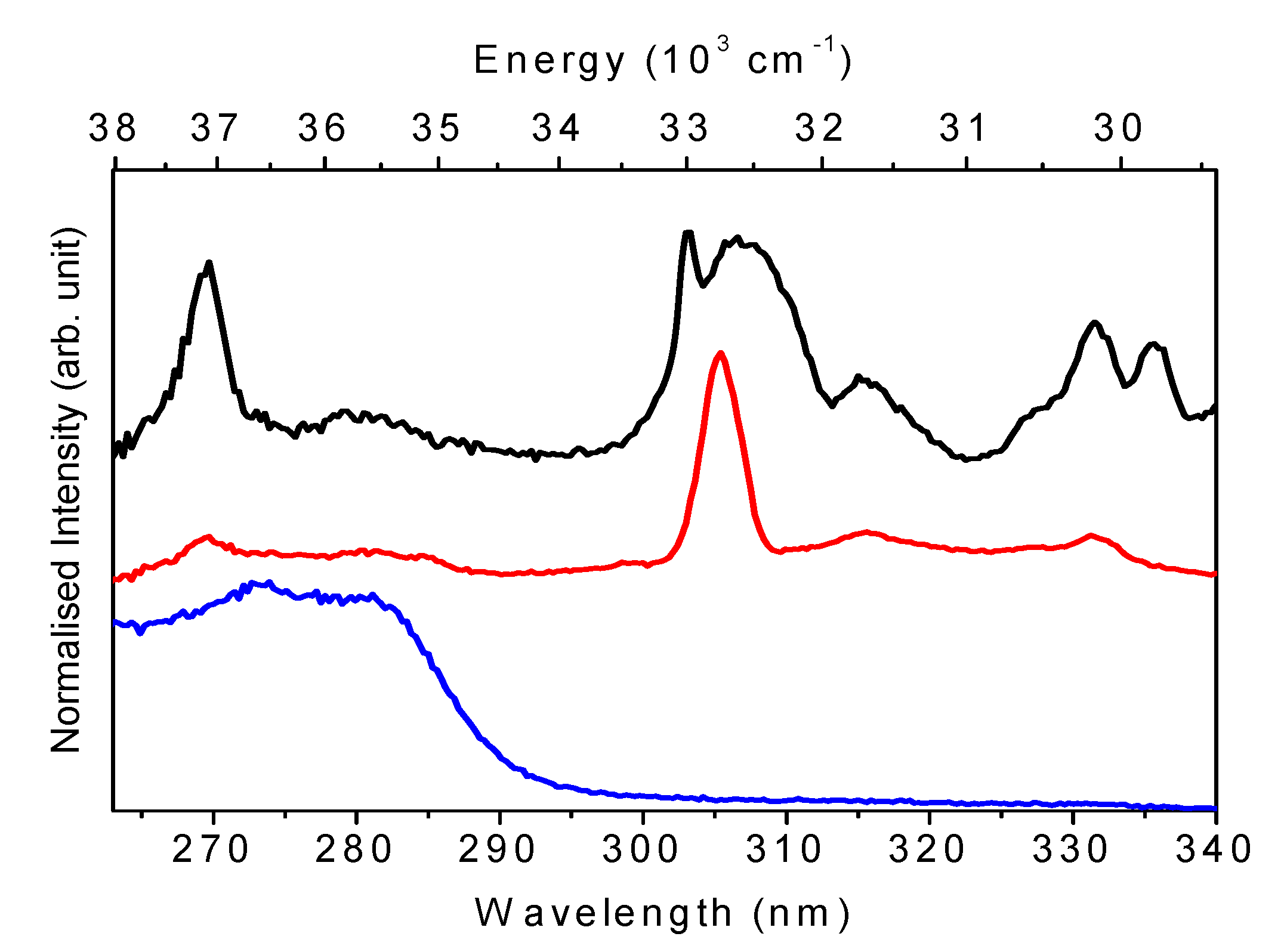
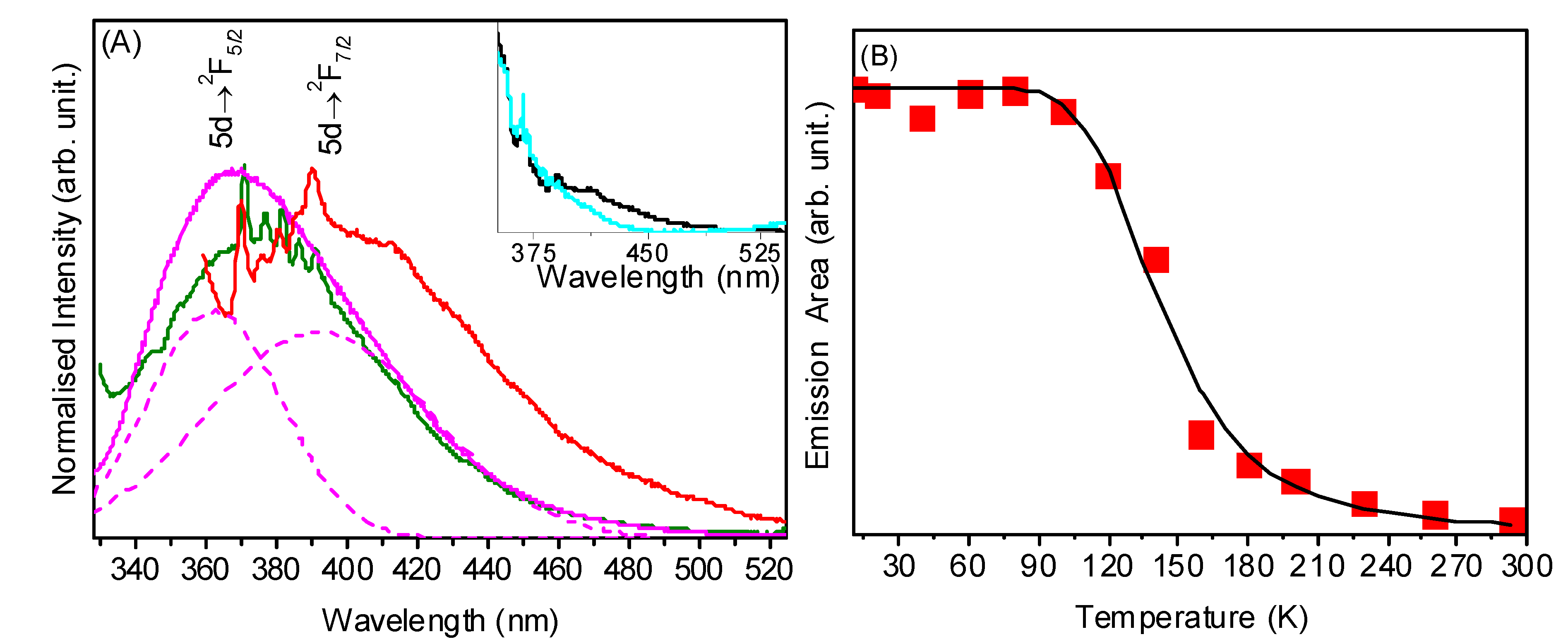
2.3. UV-Vis and Photoluminescence Spectroscopy



3. Experimental Section
3.1. Samples
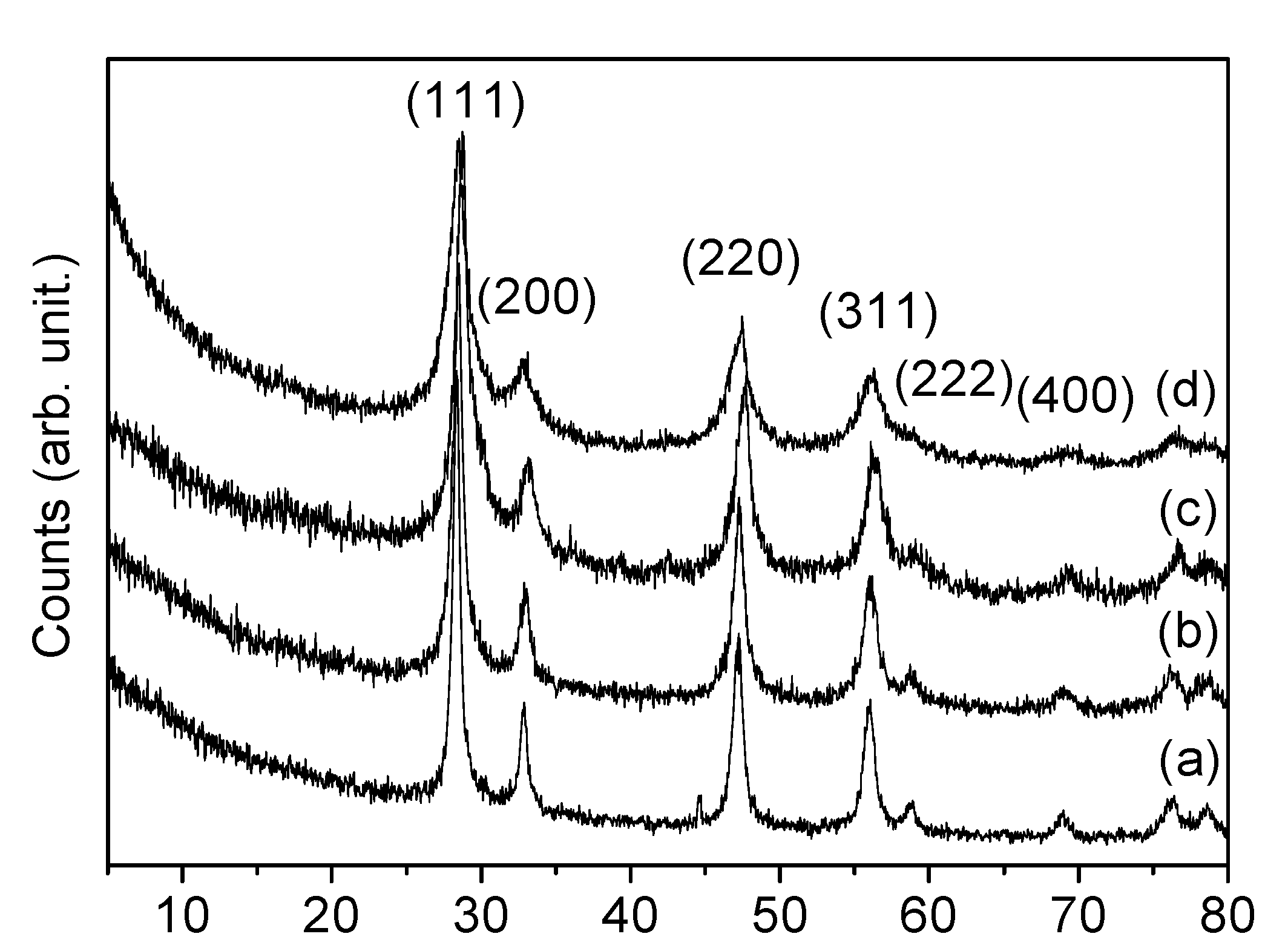
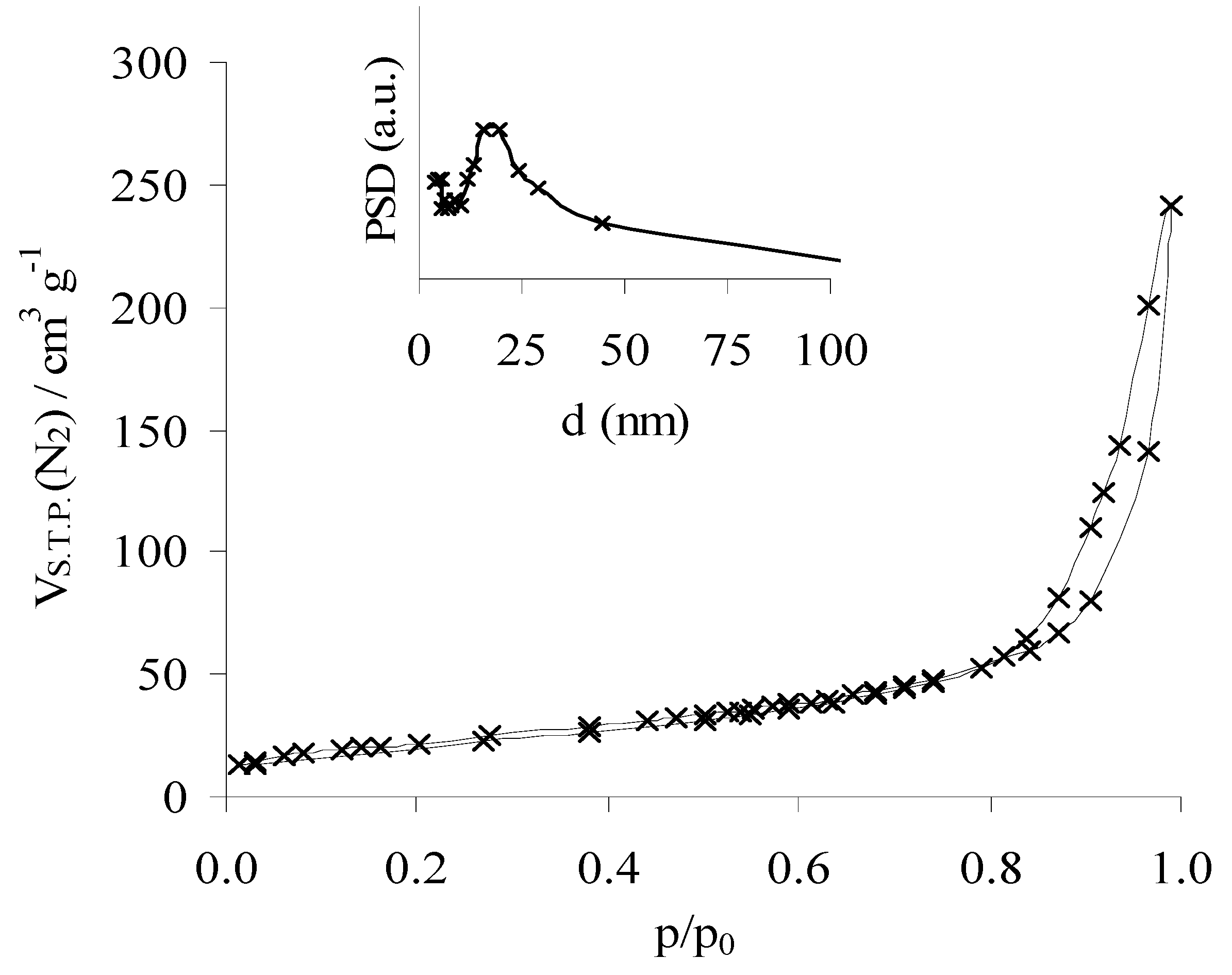
3.2. Structure and Texture Characterisation
3.3. Catalysis
3.4. UV/Vis Spectroscopy
3.5. Photoluminescence Spectroscopy
4. Conclusions
Acknowledgements
- Samples Availability: Samples of the compounds CeO2 nanorods are available from the authors (or from MDPI).
References and Notes
- Trovarelli, A.; Leitenburg, C.; Boaro, M.; Dolcetti, G. The utilization of ceria in industrial catalysis. Catal. Today 1999, 50, 353–367. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, K.; Wang, L.; Wang, B.; Li, Y. Oxygen vacancy clusters promoting reducibility and activity of ceria nanorods. J. Am. Chem. Soc. 2009, 131, 3140–3141. [Google Scholar] [CrossRef]
- Chen, J.; Patil, S.; Seal, S.; Mcginnis, J.F. Rare earth nanoparticles prevent retinal degeneration induced by intracellular peroxides. Nat. Nanotechnol. 2006, 1, 142–150. [Google Scholar] [CrossRef]
- Tang, C.; Band, Y.; Liu, B.; Golberg, D. Cerium oxide nanotubes prepared from cerium hydroxide nanotubes. Adv. Mater. 2005, 17, 3005–3009. [Google Scholar] [CrossRef]
- Pan, C.; Zhang, D.; Shi, L.; Jianhui, F. Template-free synthesis, controlled conversion, and CO oxidation properties of CeO2 nanorods, nanotubes, nanowires, and nanocubes. Eur. J. Inorg. Chem. 2008, 15, 2429–2436. [Google Scholar]
- Pan, C.; Zhang, D.; Shi, L. CTAB assisted hydrothermal synthesis, controlled conversion and CO oxidation properties of CeO2 nanoplates, nanotubes, and nanorods. J. Sol. Stat. Chem. 2008, 181, 1298–1306. [Google Scholar] [CrossRef]
- Chen, G.; Xu, C.; Song, X.; Zhao, W.; Ding, Y.; Sun, S. Interface reaction route to two different kinds of CeO2 nanotubes. Inorg. Chem. 2008, 47, 723–728. [Google Scholar]
- Zhou, K.; Yang, Z.; Yang, S. Highly Reducible CeO2 Nanotubes. Chem. Mater. 2007, 19, 1215–1217. [Google Scholar] [CrossRef]
- Zhang, D.; Fu, H.; Shi, L.; Fang, J.; Li, Q. Carbon nanotube assisted synthesis of CeO2 nanotubes. J. Sol. Stat.Chem. 2007, 180, 654–660. [Google Scholar] [CrossRef]
- Zhang, D.; Yana, T.; Panc, C.; Shia, L.; Zhang, J. Carbon nanotube-assisted synthesis and high catalytic activity of CeO2 hollow nanobeads. Mat. Chem. Phys. 2009, 113, 527–530. [Google Scholar] [CrossRef]
- Zhang, D.; Pan, C.; Shi, L.; Huang, L.; Fang, J.; Fu, H. A highly reactive catalyst for CO oxidation: CeO2 nanotubes synthesized using carbon nanotubes as removable templates. Microporous Mesoporous Mater. 2009, 117, 193–200. [Google Scholar] [CrossRef]
- Blasse, G.; Bril, A. Investigation of Some Ce3+-Activated Phosphors. J. Chem. Phys. 1967, 47, 5139–5145. [Google Scholar] [CrossRef]
- Zhou, H.P.; Zhang, Y.W.; Mai, H.X.; Sun, X.; Liu, Q.; Song, W.G.; Yan, C.H. Spontaneous organization of uniform CeO2 nanoflowers by 3D oriented attachment in hot surfactant solutions monitored with an in situ electrical conductance technique. Chem. Eur. J. 2008, 14, 3380–3390. [Google Scholar] [CrossRef]
- Kornblum, N.; DeLaMare, H.E. The base catalyzed decomposition of a dialkyl peroxide. J. Am. Chem. Soc. 1951, 73, 880–881. [Google Scholar] [CrossRef]
- Wang, Y.W.; Duh, Y.S.; Shua, C.M. Characterization of the self-reactive decomposition of tert-butyl hydroperoxide in three different diluents. Process. Saf. Prog. 2007, 26, 299–303. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Kochi, J.K. Metal-Catalyzed Oxidations of Organic Compounds; Academic Press: New York, NY, USA, 1981. [Google Scholar]
- Danoczy, E.; Paallukacs, J.; Gal, D. On the reactivity of alkoxy radicals - interaction between 1-phenylethoxy radical and ethylbenzene in the absence of oxygen. Ber. Bunsenges. Phys. Chem. 1993, 97, 554–558. [Google Scholar] [CrossRef]
- Ganin, E.; Amer, I. Selective cerium-catalyzed oxidation of alkyl benzenes to benzyl esters by bromate salts. J. Mol. Catal. A: Chem. 1997, 116, 323–327. [Google Scholar]
- Murugan, B.; Ramaswamy, A.V. Defect-site promoted surface reorganization in nanocrystalline ceria for the low-temperature activation of ethylbenzene. J. Am. Chem. Soc. 2007, 129, 3062–3063. [Google Scholar]
- Radhika, T.; Sugunan, S. Vanadia supported on ceria: Characterization and activity in liquid-phase oxidation of ethylbenzene. Catal. Commun. 2007, 8, 150–156. [Google Scholar] [CrossRef]
- Edwards, J.O.; Curci, R. Catalytic Oxidations with Hydrogen Peroxide as Oxidant; Strukul, G., Ed.; Kluwer Academic Press: Dordrecht, The Netherlands, 1992. [Google Scholar]
- Timofeeva, M.N.; Jhung, S.H.; Hwang, Y.K; Kim, D.K.; Panchenko, V.N.; Melgunov, M.S.; Chesalov, Y.A.; Chang, J.S. Ce-silica mesoporous SBA-15-type materials for oxidative catalysis: Synthesis, characterization, and catalytic application. Appl. Catal. A: Gen. 2007, 317, 1–10. [Google Scholar]
- Salavati-Niasari, M.; Salemi, P.; Davar, F. Oxidation of cyclohexene with tert-butylhydroperoxide and hydrogen peroxide catalysted by Cu(II), Ni(II), Co(II) and Mn(II) complexes of N,N′-bis-(α-methylsalicylidene)-2,2-dimethylpropane-1,3-diamine, supported on alumina. J. Mol. Catal. A: Chem. 2005, 238, 215–222. [Google Scholar]
- Koola, J. D.; Kochi, J.K. Cobalt-catalyzed epoxidation of olefins. Dual pathways for oxygen-atom transfer. J. Org. Chem. 1987, 52, 4545–4553. [Google Scholar]
- Salavati-Niasari, M.; Farzaneh, F.; Ghandi, M. Oxidation of cyclohexene with tert-butylhydroperoxide and hydrogen peroxide catalyzed by alumina-supported manganese(II) complexes. J. Mol. Catal. A: Chem. 2002, 186, 101–107. [Google Scholar]
- Tang, C.; Bando, Y.; Golberg, D.; Ma, R. Cerium Phosphate Nanotubes: Synthesis, Valence State, and Optical Properties. Angew. Chem.Int. Ed. 2005, 44, 576–579. [Google Scholar] [CrossRef]
- Marabelli, F.; Wachter, P. Covalent insulator CeO2: Optical reflectivity measurements. Phys. Rev. Biol 1987, 36, 1238–1243. [Google Scholar] [CrossRef]
- Alonso, M.D. H.; Hungrıa, A.B.; Árias, A.M.; Coronado, J.M.; Conesa, J.C.; Soria, J.; Garcia, M.F. Confinement effects in quasi-stoichiometric CeO2 nanoparticles. Phys. Chem. Chem. Phys. 2004, 6, 3524–3529. [Google Scholar]
- Tsunekawa, S.; Fukuda, T. Blue shift in ultraviolet absorption spectra of monodisperse CeO2–x nanoparticles. J. Appl. Phys. 2000, 87, 1318–1322. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Si, R.; Liao, C.S.; Yan, C.H.; Xiao, C.X.; Kou, Y. Facile alcohothermal synthesis, size-dependent ultraviolet absorption, and enhanced CO conversion activity of ceria nanocrystals. J. Phys. Chem. Biol. 2003, 107, 10159–10167. [Google Scholar] [CrossRef]
- Patsalas, P.; Logothetidis, S.; Sygellou, L.; Kennou, S.S. Structure-dependent electronic properties of nanocrystalline cerium oxide films. Phys. Rev. Biol. 2003, 68, 035104–035117. [Google Scholar]
- Imanaka, N.; Toshiyuki, H.; Adachi, G.Y. Amorphous cerium−titanium solid solution phosphate as a novel family of band gap tunable sunscreen materials. Chem. Mater. 2003, 15, 2289–2291. [Google Scholar] [CrossRef]
- Rocha, J.; Ferreira, P.; Carlos, L.D.; Ferreira, A. The first microporous framework cerium silicate. Angew. Chem. Int. Ed. 2000, 39, 3276–3279. [Google Scholar] [CrossRef]
- Kostova, M.H.; Ferreira, R.A.S.; Ananias, D.; Carlos, L.D.; Rocha, J. Photoluminescent layered Y(III) and Tb(III) silicates doped with Ce(III). J. Phys. Chem. Biol. 2006, 110, 15312–15316. [Google Scholar] [CrossRef]
- Sun, C.; Li, H.; Zhang, H.; Wang, Z; Chen, L. Controlled synthesis of CeO2 nanorods by a solvothermal method. Nanotechnology 2005, 16, 1454–1463. [Google Scholar] [CrossRef]
- Blasse, G.; Schipper, W.; Hamelink, J. On the quenching of the luminescence of the trivalent cerium ion. Inorg. Chim. Acta 1991, 189, 77–80. [Google Scholar] [CrossRef]
- Morshed, A. Violet/blue emission from epitaxial cerium oxide films on silicon substrates. Appl. Phys. Lett. 1997, 70, 1647–1649. [Google Scholar] [CrossRef]
- Chunlin, C.; Shayan, Y.; Zhikai, L.; Meiyong, L.; Nuofu, M. Violet/blue photoluminescence from CeO2 thin film. Chin. Sci. Bull. 2003, 48, 1198–1200. [Google Scholar]
- Nolan, M.; Parker, S.C.; Watson, G.W. The electronic structure of oxygen vacancy defects at the low index surfaces of ceria. Surf. Sci. 2003, 595, 223–232. [Google Scholar]
- Carlos, L.D.; Ferreira, R.A.S.; Rainho, J.P.; Bermudez, V.Z. Fine-tuning of the chromaticity of the emission color of organic-inorganic hybrids co-doped with EuIII, TbIII, and TmIII. Adv. Funct. Mater. 2002, 12, 819–823. [Google Scholar] [CrossRef]
- Cooke, D.W.; Bennett, B.L.; Muenchausen, R.E.; Lee, J.K.; Nastasi, M.A. Intrinsic ultraviolet luminescence from Lu2O3, Lu2SiO5 and Lu2SiO5:Ce3+. J. Luminescence 2004, 106, 125–132. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Macedo, A.G.; Fernandes, S.E.M.; Valente, A.A.; Ferreira, R.A.S.; Carlos, L.D.; Rocha, J. Catalytic Performance of Ceria Nanorods in Liquid-Phase Oxidations of Hydrocarbons with tert-Butyl Hydroperoxide. Molecules 2010, 15, 747-765. https://doi.org/10.3390/molecules15020747
Macedo AG, Fernandes SEM, Valente AA, Ferreira RAS, Carlos LD, Rocha J. Catalytic Performance of Ceria Nanorods in Liquid-Phase Oxidations of Hydrocarbons with tert-Butyl Hydroperoxide. Molecules. 2010; 15(2):747-765. https://doi.org/10.3390/molecules15020747
Chicago/Turabian StyleMacedo, Andreia G., Sílvia E. M. Fernandes, Anabela A. Valente, Rute. A. S. Ferreira, Luís D. Carlos, and João Rocha. 2010. "Catalytic Performance of Ceria Nanorods in Liquid-Phase Oxidations of Hydrocarbons with tert-Butyl Hydroperoxide" Molecules 15, no. 2: 747-765. https://doi.org/10.3390/molecules15020747