Synthesis of N-(Methoxycarbonylthienylmethyl)thioureas and Evaluation of Their Interaction with Inducible and Neuronal Nitric Oxide Synthase

Abstract
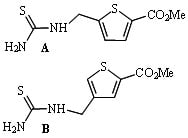
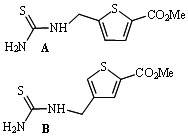
:
1. Introduction
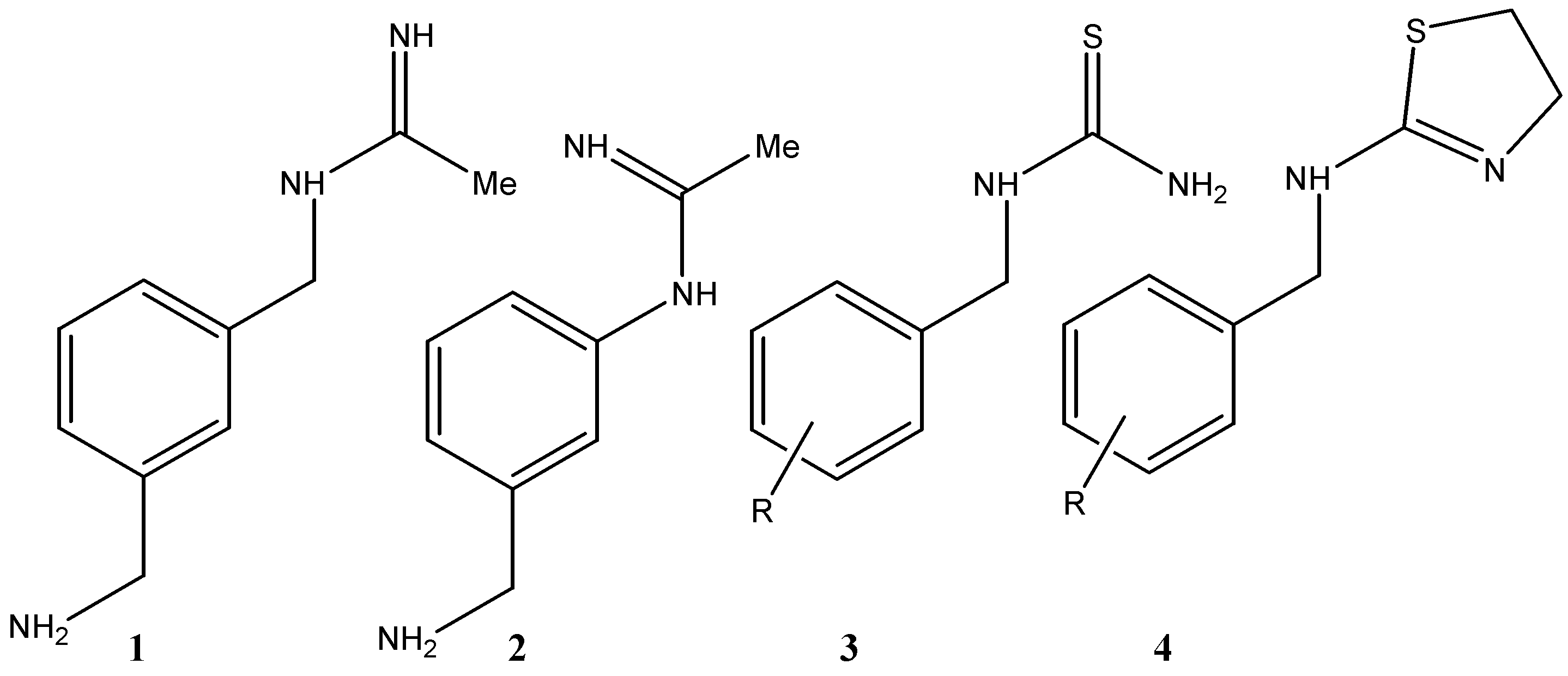
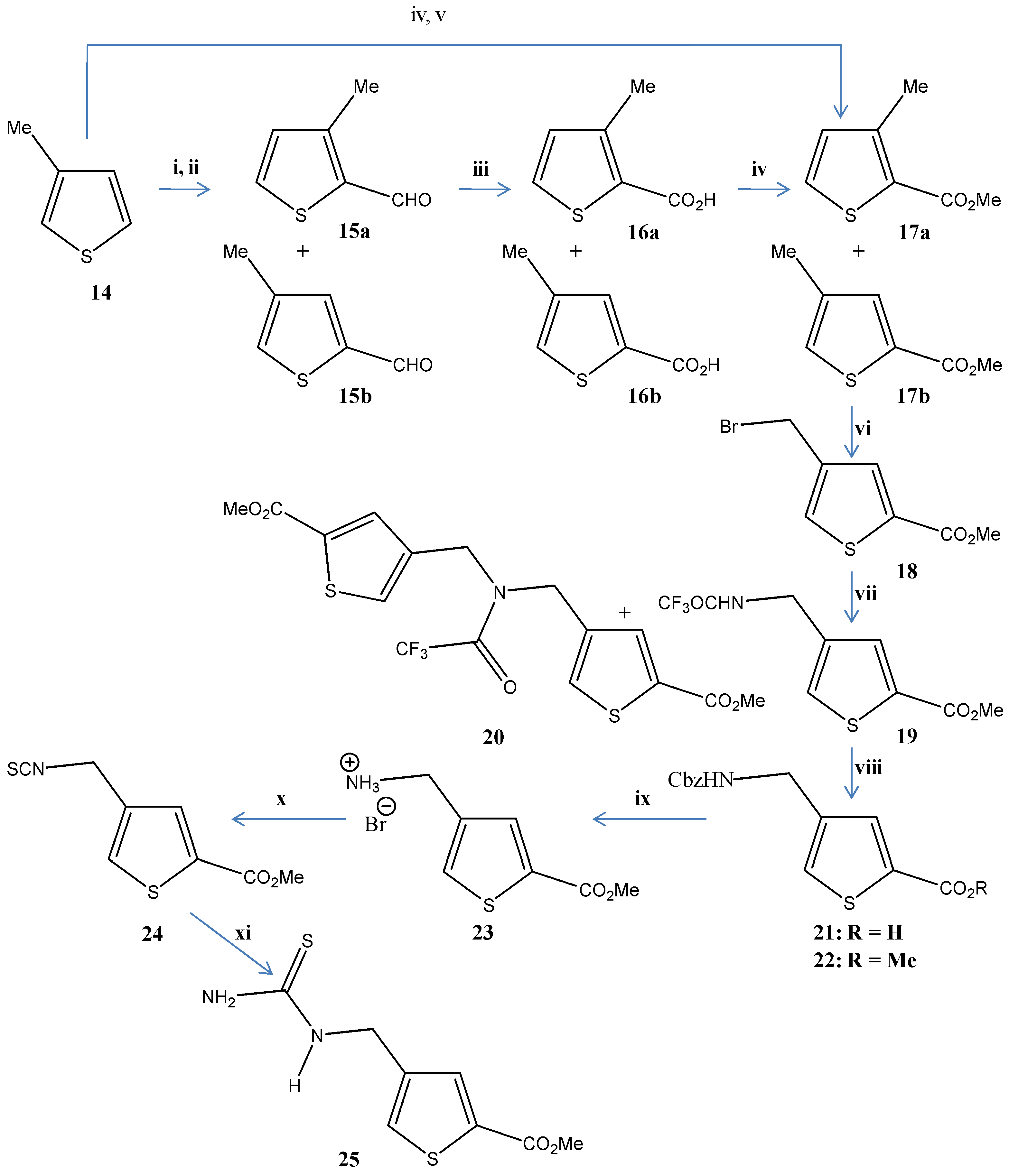
2. Results and Discussion
Biological Evaluation
3. Experimental
3.1. General
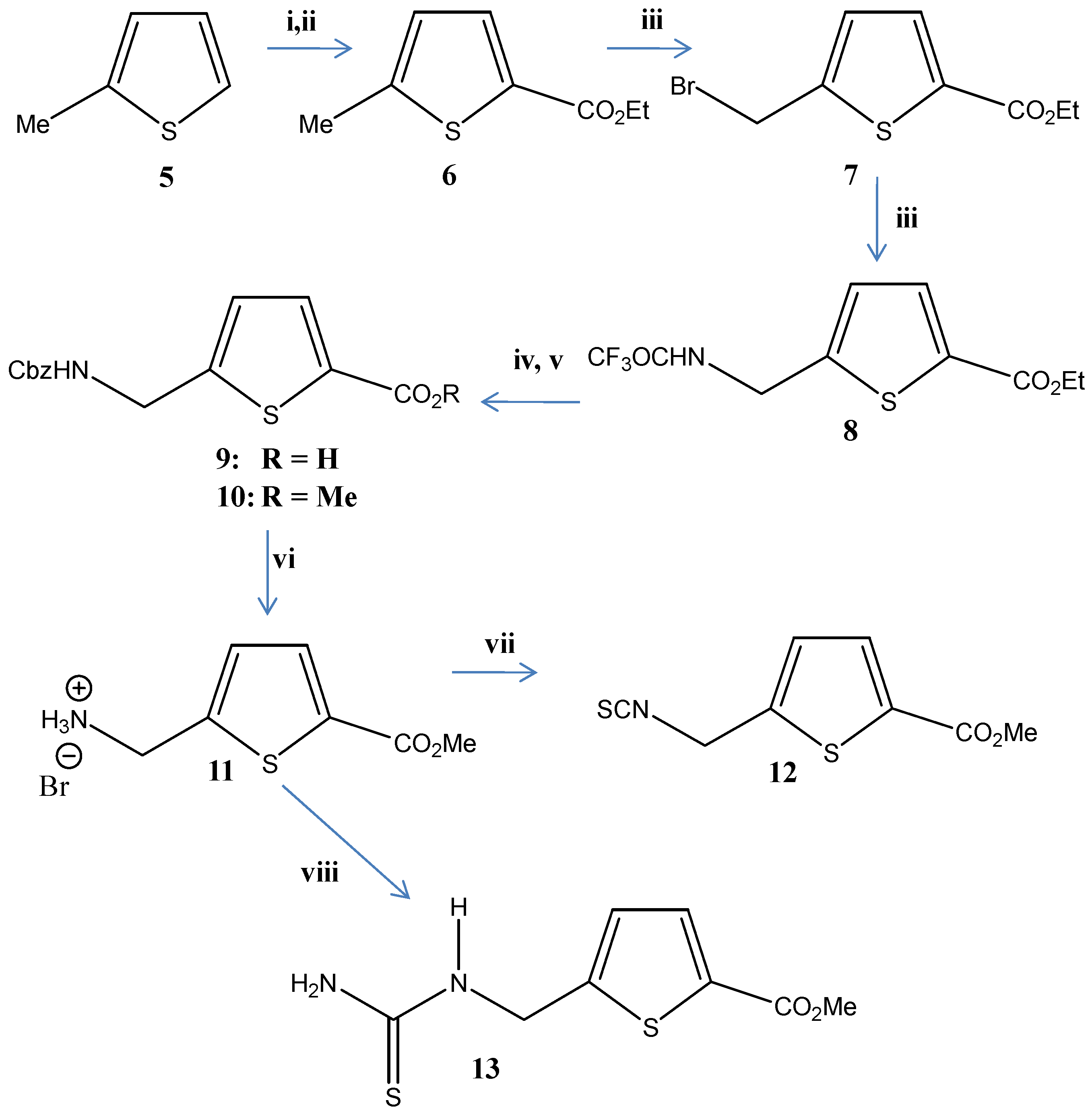
3.2. Chemistry
3.3. Enzyme study
4. Conclusions
Acknowledgements
References and Notes
- Napoli, C.; de Nigris, F.; Williams-Ignarro, S.; Pignalosa, O.; Sica, V.; Ignarro, L.J. Nitric Oxide and atherosclerosis: An update. Nitric Oxide 2006, 15, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Ignarro, L.J. Nitric oxide and atherosclerosis. Nitric Oxide 2001, 5, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Gatto, E.M.; Riobo, N.A.; Carreras, M.C.; Chernavsky, A.; Rubio, A.; Satz, M.L.; Poderoso, J.J. Overexpression of neutrophil neuronal nitric oxide synthase in Parkinson’s Disease. Nitric Oxide 2000, 4, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible nitric oxide synthase (iNOS) in tumor biology: The two sides of the same coin. Seminars Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, M.L.; Kwon, G.; Hill, J.R.; Marshall, C.A.; Corbett, J.A. Cytokines and nitric oxide in islet inflammation and diabetes. Proc. Soc. Exp. Biol. Med. 1996, 211, 24–32. [Google Scholar] [CrossRef] [PubMed]
- McCall, T.B.; Feelisch, M.; Palmer, R.M.J.; Moncada, S. Identification of N-iminoethyl-L-ornithine as an irreversible inhibitor of nitric oxide synthase in phagocytic cells. Br. J. Pharmacol. 1991, 102, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Kilbourn, R.G. Nitric Oxide Synthase inhibitors: Amino acids. Methods Enzymol. 1996, 268, 375–392. [Google Scholar] [PubMed]
- Tinker, A.C.; Beaton, H.C.; Boughton-Smith, N.; Cook, T.R.; Cooper, S.L.; Fraser-Rae, L.; Hallam, K.; Hamley, P.; McInally, T.; Nicholls, D.J.; Pimm, A.D.; Wallace, A.V. 1,2-Dihydro-4-quinazolinamines: Potent, highly selective inhibitors of inducible nitric oxide synthase which show antiinflammatory activity in vivo. J. Med. Chem. 2003, 46, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Webber, R.K.; Metz, S.; Moore, W.M.; Connor, J.R.; Currie, M.G.; Fok, K.F.; Hagen, T.J.; Hansen, D.W.; Jerome, G.M.; Manning, P.T.; Pitzele, B.S.; Toth, M.V.; Trivedi, M.; Zupec, M.E.; Tjoeng, F.S. Substituted 2-iminopiperidines as inhibitors of human nitric oxide synthase isoforms. J. Med. Chem. 1998, 41, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Garvey, E.P.; Oplinger, J.A.; Furfine, E.S.; Kiff, R.J.; Laszlo, F.; Whittle, B.J.R.; Knowles, R.G. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997, 272, 4959–4963. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.L.; Shearer, B.G.; Oplinger, J.A.; Lee, S.; Garvey, E.P.; Salter, M.; Duffy, C.; Burnette, T.C.; Furfine, E.S. N-Phenylamidines as selective inhibitors of human neuronal nitric oxide synthase: Structure-activity studies and demonstration of in vivo activity. J. Med. Chem. 1998, 41, 2858–2871. [Google Scholar] [CrossRef] [PubMed]
- Goodyer, C.L.M.; Chinje, E.C.; Jaffar, M.; Stratford, I.J.; Threadgill, M.D. Synthesis of N-benzyl- and N-phenyl-2-amino-4,5-dihydrothiazoles and thioureas and evaluation as modulators of the isoforms of nitric oxide synthase. Bioorg. Med. Chem. 2003, 11, 4189–4206. [Google Scholar] [CrossRef]
- Ulhaq, S.; Chinje, E.C.; Naylor, M.A.; Jaffar, M.; Stratford, I.J.; Threadgill, M.D. Heterocyclic analogues of L-citrulline as inhibitors of the isoforms of nitric oxide synthase (NOS) and identification of Nδ-(4,5-dihydrothiazol-2-yl)ornithine as a potent inhibitor. Bioorg. Med. Chem. 1999, 7, 1787–1796. [Google Scholar] [CrossRef]
- Goodyer, C.L.M.; Chinje, E.C.; Jaffar, M.; Stratford, I.J.; Threadgill, M.D. Time-dependence and preliminary SAR studies in inhibition of nitric oxide synthase isoforms by homologues of thiocitrulline. Bioorg. Med. Chem. Lett. 2003, 13, 3679–3680. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.; Roth, B. Radical ions. 49. Redox reactions of some thiophene derivatives. Phosphor. Sulfur Silicon 1983, 14, 211–224. [Google Scholar]
- Ulhaq, S.; Chinje, E.C.; Naylor, M.A.; Jaffar, M.; Stratford, I.J.; Threadgill, M.D. S-2-amino-5-azolylpentanoic acids related to L-ornithine as inhibitors of the isoforms of nitric oxide synthase (NOS). Bioorg. Med. Chem. 1998, 6, 2139–2149. [Google Scholar] [CrossRef]
- Beaton, H.; Boughton-Smith, N.; Hamley, P.; Ghelani, A.; Nicholls, D.J.; Tinker, A.C.; Wallace, A.V. Thienopyridines: Nitric oxide synthase inhibitors with potent in vivo activity. Bioorg. Med. Chem. Lett. 2001, 11, 1027–1030. [Google Scholar] [CrossRef]
- Ogden, J.E.; Moore, P.K. Inhibition of nitric oxide synthase potential for a novel class of therapeutic agent? Trends Biotechnol. 1995, 13, 70–78. [Google Scholar] [CrossRef]
- Mansuy, D.; Boucher, J.-L. Alternative nitric oxide-producing substrates for NO synthases. Free Radical Biol. Med. 2004, 37, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Lefévre-Groboillot, D.; Boucher, J.-L.; Mansuy, D.; Stuehr, D.J. Reactivity of the heme-dioxygen complex of the inducible nitric oxide synthase in the presence of alternative substrates. FEBS J. 2006, 273, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.B.; Lu, D.N.; Wang, P.G. N-Hydroxyguanidines as substrates of nitric oxide synthases. Curr. Topics Med. Chem. 2005, 5, 21–36. [Google Scholar] [CrossRef]
- Zhu, Y.; Nikolic, D.; van Breemen, R.B.; Silverman, R.B. Mechanism of Inactivation of Inducible Nitric Oxide Synthase by Amidine. Irreversible enzyme inactivation without inactivator modification. J. Am. Chem. Soc. 2005, 127, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Muzaffar, S.; Shukla, N.; Jeremy, J.Y. Nicotinamide adenine dinucleotide phosphate oxidase: A promiscuous therapeutic target for cardiovascular drugs? Trends Cardiovasc. Med. 2005, 15, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.; Jones, R.; Persad, R.; Angelini, G.D.; Jeremy, J.Y. Effect of sildenafil citrate and a nitric oxide donating sildenafil derivative, NCX 911, on cavernosal relaxation and superoxide formation in hypercholesterolaemic rabbits. Eur. J. Pharmacol. 2005, 517, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Muzaffar, S.; Shukla, N.; Srivastava, A.; Angelini, G.D.; Jeremy, J.Y. Sildenafil citrate and sildenafil nitrate (NCX 911) are potent inhibitors of superoxide formation and gp91(phox) expression in porcine pulmonary artery endothelial cells. Br. J. Pharmacol. 2005, 146, 109–117. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % Stimulation of hiNOS activity a | IC50 (µM) hiNOS | % Stimulation of nNOS activitya | ||
|---|---|---|---|---|---|
| t = 0 min b | t = 10 min b | t = 15 min b | t = 0 min b | t = 10 min b | |
| 1 | -79 ± 1 | -82 ± 1 | <4 | ND | ND |
| 3 (R = 3-NH2) | +58 ± 1 | +9 ± 1 | +1 ± 6 | -9 ± 6 | |
| 13 | +62 ± 2 | -4 ± 3 | -1 ± 4 | +3 ± 3 | |
| 25 | +37 ± 27 | -8 ± 11 | +6 ± 1 | +6 ± 7 | |
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suaifan, G.A.R.Y.; Goodyer, C.L.M.; Threadgill, M.D. Synthesis of N-(Methoxycarbonylthienylmethyl)thioureas and Evaluation of Their Interaction with Inducible and Neuronal Nitric Oxide Synthase. Molecules 2010, 15, 3121-3134. https://doi.org/10.3390/molecules15053121
Suaifan GARY, Goodyer CLM, Threadgill MD. Synthesis of N-(Methoxycarbonylthienylmethyl)thioureas and Evaluation of Their Interaction with Inducible and Neuronal Nitric Oxide Synthase. Molecules. 2010; 15(5):3121-3134. https://doi.org/10.3390/molecules15053121
Chicago/Turabian StyleSuaifan, Ghadeer A.R.Y., Claire L.M. Goodyer, and Michael D. Threadgill. 2010. "Synthesis of N-(Methoxycarbonylthienylmethyl)thioureas and Evaluation of Their Interaction with Inducible and Neuronal Nitric Oxide Synthase" Molecules 15, no. 5: 3121-3134. https://doi.org/10.3390/molecules15053121