Synthesis and Characterization of Novel Organotin-Phosphorous Compounds II

Abstract
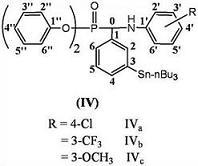
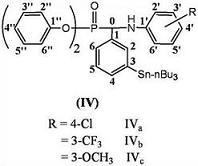
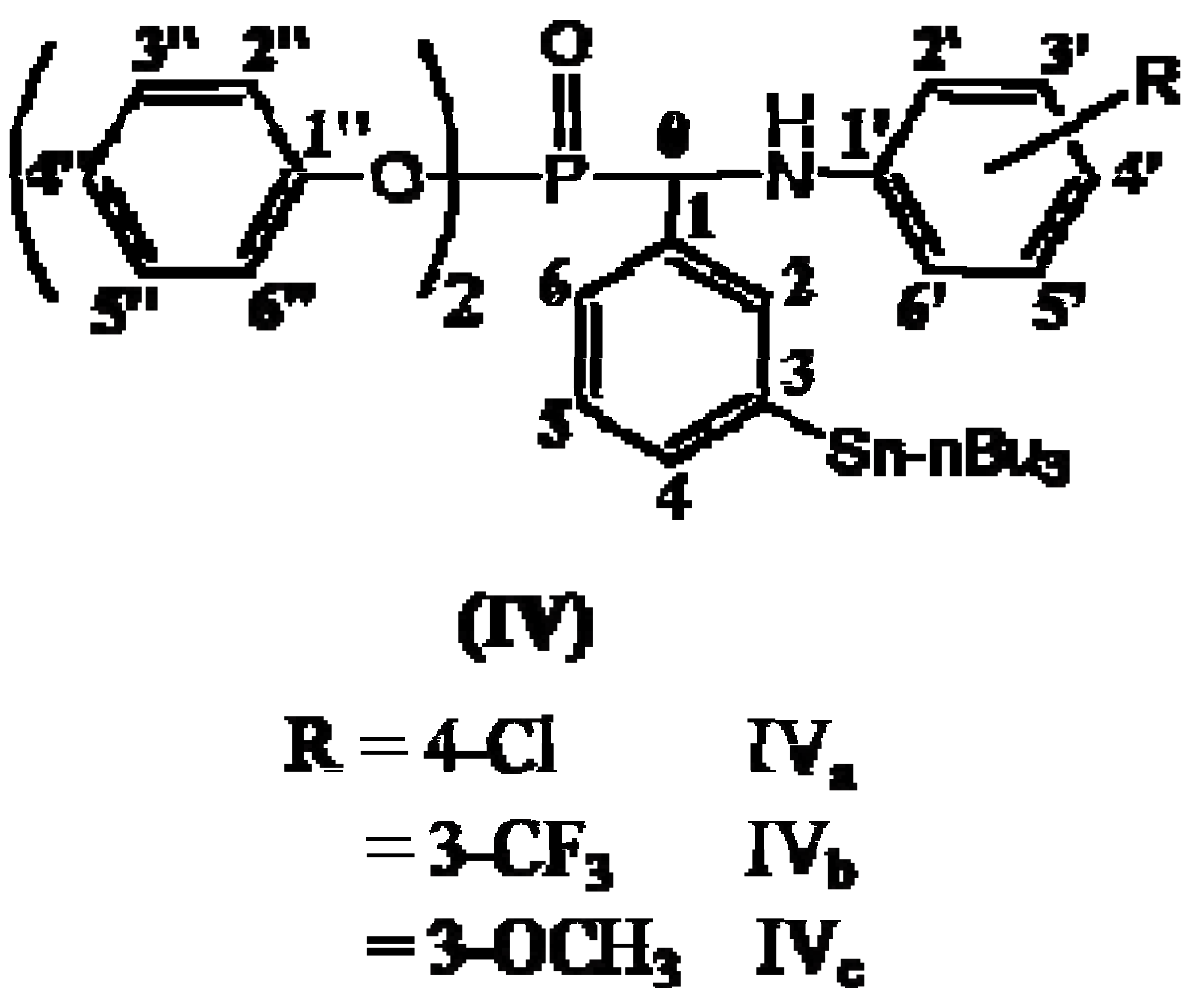
:
1. Introduction
2. Results and Discussion



{kind=link}
{kind=link}
{kind=link}
| Compound | R | Melting point (°C) | Yield (%) |
|---|---|---|---|
| IVa | 4-Cl | 81-82 | 87 |
| IVb | 3-CF3 | 87.5 | 58 |
| IVc | 3-OCH3 | 50-53 | 53 |
| Compound | Calculated | Measured | ||||
|---|---|---|---|---|---|---|
| %C | %H | %N | %C | %H | %N | |
| II | 57.43 | 8.26 | - | 57.63 | 8.33 | - |
| III | 61.09 | 8.72 | - | 61.18 | 8.64 | - |
| IVa | 60.14 | 6.41 | 1.90 | 60.02 | 6.37 | 1.93 |
| IVb | 59.09 | 6.13 | 1.81 | 58.79 | 6.21 | 1.87 |
| IVc | 62.14 | 6.86 | 1.91 | 62.21 | 6.93 | 1.94 |
2.1. 13C-NMR Spectra
| R group | δ (ppm) | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sn-CH2- | -CH2CH2- | -CH3 | C0 | C1 | C2 | C3 | C4 | C5 | C6 | ||||||||||
| 4-Cl | 29.0 | 27.8 | 9.6 | 59.2 | 136.0 | 136.5 | 146.8 | 143.3 | 128.2 | 129.5 | |||||||||
| 13.6 | 53.0 | ||||||||||||||||||
| 3-CF3 | 29.0 | 27.3 | 9.6 | 59.4 | 136.4 | 137.0 | 142.4 | 142.4 | 128.3 | 129.5 | |||||||||
| 13.6 | 53.2 | ||||||||||||||||||
| 3-OCH3 | 29.0 | 27.3 | 9.6 | 60.1 | 134.3 | 136.3 | 142.4 | 142.4 | 127.9 | 128.3 | |||||||||
| 13.5 | 54.0 | ||||||||||||||||||
| R group | δ (ppm) | ||||||||||||||||||
| C'1 | C'2 | C'3 | C'4 | C'5 | C'6 | ||||||||||||||
| 4-Cl | 147.4 | 113.9 | 135.0 | 118.1 | 129.7 | 112.2 | |||||||||||||
| 3-CF3 | 146.4 | 115.1 | 139.0 | 119.3 | 129.0 | 120.8 | |||||||||||||
| 3-OCH3 | 142.6 | 91.9 | 153.8 | 120.4 | 153.8 | 91.9 | |||||||||||||
| R group | δ (ppm) | ||||||||||||||||||
| C''1 | C''2,6a | C''3,5a | C''4 | ||||||||||||||||
| 4-Cl | 150.4 | 120.7 | 130.2 | 127.3 | |||||||||||||||
| 149.8 | 120.5 | 130.2 | |||||||||||||||||
| 3-CF3 | 150.5 | 120.6 | 129.5 | 127.9 | |||||||||||||||
| 150.1 | 120.4 | 129.5 | |||||||||||||||||
| 3-OCH3 | 150.5 | 120.7 | 130.9 | 127.8 | |||||||||||||||
| 150.4 | 120.6 | 130.9 | |||||||||||||||||
| Compound | δ (ppm) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sn-CH2- | -CH2CH2- | -CH3 | C0 | C1 | C2 | C3 | C4 | C5 | C6 | |||||
| Sn-(CH2-CH2-CH2-CH3)3 | 27.8 | 26.8 | 13.6 | - | - | - | - | - | - | - | ||||
| 18.0 | ||||||||||||||
| III | 29.1 | 27.4 | 9.7 | 192.2 | 135.6 | 137.7 | 143.6 | 142.6 | 128.4 | 129.5 | ||||
| 13.7 | ||||||||||||||
| Compound | δ (ppm) | |||||||||||||
| C'1 | C'2 | C'3 | C'4 | C'5 | C'6 | |||||||||
| 4-Chloroaniline | 147.7 | 114.8 | 134.7 | 118.2 | 130.3 | 113.2 | ||||||||
| 3-Trifluoromethaneaniline | 146.4 | 115.8 | 138.9 | 119.3 | 129.7 | 112.2 | ||||||||
| 3-Methoxyaniline | 143.3 | 98.8 | 152.9 | 119.0 | 152.9 | 98.8 | ||||||||
| Compound | δ (ppm) | |||||||||||||
| C''1 | C''2,6a | C''3,5a | C''4 | |||||||||||
| Diphenyl phosphite | 149.3 | 120.8 | 131.8 | 127.6 | ||||||||||
| 149.0 | 120.5 | 131.8 | ||||||||||||
2.2. 1H-NMR Spectra
| Compound | δ (ppm) | ||||
|---|---|---|---|---|---|
| 3(-(C H2)3CH3) | Ar-H | -O-CH-O- | -O(C H2)2O- | -C HO | |
| II | 1.2 (m, 27H) | 7.5 (m, 4H) | 5.8 (s, 1H) | 4.1 (d, 4H) | - |
| III | 1.2 (m, 27H) | 7.7 (m, 4H) | - | - | 10.3 (s, 1H) |
| Compound | δ (ppm) | ||||
| 3(-(CH2)3CH3) | -P-CH-N- | -NH- | Ar-H | ||
| IVa | 0.6-1.8 (m, 27H) | 4.7 (m, 1H) | 5.4 (d, 1H) | 6.5-6.7 (m, 18H) | |
| IVb | 0.6-1.7 (m, 27H) | 4.8-5.0 (d, 1H) | 5.3-5.4 (m, 1H) | 6.6-7.6 (m, 18H) | |
| IVc | 0.6-1.6 (m, 27H) | 3.8 (s, 1H) | 5.4 (d, 1H) | 6.25-7.6 (s, 18H) | |
2.3. FT-IR Spectra
| Compound | Wavenumber (cm-1) | |||||||
|---|---|---|---|---|---|---|---|---|
| -(CH2)3CH3 | Aromatic ring | P-O-Aryl | -P=O | C-O-C | -C=O | |||
| C-H stretching | C-H bending | C=C stretching | Stretching | |||||
| II | 2860, 2910, 2940 | 1350, 1370 | 1420, 1455 | - | - | 1080 | - | |
| III | 2840, 2910, 2950 | 1360 | 1450 | - | - | - | 1700 | |
| IVa | 2900, 2940 | 1290 | 1480 | 1180 | 1200 | - | - | |
| IVb | 2840, 2900, 2960 | 1340 | 1480 | 1100 | 1200 | - | - | |
| IVc | 2920, 2940 | 1300 | 1470 | 1040 | 1200 | - | - | |
3. Experimental
3.1. Instruments
3.2. Methods
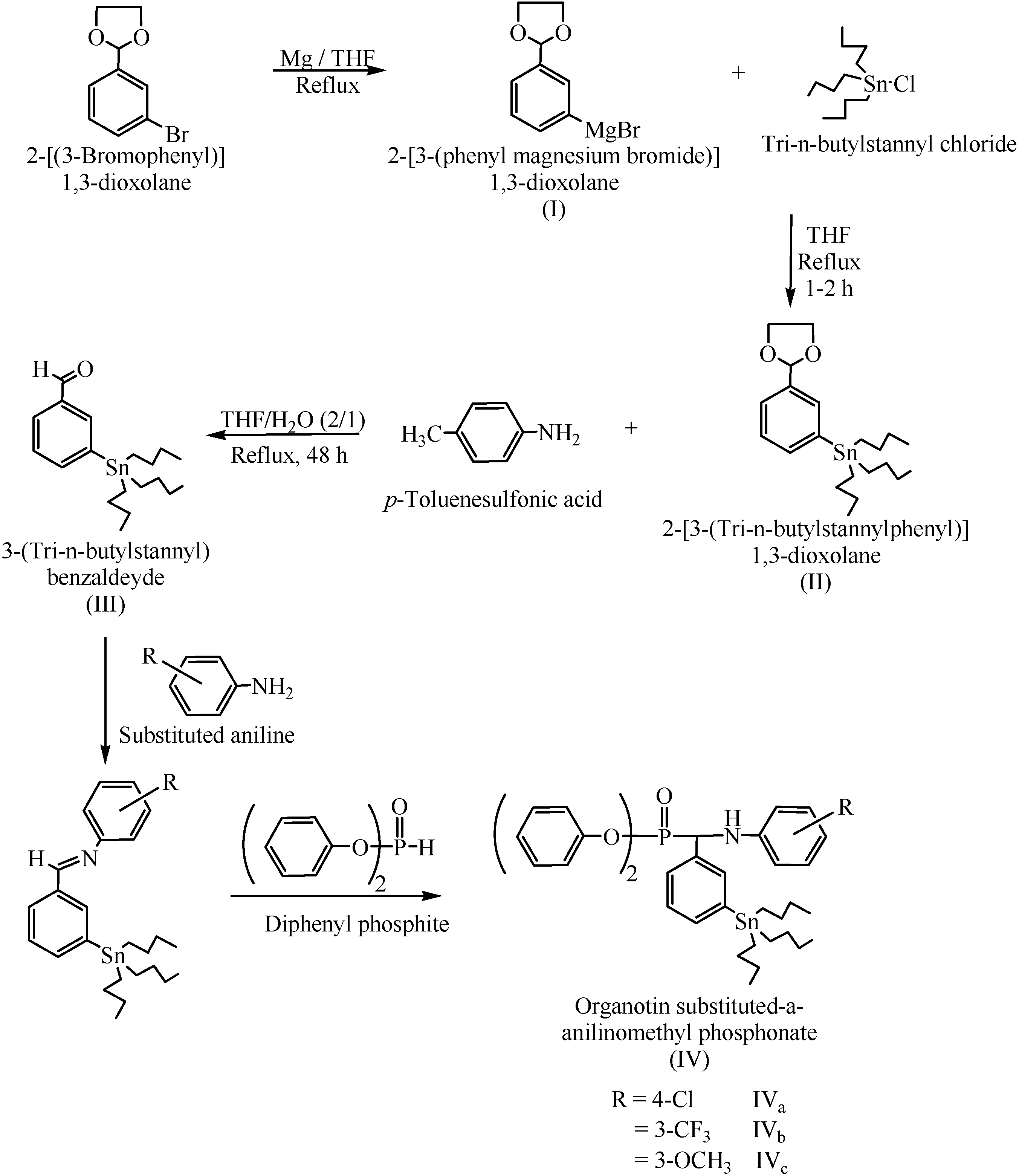
3.2.1. Preparation of 2-[3-(tri-n-butylstannyl)]1,3-dioxolane (II)
3.2.2. Preparation of 3-(tri-n-butylstannyl)-benzaldehyde (III)
3.2.3. Preparation of organotin substituted α-anilinomethyl phosphonates IV
4. Conclusions
References
- Chaudhary, A.; Agarwal, M.; Singh, R.V. Organotin(IV) and organolead(IV) complexes as biocides and fertility regulators: synthetic, spectroscopic and biological studies. Appl. Organomet. Chem. 2006, 20, 295–303. [Google Scholar]
- Michell, T.N. Organotin reagents in cross-coupling. In Metal Catalysis Cross-Coupling Reactions; Diederich, F., Stang, P.J., Eds.; Wiley-VCH: Weinheim, Germany, 1998; p. 157. [Google Scholar]
- Marton, D.; Russo, R.; Stivanello, D.; Tagliavini, G. Preparation of Benzylstannanes by Zinc-Mediated Coupling of Benzyl Bromides with Organotin Derivatives. Physicochemical Characterization and Crystal Structures. Organometallics 1996, 15, 1645–1650. [Google Scholar]
- Singh, M.S.; Tawade, K. Synthesis and characterization of some organotin(iv) complexes of α-benzoin oxime. Synth. React. Inorg. Met.-Org.Chem. 2001, 31, 157–165. [Google Scholar] [CrossRef]
- Ma, C.; Wang, Y.; Zhang, R. New organotin complexes with trans(cis)-1,4-cyclohexanedicarboxylic acid: Synthesis, characterization and crystal structures of mononuclears, 2D network polymers and a tetratin macrocycle. Inorg. Chim. Acta 2009, 362, 4137–4144. [Google Scholar] [CrossRef]
- Pellerito, L.; Nagy, L. Organotin(IV)n+ complexes formed with biologically active ligands: equilibrium and structural studies, and some biological aspects. Coord. Chem. Rev. 2002, 224, 111–150. [Google Scholar]
- Singh, R.; Kaushik, N.K. Spectral and thermal studies with anti-fungal aspects of some organotin(IV) complexes with nitrogen and sulphur donor ligands derived from 2-phenylethylamine. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2008, 71, 669–675. [Google Scholar] [CrossRef]
- Evans, C.J. Industrial uses of tin chemicals. In Chemistry of Tin, 2nd ed.; Smith, P.J., Ed.; Blackie Academic & Professional: Glasgow, UK, 1998; p. 388. [Google Scholar]
- Al-Diab, S.S. Synthesis and characterization of novel organotin-phosphorous compounds. Inorg. Chim. Acta 1989, 160, 93–97. [Google Scholar] [CrossRef]
- Al-Diab, S.S. Synthesis of Novel Organotin Copolymers. J. Chem. Res. 1986, 306–307. [Google Scholar]
- Coulson, D.R. Correlation analysis of carbon-13 and fluorine-19 NMR substituent effects in arylplatinum complexes. J. Am. Chem. Soc. 1976, 98, 3111–3119. [Google Scholar] [CrossRef]
- Lynch, B.M. Proportionality relationships in the carbon-13 nuclear magnetic resonance spectra of para-disubstituted benzenes: a new interpretation of non-additive behavior. Can. J. Chem. 1977, 55, 541–547. [Google Scholar] [CrossRef]
- Bonder, G.M.; Gaul, M.M. A carbon-13 NMR study of phosphine, phosphite, arsine and stibine ligands and their LNi(CO)3, LCr(CO)5 and η-(C5H5)Mn-(CO)2L complexes. J. Organomet. Chem. 1975, 101, 63–69. [Google Scholar] [CrossRef]
- Bullpitt, M.; Kitching, W.; Adcock, W.; Doddrell, D. Group IVb metalloidal substituent effects studied by carbon-13 nuclear magnetic resonance spectroscopy. J. Organomet. Chem. 1976, 116, 161–185. [Google Scholar]
- Al-Najjar, I.M.; Amin, H.B. The carbon-13 chemical shifts and the analysis of the relaxation times T1 and long range 13C–1H coupling constant of quinoline and of 1-(X-quinolyl)ethyl acetate derivatives. Spectrochim. Acta 1987, 43A, 1307–1315. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Deyab, S.S.; El-Newehy, M.H. Synthesis and Characterization of Novel Organotin-Phosphorous Compounds II. Molecules 2010, 15, 1425-1432. https://doi.org/10.3390/molecules15031425
Al-Deyab SS, El-Newehy MH. Synthesis and Characterization of Novel Organotin-Phosphorous Compounds II. Molecules. 2010; 15(3):1425-1432. https://doi.org/10.3390/molecules15031425
Chicago/Turabian StyleAl-Deyab, Salem S., and Mohamed H. El-Newehy. 2010. "Synthesis and Characterization of Novel Organotin-Phosphorous Compounds II" Molecules 15, no. 3: 1425-1432. https://doi.org/10.3390/molecules15031425
APA StyleAl-Deyab, S. S., & El-Newehy, M. H. (2010). Synthesis and Characterization of Novel Organotin-Phosphorous Compounds II. Molecules, 15(3), 1425-1432. https://doi.org/10.3390/molecules15031425