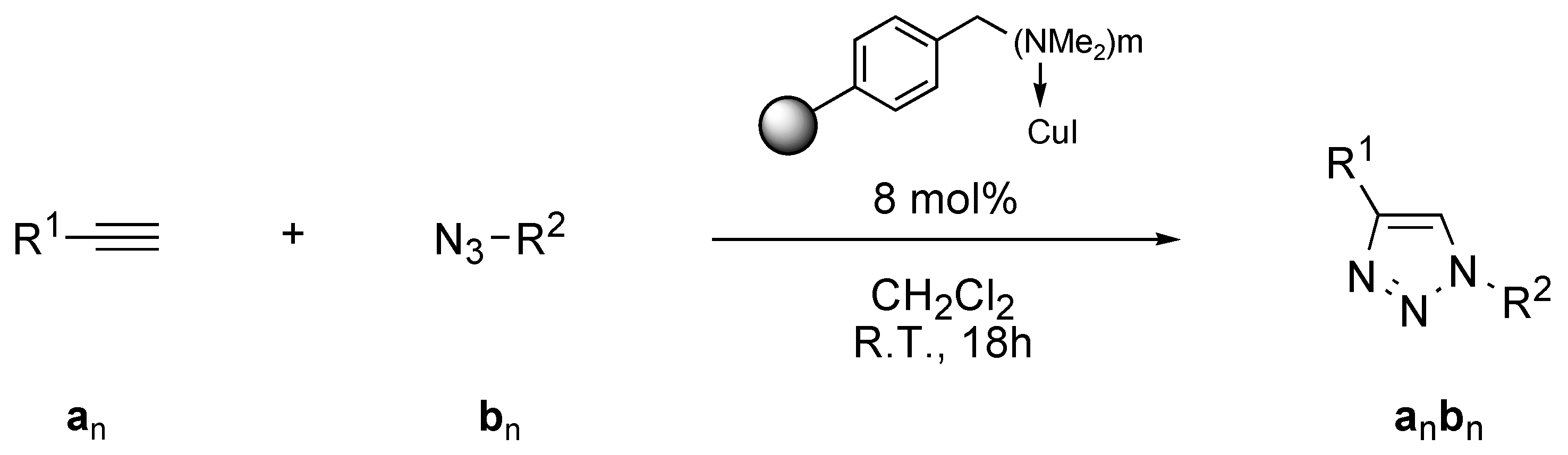
3.2. Procedures
Dry Amberlyst A-21 [
43]: Commercial wet Amberlyst A-21 resin (Aldrich, 20–50 mesh, 100 g) was suspended in MeOH (500 mL) for 0.5 h and filtered (3 times) and then soaked in methylene chloride (500 mL) for 0.5 h and again filtered (3 times). The resulting resin was placed in a round-bottom flask on a rotatory evaporator and dried at 50 ºC under 10 mm Hg until it was free flowing. The dried resin was then kept overnight
in vacuo in a desiccator over P
2O
5. Specifications from the manufacturer indicate that the polymer contains 4.8 mequiv of amine/g of dry resin.
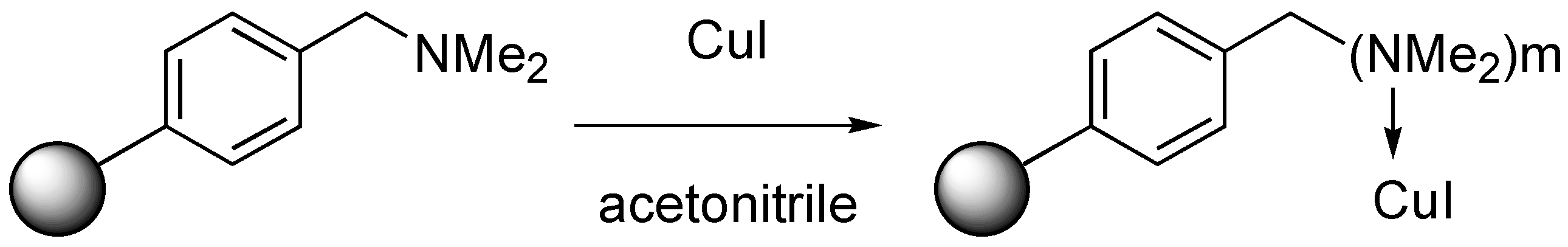
Preparation of the supported catalyst (A-21.CuI) [
43]: Dry Amberlyst A-21 (1.0 g, 4.8 mmol amine) was added to a solution of copper (I) iodide (381 mg, 2.00 mmol) in acetonitrile (15 mL) and gently shaken on an orbital stirrer for 17 h. The solvent was drawn off and the resin washed with CH
3CN (2 × 15 mL), CH
2Cl
2 (2 × 15 mL) and dried in vacuo (0.01 mm Hg) at 40 ºC. The weight increase was of 0,307 g (1.61 mmol CuI) that gave a polymer loading of 1.23 mmol CuI.g
−1. Elemental analyses (Service Central d’Analyses du CNRS, Solaize, France) gave a copper content of 8.64%, indicative of a loading of 1.35 mmol CuI.g
−1.
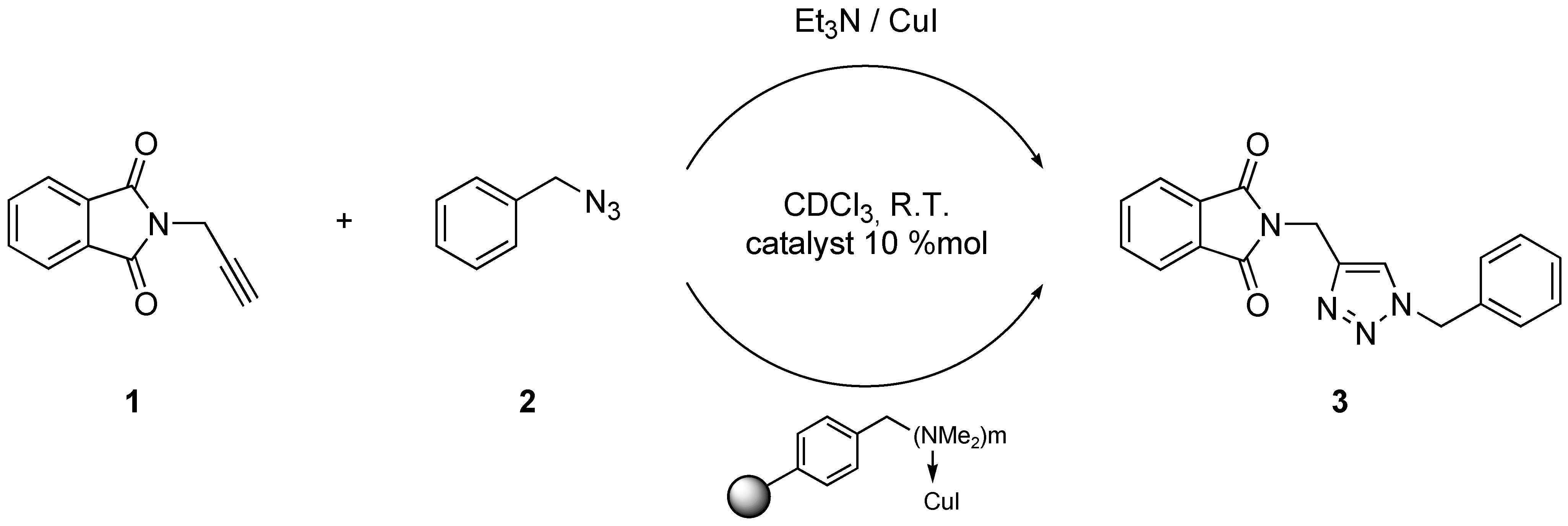
General procedure for automated synthesis of triazoles
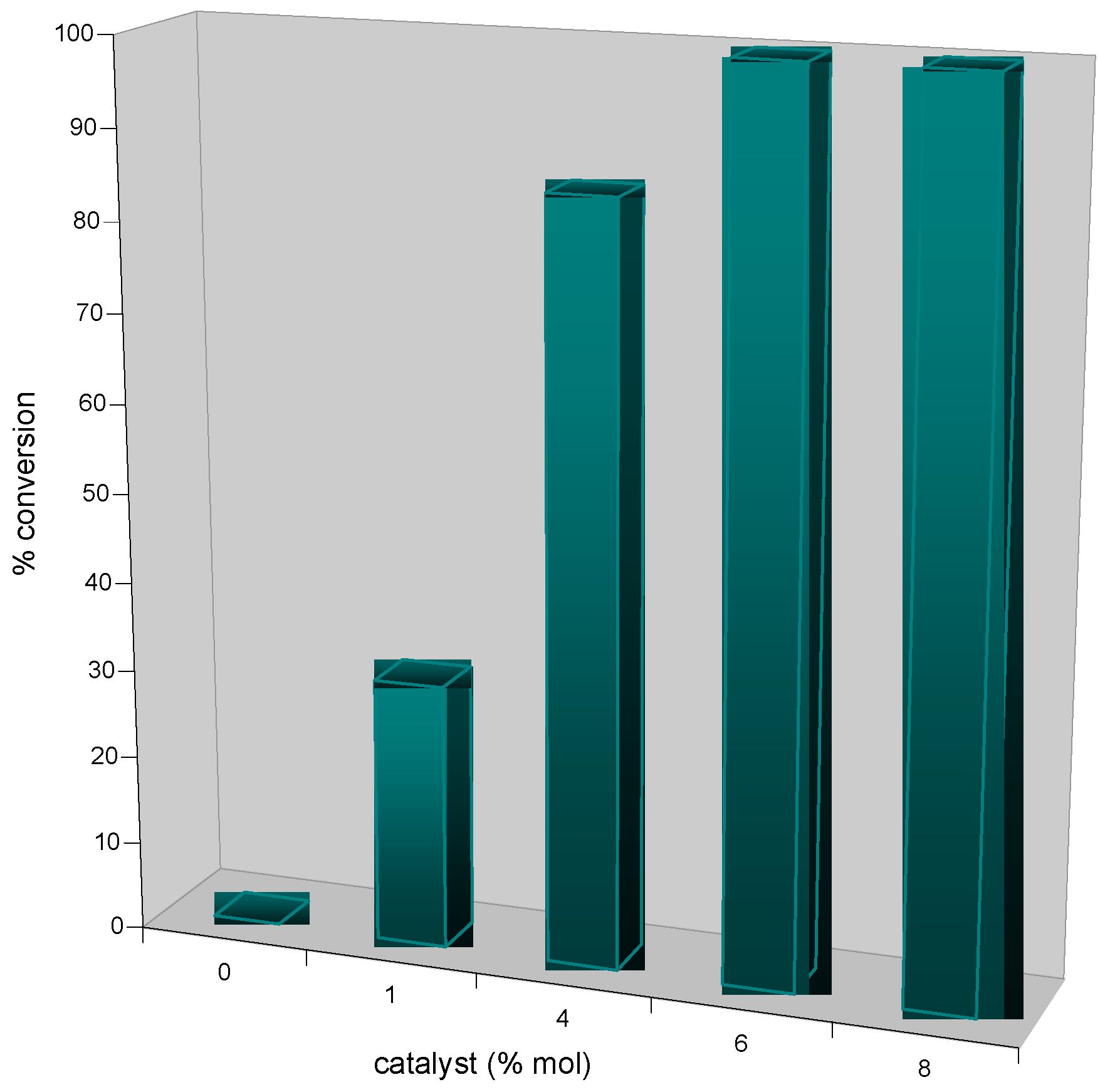
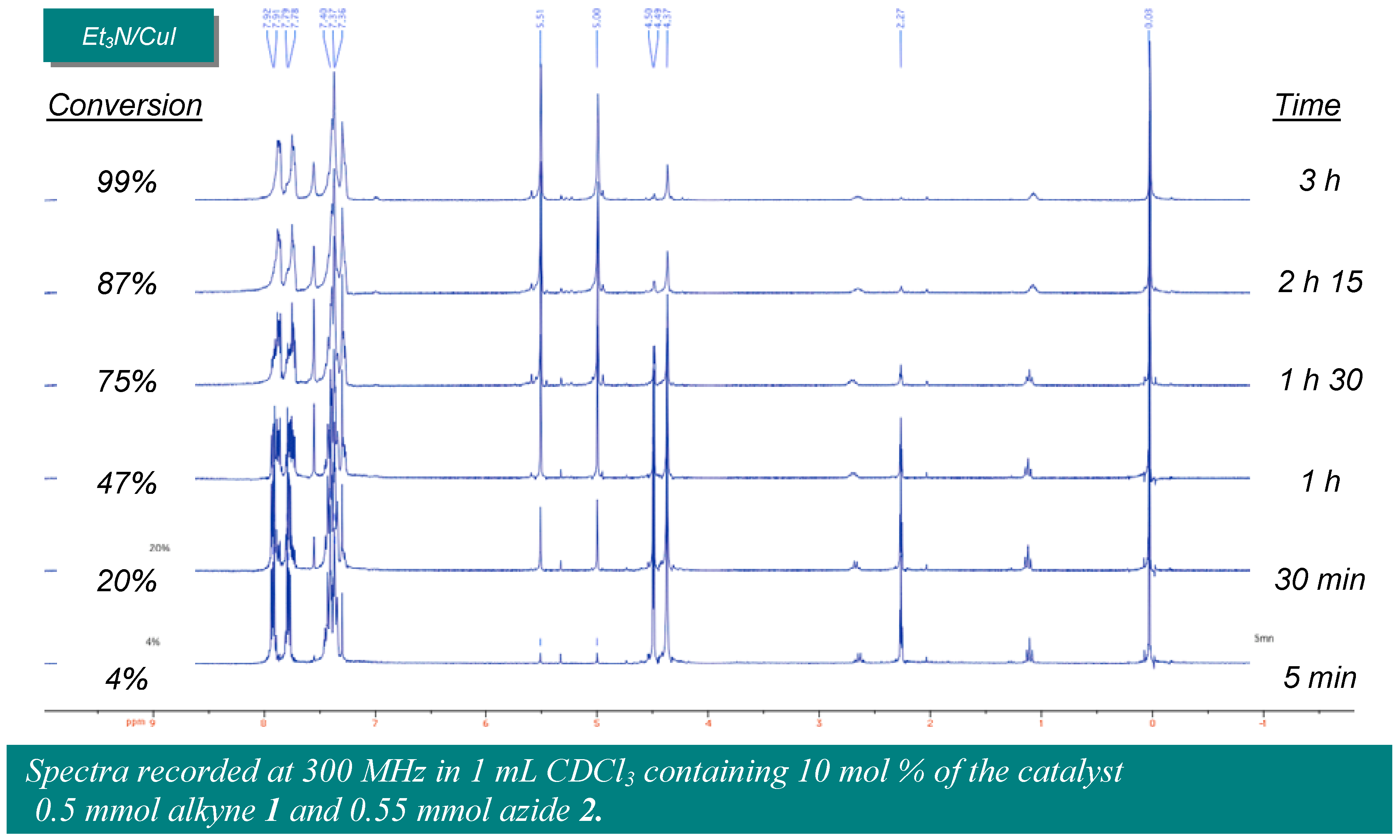
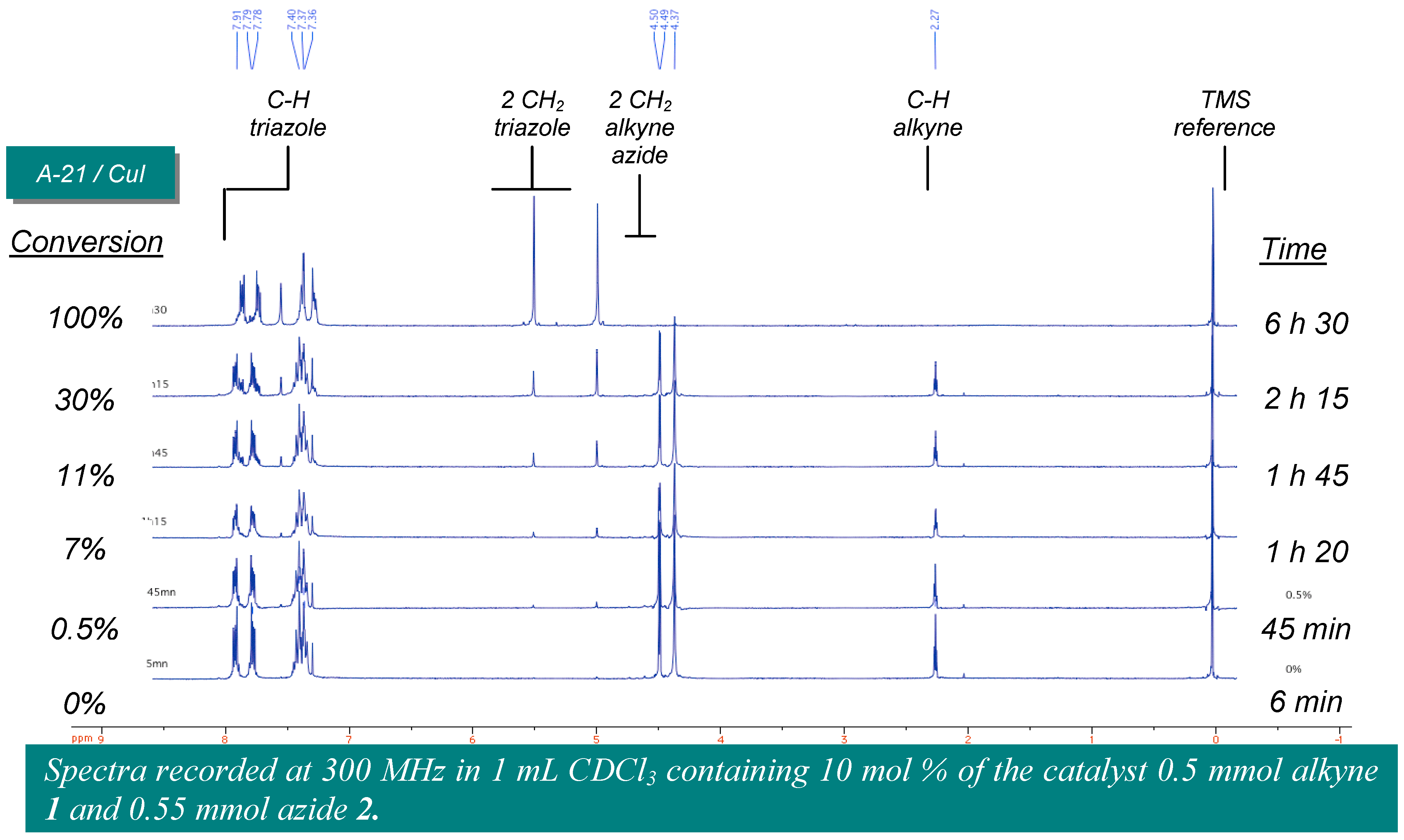
Amberlyst A-21·CuI (1.35 mmol/g, 30 mg, 0.040 mmol, 8 mol %) was placed in one of the Chemspeed ASW-2000 reactor equipped with a plunging filter, leaving the paired reactor empty. The azide (0.55 mmol) and alkyne (0.50 mmol), both dissolved in 1 mL dichloromethane were sequentially added at 1mL·min−1. The reactors were orbitally shaken at 600 rpm for 12 hours at room temperature. The product’s solution was separated from the catalyst by filtration followed by washing of the catalyst by dichloromethane (2 × 2.5 mL). The combined extracts were evaporated to obtain the product.
CAUTION! Organic azides are potentially explosive and should be handle with care. Even if no incident occurred in this reaction on this scale, the cycloaddition can be exothermic and should not be attempted on a larger scale, without being aware of explosion risks.
1-Benzyl-4-hydroxymethyl-1,2,3-triazole (a1b1). Prepared from 28 mg (0.50 mmol) of a1 and 73 mg (0.55 mmol) of b1. The product was obtained as a white solid (92 mg, 97%). C10H11N3O, M = 189.22 g.mol−1. m.p. 76–78 ºC. FTIR: ν 3257, 3144, 3091, 1451, 1040 cm−1. 1H-NMR (CDCl3): δ 4.47 (s, 1H), 4.70 (s, 2H), 5.46 (s, 2H), 7.17–7.45 (m, 5 H), 7.91 (s, 1H) ppm. 13C-NMR (CDCl3): δ 54.1, 56.0, 122.0, 128.1, 128.7, 129.1, 134.5, 148.0 ppm. LC-MS: ELSD pur. 97%, UV pur. 100%, Rt = 3.41 min, m/z: 190 ([M+H]+, 100%).
1-Ethoxycarbonylmethyl-4-hydroxymethyl-1,2,3-triazole (a1b2). Prepared from 28 mg (0.50 mmol) of a1 and 71 mg (0.55 mmol) of b2. The product was obtained as a pale yellow oil (92 mg, 99%). C7H11N3O3, M = 185.18 g.mol−1. FTIR: ν 3110, 3076, 3038, 2849, 1708 cm−1. 1H-NMR (CDCl3): δ 1.26 (t, J = 7.2 Hz, 3H), 4.21 (q, J = 7.2 Hz, 2H), 4.72 (s, 2H), 5.12 (s, 2H), 7.67 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.0, 50.8, 56.1, 62.4, 123.8, 148.3, 166.5 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%, Rt = 3.02 min, m/z: 186 ([M+H]+, 81%).
4-Hydroxymethyl-1-(3-hydroxypropyl)-1,2,3-triazole (a1b3). Prepared from 28 mg (0.50 mmol) of a1 and 56 mg (0.55 mmol) of b3. The product was obtained as a viscous colorless oil (37.7 mg, 48%). C6H11N3O2, M = 157.17 g.mol−1. FTIR: ν 3382, 3142, 2944, 2881, 1658, 1437, 1344, 1219, 1138, 1056 cm−1. 1H-NMR (CD3)2CO): δ 2.02 (q, J = 6.0 Hz, 2H), 3.24 (s, 1H), 3.49 (t, J = 6.0 Hz, 2H), 4.41 (t, J = 6.0 Hz, 2H), 4.63 (s, 2H), 7.66 (s, 1H) ppm. 13C-NMR (CD3)2CO): δ 33.8, 47.5, 56.5, 58.8, 122.8, 148.9 ppm. LC-MS: ELSD pur. 90%, UV pur. 100%, Rt = 2.67 min, m/z: 158 ([M+H]+, 100%).
1-(3-Trifluoroacetamidopropyl)-4-hydroxymethyl-1,2,3-triazole (a1b4). Prepared from 28 mg (0.50 mmol) of a1 and 108 mg (0.55 mmol) of b4. The product was obtained as a grey solid (53 mg, 42%). C8H11F3N4O2, M = 252.20 g.mol−1. m.p. 91 ºC. FTIR: ν 3306, 3219, 3061, 2946, 1721, 1576, 1467, 1181, 1069 cm−1. 1H-NMR (CD3)2SO): δ 2.04 (m, 2H), 3.20 (q, J = 6.0 Hz, 2H), 4.37 (t, J = 6.9 Hz, 2H), 4.50 (d, J = 5.4 Hz, 2H), 5.14 (t, J = 5.7 Hz, 1H), 7,98 (s, 1H), 8.49 (s, 1H) ppm. 13C-NMR (CD3)2SO): δ 28.9, 36.6, 38.6, 46.8, 55.0, 122.7 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%, Rt = 2.07 min, m/z: 253 ([M+H]+, 100%).
4-Hydroxymethyl-1-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl-1,2,3-triazole (a1b5). Prepared from 28 mg (0.50 mmol) of a1 and 86 mg (0.55 mmol) of b5. The product was obtained as a yellow oil (89 mg, 90%). C9H15N3O3, M = 213.24 g.mol−1. FTIR: ν 3379, 3146, 2986, 2936, 2881, 1647, 1452, 1379, 1223, 1149, 1060 cm−1. 1H-NMR (CDCl3): δ 1.32 (s, 3H), 1.37 (s, 3H), 3.73 (dd, J = 5.3, 8.7 Hz, 1H), 4.10 (dd, J = 6.1, 8.7 Hz, 1H), 4.36–4.56 (m, 3H), 4.74 (s, 2H), 7.73 (s, 1H) ppm. 13C-NMR (CDCl3); δ 25.2, 26.7, 52.5, 56.6, 66.5, 74.1, 110.3, 123.0, 147.6 ppm. LC-MS: ELSD pur. 95%, UV pur. 100%, Rt = 2.18 min, m/z: 214 ([M+H]+, 100%).
1-[(Cyclohex-3-en-1-yl)methyl]-4-hydroxymethyl-1,2,3-triazole (a1b6). Prepared from 28 mg (0.50 mmol) of a1 and 75 mg (0.55 mmol) of b6.The product was obtained as a brown oil (96,7 mg, 82%). C10H15N3O, M = 193.35 g.mol−1. FTIR: ν 3286, 3124, 3070, 3029, 2929, 2838, 1654, 1040, 1015 cm−1. 1H-NMR (CDCl3): δ 1.32 (m, 2H), 1.75 (m, 2H), 2.02 (m, 2H), 2.20 (m, 1H), 4.27 (d, J = 7.3 Hz, 2H), 4.78 (s, 2H), 5.60–5.72 (m, 2H), 7.58 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.2, 25.2, 28.9, 29.7, 34.8, 52.8, 55.5, 56.2, 122.5, 127.1, 147.9 ppm. LC-MS: ELSD pur. 80%, UV pur. 100%, Rt = 3.06 min, m/z: 194 ([M+H]+, 100%).
4-Hydroxymethyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a1b7). Prepared from 28 mg (0.50 mmol) of a1 and 84 mg (0.55 mmol) of b7. The product was obtained as a yellow oil (61 mg, 58%). C9H11N3OS, M = 209.27 g.mol−1. FTIR: ν 3341, 3147, 2928, 2862, 1548, 1433, 1214, 1045, 1008 cm−1. 1H-NMR (CDCl3): δ 3.42 (t, J = 7.1 Hz, 2H), 4.59 (t, J = 7.1 Hz, 2H), 4.73 (s, 2H), 6.74 (d, J = 3.3 Hz, 1H), 6.91 (dd, J = 3.3 and 5.0 Hz, 1H), 7.16 (d, J = 5.1 Hz, 1H), 7.47 (s, 1H) ppm. 13C-NMR (CDCl3) ; δ 30.8, 51.7, 56.1, 122.3, 124.7, 126.2, 127.3, 138.8, 147.8 ppm. LC-MS: ELSD pur. 95%, UV pur. 100%, Rt = 2.74 min, m/z: 210 ([M+H]+, 100%).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-hydroxymethyl-1,2,3-triazole (a1b8). Prepared from 28 mg (0.50 mmol) of a1 and 126 mg (0.55 mmol) of b8. The product was obtained as a yellow oil (115.5 mg, 81%). C12H19N3O5, M = 285.30 g.mol−1. FTIR: ν 3432, 2986, 2928, 2870, 1379, 1268, 1214, 1103 cm−1. 1H-NMR (CDCl3): δ 1.31 (s, 3H), 1.47 (s, 3H), 3.19 (dd, J = 3.4 and 6.3 Hz, 1H), 3.43 (dd, J = 3.0, 5.4 Hz, 1H), 3.45–3.53 (m, 3H), 4.09 (dd, J = 3.9, 10.7 Hz, 1H), 4.57 (dd, J = 3,0 and 14,2 Hz, 1H), 4.67–4.73 (m, 1H), 4.78–4.86 (m, 2H), 7.78 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.42, 25.93, 53.19, 69.63, 72.28, 80.77, 81.42, 81.94, 88.11, 112.68 ppm. LC-MS: ELSD pur. 95%, UV pur. 100%, Rt = 2.74 min, m/z: 210 ([M+H]+, 100%).
1-Benzyl-4-phenoxymethyl-1,2,3-triazole (a2b1). Prepared from 66 mg (0.50 mmol) of a2 and 73 mg (0.55 mmol) of b1. The product was obtained as a white solid (136 mg, 99%). C16H15N3O, M = 265.31 g.mol−1. m.p. 125 ºC. FTIR: ν 3132, 3016, 2970, 2920, 2866, 1588, 1488, 1239, 1222, 1052 cm−1. 1H-NMR (CDCl3): δ 5.18 (s, 2H), 5.51 (s, 2H), 6.90–7.39 (m,10H), 7.52 (s, 1H) ppm. 13C-NMR (CDCl3): δ 54.2, 62.0, 114.8, 121.3, 128.1, 128.8, 129.1, 129.5, 134.5, 144.6, 158.2 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 9.10 min; m/z: 266 ([M+H]+, 100%).
1-Ethoxycarbonylmethyl-4-phenoxymethyl-1,2,3-triazole (a2b2). Prepared from 66 mg (0.50 mmol) of a2 and 71 mg (0.55 mmol) of b2. The product was obtained as a beige solid (104 mg, 80%). C13H15N3O3, M = 261.28 g.mol−1. m.p. 116–118 ºC. FTIR: ν 3153, 2944, 2962, 2879, 1746, 1596, 1483, 1471, 1401, 1235, 1210, 1177, 1031 cm−1. 1H-NMR (CDCl3): δ 1.25 (t, J = 7.1 Hz, 3H), 4.23 (q, J = 6.8 Hz, 2H), 5.11 (s, 2H), 5.18 (s, 2H), 6.92–7.29 (m, 5H), 7.73 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.0, 50.8, 61.8, 62.4, 114.8, 121.2, 124.3, 129.5, 144.5, 158.2, 166.2 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 8.30 min; m/z: 262 ([M+H]+, 100%).
1-(3-Hydroxypropyl)-4-phenoxymethyl-1,2,3-triazole (a2b3): Prepared from 66 mg (0.50 mmol) of a2 and 56 mg (0.55 mmol) of b3. The product was obtained as a yellow oil (120 mg, 99%). C12H15N3O2, M = 233.27 g.mol−1. FTIR: ν 3304, 3132, 3107, 2945, 2870, 1600, 1488, 1239, 1218, 1052 cm−1. 1H-NMR (CDCl3): δ 2.10 (q, J = 6.1 Hz, 2H), 3.23 (s, 1H), 4.49 (t, J = 6.8 Hz, 2H), 5.16 (s, 2H), 6.94–6.97 (m, 3H), 7.24–7.30 (m, 2H), 7.66 (s, 1H) ppm. 13C-NMR (CDCl3): δ 32.6, 47.1, 58.5, 61.9, 114.7, 121.3, 123.2, 129.5, 144.1, 158.2 ppm. LC-MS: ELSD pur. 97%, UV pur. 100%; Rt = 3.54 min; m/z: 234 ([M+H]+, 100%).
1-(3-Trifluoroacetamidopropyl)-4-phenoxymethyl-1,2,3-triazole (a2b4). Prepared from 66 mg (0.50 mmol) of a2 and 108 mg (0.55 mmol) of b4. The product was obtained as a yellow solid (111 mg, 68%). C14H15F3N4O2, M = 382.30 g.mol−1. m.p. 79 ºC. FTIR: ν 3356, 3140, 3102, 2958, 2883, 1704, 1600, 1559, 1488, 1243, 1206, 1168 cm−1. 1H-NMR (CDCl3): δ 2.22 (m, 2H), 3.41 (q, J = 6.5 Hz, 2H), 4,42 (t, J = 6.5 Hz, 2H), 5.17 (s, 2H), 6.95–7.00 (m, 3H), 7.26–7.31 (m, 2H), 7.68 (s, 1H) ppm. 13C-NMR (CDCl3): δ 29.1, 37.0, 47.7, 61.7, 114.7, 117.7, 121.4, 123.3, 129.6, 144.5, 157.6, 158.1 ppm. LC-MS: ELSD pur. 98%, UV pur. 100%; Rt = 8.37 min; m/z: 329 ([M+H]+, 100%).
1-(2,2-Dimethyl-1,3-dioxolan-4-yl)methyl-4-phenoxymethyl-1,2,3-triazole (a2b5). Prepared from 66 mg (0.50 mmol) of a2 and 86 mg (0.55 mmol) of b5. The product was obtained as a yellow solid (145 mg, 99%). C14H17N3O3, M = 289.34 g.mol−1. m.p. 99 ºC. FTIR: ν 3132, 3082, 2987, 2929, 2870, 1604, 1492, 1380, 1226, 1035 cm−1. 1H-NMR (CDCl3): δ 1.34 (s, 6H), 3.73 (dd, J = 5.8, 8.9 Hz, 1H), 4.12 (dd, J = 6.2, 8.8 Hz, 1H), 5.22 (s, 2H), 6.97–7.00 (m, 3H), 7.26–7.32 (m, 2H), 7.76 (s, 1H) ppm. 13C-NMR (CDCl3): δ 25.2, 26.6, 52.4, 61.9, 66.4, 73.9, 110.2, 114.7, 121.2, 124.0, 129.6, 144.3, 158.2 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 8.38 min; m/z: 290 ([M+H]+, 100%).
1-[(Cyclohex-3-en-1-yl)methyl]-4-phenoxymethyl-1,2,3-triazole (a2b6). Prepared from 66 mg (0.50 mmol) of a2 and 75 mg (0.55 mmol) of b6. The product was obtained as a brown oil (140 mg, 99%). C16H19N3O, M = 269.35 g.mol−1. FTIR: ν 3136, 3095, 3028, 2945, 2916, 2841, 1654, 1604, 1501, 1256, 1040 cm−1. 1H-NMR (CDCl3): δ 1.25–1.39 (m, 1H), 1.66–1.84 (m, 2H), 1.98–2.28 (m, 4H), 4.29 (d, J = 7.3 Hz, 2H), 5.23 (s, 2H), 5.64–5.70 (m, 2H), 6.97-7.00 (m, 3H), 7.27–7.33 (m, 2H), 7.59 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.1, 25.8, 28.8, 34.7, 55.5, 62.1, 114.8, 121.2, 122.9, 124.8, 127.1, 129.5, 144.3, 158.2. LC-MS: ELSD pur. 90% ; Rt = 9.45 min; m/z: 270 ([M+H]+, 100%).
4-Phenoxymethyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a2b7): Prepared from 66 mg (0.50 mmol) of a2 and 84 mg (0.55 mmol) of b7. The product was obtained as a yellow solid (142 mg, 99%). C15H15N3OS, M = 285.37 g.mol−1. m.p. 98 ºC. FTIR: ν 3132, 3086, 2953, 2916, 2879, 1584, 1492, 1243, 1040 cm−1. 1H-NMR (CDCl3): δ 3,44 (t, J = 7.0 Hz, 2H), 4.61 (t, J = 7.0 Hz, 2H), 5.20 (s, 2H), 6.68–6.69 (m, 1H), 6.86–6.89 (m, 1H), 6.95–6.99 (m, 3H), 7.15–7.17 (m, 1H), 7.26–7.32 (m, 2H), 7.40 (s, 1H) ppm. 13C-NMR (CDCl3): δ 30.8, 51.7, 61.9, 114.8, 121.2, 123.0, 124.6, 126.2, 127.2, 129.5, 138.7, 158.1 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 9.00 min; m/z: 286 ([M+H]+, 100%).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-phenoxymethyl-1,2,3-triazole
(a2b8): Prepared from 66 mg (0.50 mmol) of a2 and 126 mg (0.55 mmol) of b8. The product was obtained as a yellow oil (172 mg, 95%). C18H23N3O5, M = 361.40 g.mol−1. FTIR: ν 3443, 2986, 2928, 2857, 1601, 1494, 1378, 1210, 1103, 1032 cm−1. 1H-NMR (CDCl3): δ 1.36 (s, 3H), 1.53 (s, 3H), 3.13 (dd, J = 3.6, 5 Hz, 1H), 3.40–3.54 (m, 3H), 4.07 (dd, J = 3.9 Hz, 10.7Hz, 1H), 4.57 (dd, J = 3.6 Hz, 14.2Hz, 1H), 4.66–4.73 (m, 1H), 4.77–4.85 (m, 2H), 5.24 (s, 2H), 7.00–7.05 (m, 3H), 7.30–7.37 (m, 2H), 7.87 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.3, 25.8, 52.5, 61.9, 69.2, 72.6, 80.6, 81.3, 81.5, 112.6, 114.8, 121.2, 124.8, 129.5, 144.0, 158.2 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 6.37 min; m/z: 362 ([M+H]+, 100%).
1-Benzyl-4-diethoxymethyl-1,2,3-triazole (a3b1). Prepared from 72 µL (0.50 mmol) of a3 and 73 mg (0.55 mmol) of b1. The product was obtained as a yellow solid (140 mg, 99%). C14H19N3O2, M = 261.33 g.mol−1. m.p. 60 ºC. FTIR: ν 3120, 3073, 2983, 2929, 2891, 1455, 1267, 1094, 1052 cm−1. 1H-NMR (CDCl3): δ 1.20 (t, J = 7.0 Hz, 6H), 3.62 (2q, J = 7.1 Hz, 4H), 5.50 (s, 2H), 5.68 (s, 1H), 7.25–7.35 (m, 5H), 7.49 (s, 1H) ppm. 13C-NMR (CDCl3): δ 15.1, 54.2, 61.6, 96.8, 121.8, 128.1, 128.7, 129.1, 134.5, 147.5 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 8.35 min; m/z: 234 ([M+H-(CH2CH2)]+, 90%), 284 ([M+Na]+, 10%).
1-Ethoxycarbonylmethyl-4-diethoxymethyl-1,2,3-triazole (a3b2). Prepared from 72 µL (0.50 mmol) of a3 and 71 mg (0.55 mmol) of b2. The product was obtained as a yellow oil (131 mg, 99%). C11H19N3O4, M = 257.29 g.mol−1. FTIR: ν 3136, 2987, 2883, 1750, 1218, 1064, 1048, 1023 cm−1. 1H-NMR (CDCl3): δ 1.12–1.22 (m, 9H), 3.57 (2q, J = 6.9 Hz, 4H), 4.51 (q, J = 7.2 Hz, 2H), 5.15 (s, 2H), 5.75 (s, 1H), 7.73 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.4, 15.5, 51.2, 61.8, 62.7, 97.0, 124.0, 148.1, 166.2 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 5.33 min; m/z: 230 ([M+H-(CH2CH2)]+, 80%), 280 ([M+Na]+, 20%).
4-Diethoxymethyl-1-(3-hydroxypropyl)-1,2,3-triazole (a3b3). Prepared from 72 µL (0.50 mmol) of a3 and 56 mg (0.55 mmol) of b3. The product was obtained as a yellow oil (119 mg, 99%). C10H19N3O3, M = 229.28 g.mol−1. FTIR: ν 3394, 2973, 2937, 2883, 1135, 1106, 1065 cm−1. 1H-NMR (CDCl3): δ 1.22 (t, J = 6.9 Hz, 6H), 2.12 (2q, J = 6.0 Hz, 2H), 2.74 (s, 1H), 3.56–3.70 (m, 6H), 4.51 (t, J = 6.0 Hz, 2H), 5.64 (s, 1H), 7.64 (s, 1H) ppm. 13C-NMR (CDCl3): δ 15.1, 32.6, 47.1, 58.4, 61.6, 96.8, 122.3, 147.0 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 2.35 min; m/z: 202 ([M+H-(CH2CH2)]+, 70%), 252 ([M+Na]+, 30%).
4-Diethoxymethyl-1-(3-trifluoroacetamidopropyl)-1,2,3-triazole (a3b4). Prepared from 72 µL (0.50 mmol) of a3 and 108 mg (0.55 mmol) of b4. The product was obtained as a yellow solid (182 mg, 99%). C12H19F3N4O3, M = 324.31 g.mol−1. m.p. 80 ºC. FTIR: ν 3219, 3078, 2974, 2937, 2895, 1717 1571, 1193, 1152, 1052 cm−1. 1H-NMR (CDCl3): δ 1.24 (t, J = 7.0 Hz, 6H), 2.24 (qn, J = 6.5 Hz, 2H), 3.44 (q, J = 6.4 Hz, 2H), 3.68 (2q, J = 7.4 Hz, 4H), 4.46 (t, J = 6.5 Hz, 2H), 5.69 (s, 1H), 7.65 (s, 1H) ppm. 13C-NMR (CDCl3): δ 15.1, 29.2, 37.1, 47.6, 61.8, 96.7, 113.9, 122.5, 147.0, 158.0 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 5.45 min; m/z: 297 ([M+H-(CH2CH2)]+, 60%),347 ([M+Na]+, 40%).
4-Diethoxymethyl-1-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl-1,2,3-triazole (a3b5). Prepared from 72 µL (0.50 mmol) of a3 and 86 mg (0.55 mmol) of b5. The product was obtained as a yellow oil (97 mg, 68%). C12H21N3O4, M = 285.35 g.mol−1. FTIR: ν 3136, 2987, 2937, 2879, 1372, 1226, 1052 cm−1. 1H-NMR (CDCl3): δ 1.27 (t, J = 7.0 Hz, 6H), 1.34 (s, 6H), 3.62–3.79 (m, 5H), 4.14 (dd, J = 6.2 Hz, 8.8 Hz, 1H), 4.45–4.59 (m, 3H), 5.75 (s, 1H), 7.77 (s, 1H) ppm. 13C-NMR (CDCl3): δ 15.1, 25.2, 26.6, 52.2, 61.8, 66.4, 74.0, 96.9, 110.2, 123.3, 147.2 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 5.32 min; m/z: 258 ([M+H-(CH2CH2)]+, 80%), 308 ([M+Na]+, 20%).
1-[(Cyclohex-3-en-1-yl)methyl]-4-diethoxymethyl-1,2,3-triazole (a3b6). Prepared from 72 µL (0.50 mmol) of a3 and 75 mg (0.55 mmol) of b6. The product was obtained as a yellow oil (126 mg, 95%). C14H23N3O2, M = 265.36 g.mol−1. FTIR: ν 3144, 2983, 2933, 1375, 1106, 1035 cm−1. 1H-NMR (CDCl3): δ 1.24 (t, J = 7.0 Hz, 6H), 1.64–1.84 (m, 3H), 1.97–2.26 (m, 4H), 3.65 (2q, J = 7.1 Hz, 4H), 4.26 (d, J = 7.3 Hz, 2H), 5.63–5.29 (m, 2H), 5.71 (s, 1H), 7.57 (s, 1H) ppm. 13C-NMR (CDCl3): δ 15.2, 24.1, 25.8, 28.9, 34.7, 55.4, 61.7, 96.9, 122.2, 124.8, 127.1, 147.3 ppm. LC-MS: ELSD pur. 100% ; Rt = 8.76 min; m/z: 238 ([M+H-(CH2CH2)]+, 100%)
4-Diethoxymethyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a3b7). Prepared from 72 µL (0.50 mmol) of a3 and 84 mg (0.55 mmol) of b7. The product was obtained as a yellow solid (101 mg, 72%). C13H19N3O2S, M = 281.38 g.mol−1. m.p. 53 ºC. FTIR: ν 3140, 3086, 2974, 2879, 1123, 1102, 1052 cm−1. 1H-NMR (CDCl3): δ 1.26 (t, J = 7.0 Hz, 6H), 3.47 (t, J = 7.1 Hz, 2H), 3.64 (2q, J = 7.1 Hz, 4H), 4.63 (t, J = 7.2 Hz, 2H), 5.73 (s, 1H), 6.76 (dd, J = 3.4, 0.7 Hz, 1H), 6.94 (dd, J = 3.4, 5.1 Hz, 1H), 7.21 (dd, J = 5.0, 1.1 Hz, 1H), 7.44 (s, 1H) ppm. 13C-NMR (CDCl3): δ 15.1, 30.8, 51.6, 61.5, 96.7, 122.3, 124.5, 126.1, 127.1, 138.7, 147.0 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 8.42 min; m/z: 254 ([M+H-(CH2CH2)]+, 80%), 304 ([M+Na]+, 20%).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-diethoxymethyl-1,2,3-triazole
(a3b8). Prepared from 72 µL (0.50 mmol) of a3 and 126 mg (0.55 mmol) of b8. The product was obtained as a yellow oil (172 mg, 95%). C16H27N3O6, M = 357.41 g.mol−1. FTIR: ν 3455, 2956, 2913, 1375, 1195, 1103, 1057 cm−1. 1H-NMR (CDCl3): δ 1.24 (t, J = 7.1 Hz, 6H), 1.32 (s, 3H), 1.50 (s, 3H), 3.15 (dd, J = 3.6, 5.7 Hz, 1H), 3.43–3.57 (m, 3H), 3.66 (2q, J = 7.5 Hz, 4H), 4.07 (dd, J = 3.1, 10.8 Hz, 1H), 4.56 (dd, J = 3.5, 14.1 Hz, 1H), 4.64–4.73 (m, 1H), 4.77–4.85 (m, 2H), 5.73 (s, 1H), 7.80 (s, 1H) ppm. 13C-NMR (CDCl3): δ 15.1, 24.4, 25.9, 52.4, 61.6, 69.2, 72.6, 80.6, 81.3, 81.5, 96.8, 112.6, 123.9, 146.9 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 3.27 min; m/z: 330 ([M+H-(CH2CH2)]+, 90%), 380 ([M+Na]+, 10%).
1-Benzyl-4-methoxycarbonylmethyl-1,2,3-triazole (a4b1). Prepared from 44 mg (0.50 mmol) of a4 and 73 mg (0.55 mmol) of b1. The product (a4b1) was obtained as a off-white solid (107 mg, 99%). C11H11N3O2, M = 217.23 g.mol−1. m.p. 116–118 ºC. FTIR: ν 3112, 3066, 2950, 1725, 1538, 1239, 1048 cm−1. 1H-NMR (CDCl3): δ 3.90 (s, 3H), 5.55 (s, 2H), 7.27–7.36 (m, 5H), 7.99 (s, 1H) ppm. 13C-NMR (CDCl3): δ 52.2, 54.5, 127.3, 128.3, 129.2, 129.3, 133.6, 140.3, 161.1 ppm. LC-MS: ELSD pur. 89%, UV pur. 100%, Rt = 8.88 min, m/z: 218 ([M+H]+, 100%)
1-Ethoxycarbonylmethyl-4-methoxycarbonylmethyl-1,2,3-triazole (a4b2). Prepared from 44 mg (0.50 mmol) of a4 and 71 mg (0.55 mmol) of b2.The product was obtained as a yellow solid (105 mg, 99%). C8H11N3O4, M = 213.19 g.mol−1. m.p. 104.5 ºC. FTIR: ν 3149, 3007, 2962, 1763, 1716, 1543, 1401, 1376, 1239, 1219, 1048, 1032 cm−1. 1H-NMR (CDCl3): δ 1.30 (t, J = 7.2 Hz, 3H), 3.95 (s, 3H), 4.28 (q, J = 7.2 Hz, 2H), 5.25 (s, 2H), 8.30 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.4, 51.4, 52.7, 63.2, 129.4, 161.4, 166.0 ppm. LC-MS: ELSD pur. 91%, UV pur. 100%, Rt = 3.68 min, m/z: 214 ([M+H]+, 100%).
1-(3-Hydroxypropyl)-4-methoxycarbonylmethyl-1,2,3-triazole (a4b3). Prepared from 44 mg (0.50 mmol) of a4 and 56 mg (0.55 mmol) of b3. The product was obtained as an orange solid (119 mg, 76%). C7H11N3O3, M = 185.18 g.mol−1. m.p. 52 ºC. FTIR: ν 3124, 2958, 2875, 1737, 1721, 1543, 1223, 1044 cm−1. 1H-NMR (CDCl3): δ 2.18 (q, J = 6.0 Hz, 2H), 3.67 (t, J = 6.0 Hz, 2H), 3.93 (s, 3H), 4.62 (t, J = 6.0 Hz, 2H), 8.22 (s, 1H) ppm. 13C-NMR (CDCl3): δ 32.4, 47.5, 52.2, 58.3, 128.2, 139.7, 161.2 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%, Rt = 2.98 min, m/z: 186 ([M+H]+, 100%).
1-(3-Trifluoroacetamidopropyl)-4-methoxycarbonylmethyl-1,2,3-triazole (a4b4). Prepared from 44 mg (0.50 mmol) of a4 and 108 mg (0.55 mmol) of b4. The product was obtained as an orange solid (125 mg, 89%). C9H11F3N4O3, M = 280.21 g.mol−1. m.p. 67 ºC. FTIR: ν 3290, 3137, 3095, 2962, 1563, 1219, 1194, 1160, 1048 cm−1. 1H-NMR (CDCl3): δ 1.90 (t, J = 6.5 Hz, 3H), 3.48 (q, J = 6.2 Hz, 2H), 3.98 (s, 2H), 4.54 (t, J = 6.6 Hz, 2H), 6.86 (s, 1H), 8.24 (s, 1H) ppm. 13C-NMR (CDCl3): δ 28.0, 37.7, 49.2, 50.6, 126.2, 128.1, 139.9, 161,1 ppm. LC-MS: ELSD pur. 61%, UV pur. 100%, Rt = 3.32 min, m/z: 181 ([M+H]+, 100%).
4-Methoxycarbonylmethyl-1-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl-1,2,3-triazole (a4b5). Prepared from 44 mg (0.50 mmol) of a4 and 86 mg (0.55 mmol) of b5. The product was obtained as a yellow solid (89 mg, 89%). C10H15N3O4, M = 241.25 g.mol−1. m.p. 103 ºC. FTIR: ν 3120, 2987, 2953, 2863, 1721, 1547, 1252, 1044 cm−1. 1H-NMR (CDCl3): δ 1.32 (s, 3H), 1.37 (s, 3H), 3.73 (dd, J = 5.3, 8.7 Hz, 1H), 4.10 (dd, J = 6.1, 8.7 Hz, 1H), 4.36–4.56 (m, 3H), 4.74 (s, 2H), 7.73 (s, 1H) ppm. 13C-NMR (CDCl3): δ 25.1, 26.6, 52.2, 53.1, 66.2, 73.7, 110.4, 128.8, 139.9, 161.1 ppm. LC-MS: ELSD pur. 95%, UV pur. 100%, Rt = 2.18 min, m/z: 214 ([M+H]+, 100%).
1-[(Cyclohex-3-en-1-yl)methyl]-4-methoxycarbonylmethyl-1,2,3-triazole (a4b6). Prepared from 44 mg (0.50 mmol) of a4 and 75 mg (0.55 mmol) of b6. The product was obtained as a grey solid (96,7 mg, 82%). C11H15N3O2, M = 221.26 g.mol−1. m.p. 94 ºC. FTIR: ν 3124, 3016, 2921, 2845, 1721, 1547, 1235, 1040 cm−1. 1H-NMR (CDCl3): δ 1.32 (m, 2H), 1.75 (m, 2H), 2.02 (m, 2H), 2.20 (m, 1H), 4.27 (d, J = 7.3 Hz, 2H), 4.78 (s, 2H), 5.60–5.72 (m, 2H), 7.58 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.0, 25.7, 28.7, 34.7, 52.2, 55.7, 124.6, 127.1, 161.3 ppm. LC-MS: ELSD pur. 80%, UV pur. 100%, Rt = 3.06 min, m/z: 194 ([M+H]+, 100%).
4-Methoxycarbonylmethyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a4b7). Prepared from 44 mg (0.50 mmol) of a4 and 84 mg (0.55 mmol) of b7. The product was obtained as a yellow solid (61 mg, 58%). C10H11N3O2S, M = 237.28 g.mol−1. m.p. 136.5 ºC. FTIR: ν 3095, 3037, 2953, 2921, 2850, 1729, 1534, 1231, 1053 cm−1. 1H-NMR (CDCl3): δ 3.42 (t, J = 7.1 Hz, 2H), 4.59 (t, J = 7.1 Hz, 2H), 4.73 (s, 2H), 6.74 (d, J = 3.3 Hz, 1H), 6.91 (dd, J = 3.3, 5.0 Hz, 1H), 7.16 (d, J = 5.1 Hz, 1H), 7.47 (s, 1H) ppm. 13C-NMR (CDCl3): δ 30.6, 52.1, 124.9, 126.4, 127.3, 127.9, 138.2, 139.7, 161.1 ppm. LC-MS: ELSD pur. 95%, UV pur. 100%, Rt = 2.74 min, m/z: 210 ([M+H]+, 100%).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-methoxycarbonylmethyl-1,2,3-triazole (a4b8). Prepared from 44 mg (0.50 mmol) of a4 and 126 mg (0.55 mmol) of b8. The product was obtained as a pale yellow oil (155,1 mg, 99%). C13H19N3O6, M = 313.31 g.mol−1. FTIR: ν 3445, 3130, 2979, 2854, 1736, 1542, 1437, 1275, 1208, 1099, 1068 cm−1. 1H-NMR (CDCl3): δ 1.33 (s, 3H), 1.50 (s, 3H), 3.13 (dd, J = 3.5, 5.5 Hz, 1H), 3.46 (dd, J = 3.5, 10.8 Hz, 1H), 3.94 (s, 3H), 4.07 (d, J = 10.8 Hz, 1H), 4.40–4.44 (m, 1H), 4.64 (dd, J = 3.4, 14.2 Hz, 1H), 4.74 (dd, J = 5.6, 14.2 Hz, 1H), 4.79–4.87 (m, 2H), 8.33 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.3, 25.9, 52.2, 52.9, 69.0, 72.7, 80.7, 81.3, 112.8, 129.5, 161.3 ppm. LC-MS: ELSD pur. 95%, UV pur. 100%, Rt = 2.74 min, m/z: 210 ([M+H]+, 100%).
1-Benzyl-4-phthalimidomethyl-1,2,3-triazole (a5b1). Prepared from 93 mg (0.50 mmol) of a5 and 73 mg (0.55 mmol) of b1. The product was obtained as a white solid (148 mg, 93%). C18H14N4O2, M = 318.34 g.mol−1. m.p. 179–181 ºC. FTIR: ν 3110, 3076, 3038, 2849, 1771, 1708, 1432, 1402, 1097 cm−1. 1H-NMR (CDCl3): δ 4.97 (s, 2H), 5.49 (s, 2H), 7.25–7.37 (m, 5H), 7.51 (s, 1H), 7.70–7.85 (m, 4H) ppm. 13C-NMR (CDCl3): δ 33.1, 54.2, 122.7, 123.4, 128.1, 128.7, 129.1, 132.0, 134.1, 134.5, 143.1, 167.6 ppm. LC-MS: ELSD pur. 98%, UV pur. 100%; Rt = 9.10 min; m/z: 319 ([M+H]+).
1-Ethoxycarbonylmethyl-4-phthalimidomethyl-1,2,3-triazole (a5b2). Prepared from 93 mg (0.50 mmol) of a5 and 71 mg (0.55 mmol) of b2. The product was obtained as a white solid (145 mg, 92%). C15H14N4O4. M =314.30 g.mol−1. m.p. 106–108 ºC. FTIR: ν 3122, 3066, 2998, 2955, 2888, 1755, 1701, 1446, 1402, 1210, 1102, 1052 cm−1. 1H-NMR (CDCl3): δ 1.27 (t, J = 7.2 Hz, 3H), 4.23 (q, J = 7.2 Hz, 2H), 4.99 (s, 2H), 5.11 (s, 2H), 7.70–7.82 (m, 5H) ppm. 13C-NMR (CDCl3): δ 14.0 5, 32.9, 50.8, 62.3, 123.4, 124.4, 131.9, 134.1, 143.0, 166.2, 167.6 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 5.63 min; m/z: 315 ([M+H]+).
1-(3-Hydroxypropyl)-4-phthalimidomethyl-1,2,3-triazole (a5b3). Prepared from 93 mg (0.50 mmol) of a5 and 56 mg (0.55 mmol) of b3. The product was obtained as a white solid (130 mg, 90%). C14H14N4O3, M = 286.29 g.mol−1. m.p. 108–109 ºC. FTIR: ν 3316, 3128, 2937, 2863, 1766, 1697, 1421, 1397, 1092, 1052 cm−1. 1H-NMR (CDCl3): δ 2.06 (q, J = 6.0 Hz, 2H), 3.61 (t, J = 6.0 Hz, 2H), 4.47 (t, J = 6.0 Hz, 2H), 4.96 (s, 2H), 7.68–7.71 (m, 2H), 7.81–7.83 (m, 2H) ppm. 13C-NMR (CDCl3): δ 32.5, 33.0, 46.9, 58.8, 123.2, 123.4, 132.0, 134.1, 167.7 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 8.23 min; m/z: 287 ([M+H]+).
1-(3-Trifluoroacetamidopropyl)-4-phthalimidomethyl-1,2,3-triazole (a5b4). Prepared from 93 mg (0.50 mmol) of a5 and 108 mg (0.55 mmol) of b4. The product was obtained as a white solid (107 mg, 57%). C16H14F3N5O3, M = 381.32 g.mol−1. m.p. 171–173 ºC. FTIR: ν 3350, 3150, 2094, 2961, 2872, 1766, 1712, 1559, 1402, 1427, 1219, 1186, 1141, 1048 cm−1. 1H-NMR (CDCl3): δ 2.18 (q, J = 6.0 Hz, 2H), 3.36 (t, J = 6.0 Hz, 2H), 4.41 (t, J = 6.0 Hz, 2H), 4.99 (s, 2H), 7.75–7.66 (m, 2H), 7.88–7.83 (m, 2H) ppm. 13C-NMR (CDCl3): δ 28.5, 32.6, 36.3, 47.0, 122.7, 131.7, 133.9, 167.4 ppm. LC-MS: ELSD pur. 91%, UV pur. 100%; Rt = 5.81 min; m/z: 382 ([M+H]+).
1-(2,2-Dimethyl-1,3-dioxolan-4-yl)methyl-4-phthalimidomethyl-1,2,3-triazole (a5b5). Prepared from 93 mg (0.50 mmol) of a5 and 86 mg (0.55 mmol) of b5. The product was obtained as a white solid (170 mg, 99%). C17H18N4O4, M = 342.36 g.mol−1. m.p.147–149 ºC. FTIR: ν 3124, 3074, 2981, 2941, 2873, 1771, 1706, 1431, 1402, 1102, 1072, 1043 cm−1. 1H-NMR (CDCl3): δ 1.33 (s, 3H), 1.34 (s, 3H), 3.73 (dd, J = 9.6 Hz, 1H), 4.11 (dd, J = 9.6 Hz, 1H), 4.54–4.39 (m, 3H), 5.00 (s, 2H), 7.75–7.71 (m, 2H), 7.76 (s, 1H), 7.86–7.83 (m, 2H) ppm. 13C-NMR (CDCl3): δ 25.6, 27.0, 33.4, 52.6, 66.8, 74.3, 110.5, 123.8, 124.5, 132.5, 134.4, 143.1, 167.9 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 5.38 min; m/z: 343 ([M+H]+).
1-[(Cyclohex-3-en-1-yl)methyl]-4-phthalimidomethyl-1,2,3-triazole (a5b6). Prepared from 93 mg (0.50 mmol) of a5 and 75 mg (0.55 mmol) of b6. The product was obtained as a beige solid (165 mg, 99%). C18H18N4O2, M =322.37 g.mol−1. m.p.135–137 ºC. FTIR: ν 3128, 3074, 3025, 2927, 2843, 1771, 1701, 1427, 1397, 1210, 1097 cm−1. 1H-NMR (CDCl3): δ 1.34–1.25 (m, 1H), 2.22–1.66 (m, 6H), 4.23 (d, J = 7.3 Hz, 2H), 5.04 (s, 2H), 5.70–5.65 (m, 2H) 7.62 (s, 1H), 7.77–7.74 (m, 2H), 7.91–7.88 (m, 2H) ppm. 13C-NMR (CDCl3): δ 24.1, 25.8, 28.9, 33.1, 34.6, 55.4, 123.4, 124.8, 127.0, 132.1, 134.1, 142.7, 167.6 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 8.60 min; m/z: 323 ([M+H]+).
4-Phthalimidomethyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a5b7). Prepared from 93 mg (0.50 mmol) of a5 and 84 mg (0.55 mmol) of b7. The product was obtained as a white solid (138 mg, 82%). C17H14N4O2S, M = 338.39 g.mol−1. m.p.149–151 ºC. FTIR: ν 3128, 3084, 2957, 2927, 1766, 1712, 1427, 1397, 1092 cm−1. 1H-NMR (CDCl3): δ 3.43 (t, J = 7 Hz, 2H), 4.60 (t, J = 7.1 Hz, 2H), 5.00 (s, 2H), 6.73 (dd, J = 3.4, 0.9 Hz, 1H), 6.91 (dd, J = 5.1, 3.5 Hz, 1H), 7.17 (dd, J = 5.1, 1.1 Hz, 1H), 7.46 (s, 1H), 7.78–7.76 (m, 2H), 7.90–7.88 (m, 2H) ppm. 13C-NMR (CDCl3): δ 31.1, 33.4, 52.0, 79.62, 123.7, 124.8, 126.5, 127.5, 132.5, 134.4, 139.1, 143.0, 167.9 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 8.20 min; m/z: 339 ([M+H]+).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-phthalimidomethyl-1,2,3-triazole (a5b8). Prepared from 93 mg (0.50 mmol) of a5 and 126 mg (0.55 mmol) of b8. The product was obtained as a white solid (205 mg, 99%). C20H22N4O6, M = 414.42 g.mol−1. m.p.189–191 ºC. FTIR: ν 3439, 3149, 2971, 2932, 2853, 1771, 1717, 1427, 1402, 1102, 1052 cm−1. 1H-NMR (CDCl3): δ 1.31 (s, 3H), 1.34 (s, 3H), 3.09–3.14 (m, 3H), 3.45 (dd, J = 10.7, 3.5 Hz, 1H), 4.07 (d, J = 10.7 Hz, 1H), 4.37–4.36 (m,1H), 4.54 (dd, J = 14.2, 3.5 Hz, 1H), 4.67 (dd, J = 14.4, 5.6 Hz, 1H), 4.83–4.75 (m, 2H), 4.99 (s, 1H) 7.73–7.70(m, 2H) 7.74 (s, 1H), 7.87–7.84 (m, 2H) ppm. 13C-NMR (CDCl3): δ 24.4, 25.9, 33.2, 52.5, 69.2, 72.7, 80.6, 81.4, 81.7, 112.6, 123.5, 124.7, 132.2, 134.1, 142,6, 167.9 ppm. LC-MS: ELSD pur. 97%, UV pur. 100%; Rt = 3.54 min; m/z: 415 ([M+H]+).
4-Acetamidomethyl-1-benzyl-1,2,3-triazole (a6b1). Prepared from 49 mg (0.50 mmol) of a6 and 73 mg (0.55 mmol) of b1. The product was obtained as a beige solid (96 mg, 83%). C12H14N4O, M = 230.27 g.mol−1. m.p. 118 ºC. FTIR: ν 3248, 3132, 3073, 2941, 1651, 1534, 1426, 1293 cm−1. 1H-NMR (CDCl3): δ 1.98 (s, 3H), 4.47 (d, J = 5.4 Hz, 2H), 5.49 (s, 2H), 5.51 (s, 2H), 6.18 (br s, 1H), 7.25–7.38 (m,15H), 7.47 (s, 1H) ppm. 13C-NMR (CDCl3): δ 23.1, 34.9, 54.3, 128.2, 128.9, 129.2, 134.4, 170.1 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 2.86 min; m/z: 231 ([M+H]+, 50%), 253 ([M+Na]+, 25%), 294 ([M+Cu]+, 25%).
4-Acetamidomethyl-1-ethoxycarbonylmethyl-1,2,3-triazole (a6b2). Prepared from 49 mg (0.50 mmol) of a6 and 71 mg (0.55 mmol) of b2. The product was obtained as a beige solid (83 mg, 73%). C9H14N4O3, M = 226.24 g.mol−1. m.p. 102 ºC. FTIR: ν 3252, 3140, 3066, 3003, 2970, 1746, 1671, 1550, 1218 cm−1. 1H-NMR (CDCl3): δ 1.31 (t, J = 7.2 Hz, 3H), 1.99 (s, 3H), 4.27 (q, J = 7.2 Hz, 2H), 4.52 (d, J = 5.4 Hz, 2H), 5.14 (s, 2H), 6.53 (br s, 1H), 7.70 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.0, 23.1, 34.9, 50.9, 362.5, 123.9, 166.2, 170.2 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 2.23 min; m/z: 227 ([M+H]+, 70%), 249 ([M+Na]+, 30%).
4-Acetamidomethyl-1-(3-hydroxypropyl)-1,2,3-triazole (a6b3). Prepared from 49 mg (0.50 mmol) of a6 and 56 mg (0.55 mmol) of b3. The product was obtained as a yellow oil (42 mg, 47%). C8H14N4O2, M = 198.23 g.mol−1. FTIR: ν 3283, 3089, 2924, 2875, 1634, 1552, 1440, 1288, 1052 cm−1. 1H-NMR (CDCl3): δ 1.99 (s, 3H), 2.08–2.16 (m, 2H), 3.63 (t, J = 5.4 Hz, 2H), 4.48–4.53 (m, 4H), 6.84 (br s, 1H), 7.67 (s, 1H) ppm. 13C-NMR (CDCl3): δ 23.1, 32.5, 34.8, 47.1, 58.5, 123.1, 170.5 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 1.67 min; m/z: 199 ([M+H]+, 75%), 221 ([M+Na]+, 25%).
4-Acetamidomethyl-1-(3-trifluoroacetamidopropyl)-1,2,3-triazole (a6b4). Prepared from 49 mg (0.50 mmol) of a6 and 108 mg (0.55 mmol) of b4. The product was obtained as a beige solid (107 mg, 73%). C10H14F3N5O2, M = 293.25 g.mol−1. m.p. 135 ºC. FTIR: ν 3277, 3232, 3086, 2953, 1728, 1650, 1559, 1202, 1148 cm−1. 1H-NMR (CDCl3): δ 1.87 (s, 3H), 2.04–2.11 (m, 2H), 3.20–3.24 (m, 2H), 4,30–4.35 (m, 4H), 7.79 (s, 1H) ppm. 13C-NMR (CDCl3): δ 22.4, 30.4, 35.7, 37.9 123.6, 173.3 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 2.22 min; m/z: 294 ([M+H]+, 60%), 316 ([M+Na]+, 40%).
4-Acetamidomethyl-1-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl-1,2,3-triazole (a6b5). Prepared from 49 mg (0.50 mmol) of a6 and 86 mg (0.55 mmol) of b5. The product was obtained as a yellow solid (110 mg, 87%). C10H16N4O3, M = 240.26 g.mol−1. m.p. 87 ºC. FTIR: ν 3269, 3078, 2999, 2929, 1629, 1546, 1375, 1210, 1056 cm−1. 1H-NMR (CDCl3): δ 1.34 (s, 3H), 1.38 (s, 3H), 3.74 (dd, J = 5.7, 9 Hz, 1H), 4.12 (dd, J = 6.3, 8.7 Hz, 1H), 4.37–4.57 (m, 5H), 6.53 (br s, 1H), 7.70 (s, 1H) ppm. 13C-NMR (CDCl3): δ 23.1, 25.1, 26.7, 34.8, 52.5, 66.4, 73.9, 110.3, 123.6, 170.1 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 2.08 min; m/z: 255 ([M+H]+, 60%), 277 ([M+Na]+, 30%), 318 ([M+Cu]+, 10%).
4-Acetamidomethyl-1-[(cyclohex-3-en-1-yl)methyl]-1,2,3-triazole (a6b6). Prepared from 49 mg (0.50 mmol) of a6 and 75 mg (0.55 mmol) of b6. The product was obtained as a brown solid (103 mg, 88%). C12H18N4O, M = 234.30 g.mol−1. FTIR: ν 3244, 3140, 3057, 2929, 2850, 1658, 1566, 1289 cm−1. 1H-NMR (CDCl3): δ 1.18–1.25 (m, 1H), 1.65–1.75 (m, 2H), 1.93 (s, 3H), 1.96–2.10 (m, 4H), 4.18 (d, J = 7.2 Hz, 2H), 4.40 (d, J = 5.7 Hz, 2H), 5.56–5.62 (m, 2H), 6.49 (br s, 1H), 7.47 (s, 1H) ppm. 13C-NMR (CDCl3): δ 22.1, 23.1, 24.8, 27.9, 33.6, 84.5, 123.7, 126.1, 170.1 ppm. LC-MS: ELSD pur. 100% ; Rt = 2.92 min; m/z: 235 ([M+H]+, 50%), 257 ([M+Na]+, 30%), 298 ([M+Cu]+, 20%).
4-Acetamidomethyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a6b7). Prepared from 49 mg (0.50 mmol) of a6 and 84 mg (0.55 mmol) of b7. The product was obtained as a beige solid (108 mg, 86%). C11H14N4OS, M = 250.32 g.mol−1. m.p. 117 ºC. FTIR: ν 3264, 3140, 3078, 2953, 2862, 1654, 1550, 1438, 1289 cm−1. 1H-NMR (CDCl3): δ 1.99 (s, 3H), 3.42 (t, J = 6.9 Hz, 2H), 4.46 (d, J = 5.4 Hz, 2H), 4.58 (t, J = 7.2 Hz, 2H), 6.49 (br s, 1H), 6.73–6.74 (m, 1H), 6.90–6.93 (m, 1H), 7.17–7.18 (m, 1H), 7.41 (s, 1H) ppm. 13C-NMR (CDCl3): δ 23.1, 30.8, 34.9, 51.7, 123.4, 124.6, 126.1, 127.2, 138.7, 170.1 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 2.46 min; m/z: 251 ([M+H]+, 50%), 273 ([M+Na]+, 30%), 314 ([M+Cu]+, 20%).
4-Acetamidomethyl-1-(3,6-anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-1,2,3-triazole (a6b8). Prepared from 49 mg (0.50 mmol) of a6 and 126 mg (0.55 mmol) of b8. The product was obtained as a yellow oil (87 mg, 53%). C14H22N4O5, M = 326.36 g.mol−1. FTIR: ν 3356, 3235, 3127, 3073, 2983, 2945, 2850, 1658, 1546, 1384, 1210, 1106 cm−1. 1H-NMR (CDCl3): δ 1.33 (s, 3H), 1.50 (s, 3H), 2.00 (s, 3H), 3.18 (dd, J = 3.3, 6.0 Hz, 1H), 3.44–3.54 (m, 3H), 4.07 (d, J = 12.4 Hz, 1H), 4.50 (d, J = 4.8 Hz, 2H), 4.56 (dd, J = 3.6, 14.2 Hz, 1H), 4.65 (dd, J = 6.0, 14.2 Hz, 1H), 4.78–4.85 (m, 2H), 6.45 (br s, 1H), 7.73 (s, 1H) ppm. 13C-NMR (CDCl3): δ 23.1, 24.3, 25.8, 34.9, 52.6, 69.2, 72.6, 80.6, 81.3, 81.5, 112.6, 124.0, 170.2 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%; Rt = 2.23 min; m/z: 327 ([M+H]+, 60%), 349 ([M+Na]+, 40%).
1-Benzyl-4-trifluoroacetamidomethyl-1,2,3-triazole (a7b1). Prepared from 76 mg (0.50 mmol) of a7 and 73 mg (0.55 mmol) of b1. The product was obtained as a white solid (140 mg, 98%). C12H11F3N4O, M = 284.24 g.mol−1. m.p. 132–134 ºC. FTIR: ν 3302, 3107, 3061, 1699, 1563, 1210, 1185, 1156, 1060 cm−1. 1H-NMR (CDCl3): δ 4.58 (d, J = 4.67 Hz, 2H), 5.51 (s, 2H), 7.46–7.20 (m, 5H), 7.54 (s, 1H), 7.70 (s, 1H) ppm. 13C-NMR (CDCl3): δ 28.3, 36.2, 54.5, 128.2, 129.03, 129.3, 134.1, 157.1 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 7.37 min; m/z: 285 ([M+H]+).
1-Ethoxycarbonylmethyl-4-trifluoroacetamidomethyl-1,2,3-triazole (a7b2). Prepared from 76 mg (0.50 mmol) of a7 and 71 mg (0.55 mmol) of b2. The product was obtained as a white solid (139 mg, 99%). C9H11F3N4O3, M = 280.21 g.mol−1. m.p. 140–142 ºC. FTIR: ν 3153, 3045, 3003, 2958, 2895, 1741, 1699, 1546, 1193, 1148, 1060 cm−1. 1H-NMR (CDCl3) : δ 1.31 (t, J = 7.1 Hz, 3H), 4.28 (q, J = 7.1 Hz, 2H), 4.62 (d, J = 5.7 Hz, 2H), 5.16 (s, 2H), 7.76 (s, 1H), 8.12 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.0, 35.1, 50.9, 62.6, 124.2, 166.4 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 2.92 min; m/z: 281 ([M+H]+).
4-Trifluoroacetamidomethyl-1-(3-hydroxypropyl)-1,2,3-triazole (a7b3). Prepared from 76 mg (0.50 mmol) of a7 and 56 mg (0.55 mmol) of b3. The product was obtained as a white solid (109 mg, 87%). C8H11F3N4O2, M = 252.20 g.mol−1. m.p. 84–86 ºC. FTIR: ν 3306, 3127, 3045, 3003, 1699, 1550, 1355, 1189, 1156, 1048 cm−1. 1H-NMR (CDCl3): δ 2.12 (q, J = 5.1 Hz, 2H), 3.62 (m, 2H), 4.52 (t, J = 6.8 Hz, 2H), 4.59 (d, J = 5.6 Hz, 2H), 4.74 (s, 1H), 7.67 (s, 1H), 8.10 (s, 1H) ppm. 13C-NMR (CDCl3): δ 32.5, 35.1, 47.1, 58.4, 87.9, 159.7 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 2.22 min; m/z: 253 ([M+H]+).
4-Trifluoroacetamidomethyl-1-(3-trifluoroacetamidopropyl)-1,2,3-triazole (a7b4). Prepared from 76 mg (0.50 mmol) of a7 and 108 mg (0.55 mmol) of b4. The product was obtained as a white solid (152 mg, 88%). C10H11F6N5O2, M = 347.22 g.mol−1. m.p. 120–122 ºC. FTIR: ν 3298, 3140, 2899, 1699, 1566, 1181, 1148, 1047 cm−1. 1H-NMR (CDCl3): δ 2.04 (p, J = 6.9 Hz, 2H), 3.20 (q, J = 6.7 Hz, 2H), 4.29 (t, J = 6.9 Hz, 2H), 4,43 (d, J = 5.6, 2H), 7.53 (s, 1H), 7.66 (s, 1H), 7.92 (s, 1H) ppm. 13C-NMR (CDCl3): δ 28.6, 34.5, 36.4, 47.1, 113.8, 122.8, 156.3 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 4.02 min; m/z: 348 ([M+H]+).
4-Trifluoroacetamidomethyl-1-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl-1,2,3-triazole (a7b5). Prepared from 76 mg (0.50 mmol) of a7 and 86 mg (0.55 mmol) of b5. The product was obtained as a pale yellow solid (138 mg, 89%). C11H15F3N4O3, M = 308.26 g.mol−1. m.p. 126–128 ºC. FTIR: ν 3149, 3057, 2991, 1708, 1555, 1428, 1202, 1185, 1152, 1064 cm−1. 1H-NMR (CDCl3): δ 1.34 (s, 3H), 1.37 (s, 3H), 3.74 (dd, J = 8.8, 5.6 Hz, 1H), 4.13 (dd, J = 8.8, 6.2 Hz, 1H), 4.51–4.34 (m, 1H), 4.74–4.52 (m, 3H), 7.79 (s, 1H), 7.99 (s, 1H) ppm. 13C-NMR (CDCl3): δ 25.1, 26.6, 34.9, 52.6, 66.3, 73.9, 110.4, 117.7, 124.4, 157.7 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 3.67 min; m/z: 309 ([M+H]+).
1-[(Cyclohex-3-en-1-yl)methyl]-4-trifluoroacetamidomethyl-1,2,3-triazole (a7b6). Prepared from 76 mg (0.50 mmol) of a7 and 75 mg (0.55 mmol) of b6. The product was obtained as a beige solid (130 mg, 90%). C12H15F3N4O, M = 288.27 g.mol−1. m.p. 124–126 ºC . FTIR: ν 3156, 3032, 2899, 1721, 1566, 1429, 1193, 1152, 1064 cm−1. 1H-NMR (CDCl3): δ 1.45–1.19 (m, 1H), 1.88–1.61 (m, 2H), 2.20–1.91 (m, 4H), 4.27 (d, J = 7.2 Hz, 2H), 4.59 (d, J = 5.5 Hz, 2H), 5.62 (dt, J = 9.9, 2.1 Hz, 1H), 5.70 (dd, J = 10.0, 1.6 Hz, 1H), 7.61 (s, 1H), 8.35 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.1, 25.8, 28.8, 34.6, 35.0, 55.5, 123.2, 124.7, 127.1, 157.8 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 8.07 min; m/z: 289 ([M+H]+).
4-Trifluoroacetamidomethyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a7b7). Prepared from 76 mg (0.50 mmol) of a7 and 84 mg (0.55 mmol) of b7. The product was obtained as a beige solid (150 mg, 98%). C11H11F3N4OS, M = 304.30 g.mol−1. m.p. 108–110 ºC. FTIR: ν 3306, 3149, 3086, 2958, 1692, 1546, 1339, 1206, 1177, 1156, 1052 cm−1. 1H-NMR (CDCl3) : δ 3.43 (t, J = 7 Hz, 2H), 4.69–4.46 (m, 4H), 6.71 (d, J = 3.2 Hz, 1H), 6.91 (dd, J = 5.1, 3.5 Hz, 1H), 7.17 (dd, J = 5.1, 1.0 Hz, 1H), 7.47 (s, 1H), 8.23 (s, 1H) ppm. 13C-NMR (CDCl3): δ 30.7, 34.8, 51.9, 124.5, 126.2, 127.2, 138.4, 157.7 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 7.42 min; m/z: 305 ([M+H]+).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-trifluoroacetamidomethyl-1,2,3-triazole (a7b8). Prepared from 76 mg (0.50 mmol) of a7 and 126 mg (0.55 mmol) of b8. The product was obtained as a pale yellow oil (170 mg, 89%). C14H19F3N4O5, M = 380.33 g.mol−1. FTIR: ν 3269, 2994, 1721, 1555,1438, 1197,1185, 1159, 1102, 1056 cm−1. 1H-NMR (CDCl3): δ 1.33 (s, 3H), 1.50 (s, 3H), 3.19 (dd, J = 5.8, 3.0 Hz, 1H), 3.47 (dd, J = 11.0, 2.8 Hz, 1H), 3.64 (s, 1H), 4.07 (d, J = 10.7 Hz, 1H), 4.39 (dt, J = 6.0, 3.69 Hz, 1H), 4.61 (d, J = 7.6 Hz, 2H), 4.74–4.65 (m, 1H), 4.86–4.78 (m, 2H), 7.81 (s, 1H), 7.90 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.3, 25.8, 29.7, 35.0, 52.8, 69.1, 72.2, 79.6, 80.3, 81.3, 84.4, 112.7, 124.6, 142.4, 157.1 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 2.76min; m/z: 381 ([M+H]+).
1-Benzyl-4-(tert-butoxycarbonylamino)methyl-1,2,3-triazole (a8b1). Prepared from 78 mg (0.50 mmol) of a8 and 73 mg (0.55 mmol) of b1. The product was obtained as a white solid (130 mg, 90%). C15H20N4O2, M = 288.35 g.mol−1. m.p. 100–102 ºC. FTIR: ν 3236, 3144, 3032, 2974, 1692, 1542, 1289, 1168, 1056 cm−1. 1H-NMR (CDCl3): δ 1.40 (s, 9H), 4.38 (d, J = 4.6 Hz, 2H), 5.21 (s, 1H), 5.49 (s,2H), 7.23–7.36 (m, 5H), 7.47 (s, 1H) ppm. 13C-NMR (CDCl3): δ 28.3, 36.2, 54.2, 79.6, 128.1, 128.7, 129.1, 134.6, 155.8 ppm. LC-MS: ELSD pur. 98%, UV pur. 100%; Rt = 8.17 min; m/z: 289 ([M+H]+
4-(tert-Butoxycarbonylamino)methyl-1-ethoxycarbonylmethyl-1,2,3-triazole (a8b2). Prepared from 78 mg (0.50 mmol) of a8 and 71 mg (0.55 mmol) of b2. The product was obtained as a pale green solid (130 mg, 91%). C12H20N4O4, M = 284.32 g.mol−1. m.p.108–110 ºC. FTIR: ν 3394, 3132, 3073, 2970, 1757, 1687, 1517, 1210, 1177, 1056 cm−1. 1H-NMR (CDCl3): δ 1.30 (t, J = 7.1 Hz, 3H), 1.44 (s, 9H), 4.26 (q, J = 7.1 Hz, 2H), 4.42 (d, J = 5.8 Hz, 2H), 5.14 (s, 2H), 5.27 (s, 1H), 7.67 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.1, 28.4, 36.1, 50.9, 62.4, 79.7, 155.9, 166.2 ppm. LC-MS: ELSD pur. 94%, UV pur. 100%; Rt = 5.33 min; m/z: 285 ([M+H]+).
4-(tert-Butoxycarbonylamino)methyl-1-(3-hydroxypropyl)-1,2,3-triazole (a8b3). Prepared from 78 mg (0.50 mmol) of a8 and 56 mg (0.55 mmol) of b3. The product was obtained as a pale green oil (127 mg, 99%). C11H20N4O3, M = 256.31 g.mol−1. FTIR: ν 3336, 3136, 2965, 2933, 2875, 1696, 1525, 1168, 1048 cm−1. 1H-NMR (CDCl3): δ 1.42 (s, 9H), 2.11 (q, J = 5.1 Hz, 2H), 3.62 (m,2H), 3.72 (s,1H), 4.50 (t, J = 6.6 Hz, 2H), 5.1 (s, 1H), 7.67 (s, 1H) ppm. 13C-NMR (CDCl3): δ 28.4, 32.7, 36.0, 47.1, 58.1, 79.7, 156.1 ppm. LC-MS: ELSD pur. 93%, UV pur. 100%; Rt = 2.38 min; m/z: 257 ([M+H]+).
4-(tert-Butoxycarbonylamino)methyl-1-(3-trifluoroacetamidopropyl)-1,2,3-triazole (a8b4). Prepared from 78 mg (0.50 mmol) of a8 and 108 mg (0.55 mmol) of b4. The product was obtained as a off-white solid (175 mg, 99%). C13H20F3N5O3, M = 351.33 g.mol−1. m.p.132–134 ºC. FTIR: ν 3323, 3127, 3099, 2974, 2933, 2883, 1728, 1704, 1674, 1530, 1210, 1181, 1168, 1048 cm−1. 1H-NMR (CDCl3): δ 1.43 (s, 9H), 2.23 (p, J = 6.6 Hz, 2H), 3.41 (q, J = 6.3 Hz, 2H), 4.64–4.18 (m, 4H), 5.29 (s, 1H), 7.62 (s, 1H), 7.72 (s, 1H) ppm. 13C-NMR (CDCl3): δ 28.4, 29.2, 36.0, 37.1, 47.7, 79.9, 156.0 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 5.73 min; m/z: 352 ([M+H]+).
4-(tert-Butoxycarbonylamino)methyl-1-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl-1,2,3-triazole (a8b5). Prepared from 78 mg (0.50 mmol) of a8 and 86 mg (0.55 mmol) of b5. The product was obtained as a pale green solid (150 mg, 96%). C14H24N4O4, M = 312.37 g.mol−1. m.p. 100–102 ºC. FTIR: ν 3394, 2983, 2899, 1687, 1512, 1384, 1368, 1272, 1251, 1210, 1168, 1143, 1069 cm−1. 1H-NMR (CDCl3): δ 1.34 (s, 3H), 1.38 (s, 3H), 1.44 (s, 9H), 3.74 (dd, J = 8.7, 5.5 Hz, 1H), 4.12 (dd, J = 8.7, 6.1 Hz, 1H), 4.65–4.28 (m, 5H), 5.25 (s, 1H), 7.70 (s, 1H) ppm. 13C-NMR (CDCl3): δ 25.2, 26.7, 28.4, 36.1, 52.4, 66.4, 74.0, 79.6, 110.2, 155.8 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 5.22 min; m/z: 313 ([M+H]+).
4-(tert-Butoxycarbonylamino)methyl-1-[(cyclohex-3-en-1-yl)methyl]-1,2,3-triazole (a8b6). Prepared from 78 mg (0.50 mmol) of a8 and 75 mg (0.55 mmol) of b6. The product was obtained as a beige solid (145 mg, 99%). C15H24N4O2, M = 292.38 g.mol−1. m.p. 86–88 ºC. FTIR: ν 3385, 3127, 3019, 2965, 2920, 1696, 1512, 1363, 1272, 1168, 1052 cm−1. 1H-NMR (CDCl3): δ 1.44 (s, 9H), 1.88–1.61 (m, 2H), 2.41–1.88 (m, 5H), 4.26 (d, J = 7.3 Hz, 2H), 4.40 (d, J = 5.5 Hz, 2H), 5.24 (s, 1H), 5.81–5.52 (m, 2H), 7.53 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.1, 25.2, 28.4, 28.9, 34.7, 36.1, 55.4, 79.6, 124.8, 127.1, 155.9 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 8.55 min; m/z: 293 ([M+H]+).
4-(tert-Butoxycarbonylamino)methyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a8b7). Prepared from 78 mg (0.50 mmol) of a8 and 75 mg (0.55 mmol) of b7. The product was obtained as a beige solid (150 mg, 97%). C14H20N4O2S, M = 308.41 g.mol−1. m.p. 92–94 ºC. FTIR: ν 3311, 3120, 3095, 3070, 2978, 2933, 2875, 1679,1534, 1447, 1363, 1251, 1164, 1056 cm−1. 1H-NMR (CDCl3): δ 1.43 (s, 9H), 3.42 (t, J = 7.0 Hz, 2H), 4.36 (d, J = 5.9 Hz, 2H), 4.59 (t, J = 7.1 Hz, 2H), 5.21 (s, 1H), 6.73 (dd, J = 3.4, 0.9 Hz, 1H), 6.91 (dd, J = 5.1, 3.5 Hz, 1H), 7.17 (dd, J = 5.1, 1.1 Hz, 1H), 7.38 (s, 1H) ppm. 13C-NMR (CDCl3): δ 28.4, 30.8, 36.0, 52.7, 79.6, 124.6, 126.1, 127.0, 138.8, 155.7 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 8.20min; m/z: 309 ([M+H]+).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-(tert-butoxycarbonylamino)methyl-1,2,3-triazole (a8b8). Prepared from 78 mg (0.50 mmol) of a8 and 75 mg (0.55 mmol) of b8. The product was obtained as a beige solid (192 mg, 99%). C17H28N4O6, M = 384.44 g.mol−1. m.p. 130–132 ºC. FTIR: ν 3550, 3406, 2937, 2854, 1683, 1511, 1455, 1375, 1272, 1159, 1106, 1052 cm−1. 1H-NMR (CDCl3): δ 1.33 (s, 3H), 1.44 (s, 9H), 1.50 (s, 3H), 3.15 (dd, J = 5.9, 3.5 Hz, 1H), 3.57–3.40 (m, 2H), 4.07 (d, J = 10.7 Hz, 1H), 4.40 (d, J = 5.7 Hz, 2H), 4.56 (dd, J = 14.2, 3.5 Hz, 1H), 4.67 (dd, J = 14.4, 6.0 Hz, 1H), 4.88–4.76 (m, 2H), 5.25 (s, 1H), 7.72 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.3, 25.9, 28.4, 36.1, 52.5, 69.2, 72.6, 79.6, 80.3, 81.3, 81.6, 84.4, 112.6, 123.8, 155.8 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 3.43 min; m/z: 385 ([M+H]+).
Tris [(1-benzyl-1,2,3-triazol-4-yl)methyl]amine (a9b1). Prepared from 69 mg (0.50 mmol) of a9 and 220 mg (1.65 mmol) of b1. The product was obtained as a mixture of mono, bis (11%) and tris (81%) derivatives. C30H30N10, M = 530.64 g.mol−1. LC-MS: ELSD pur. 81%, UV pur. 81%; Rt = 5.5 min; m/z: 531 ([M+H]+).
Tris [(1-ethoxycarbonylmethyl-1,2,3-triazol-4-yl)methyl]amine (a9b2). Prepared from 69 mg (0.50 mmol) of a9 and 213 mg (1.65 mmol) of b2. The product was obtained as a mixture of mono, bis (44%) and tris (47%) derivatives. C21H30N10O6, M = 518.53 g.mol−1. LC-MS: ELSD pur. 47%, UV pur. 50%; Rt = 1.86 min; m/z: 519 ([M+H]+).
Tris {[1-(3-hydroxypropyl)-1,2,3-triazol-4-yl]methyl}amine (a9b3). Prepared from 69 mg (0.50 mmol) of a9 and 167 mg (1.65 mmol) of b3. The product was obtained as a mixture of mono, bis (99%) and tris (1%) derivatives. C18H30N10O3, M = 434.50 g.mol−1. LC-MS: ELSD pur. 0.8%, UV pur. 0.8%; Rt = 380 min; m/z: 435 ([M+H]+).
Tris {[1-(3-trifluoroacetamidopropyl)-1,2,3-triazol-4-yl]methyl}amine (a9b4). Prepared from 78 mg (0.50 mmol) of a9 and 108 mg (1.65 mmol) of b4. The product was obtained as a mixture of mono, bis (7%) and tris (92%) derivatives. C21H30F9N13O3, M = 719.58 g.mol−1. LC-MS: ELSD pur. 92%, UV pur. 90%; Rt = 1.86 min; m/z: 720 ([M+H]+).
Tris {[1-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl-1,2,3-triazol-4-yl]methyl}amine (a9b5). Prepared from 78 mg (0.50 mmol) of a9 and 86 mg (1.65 mmol) of b5. The product was obtained as a mixture of mono, bis (11%) and tris (88%) derivatives. C27H42N10O6, M = 602.70 g.mol−1. LC-MS: ELSD pur. 88%, UV pur. 99%; Rt = 1.17 min; m/z: 603 ([M+H]+).
Tris ({1-[(cyclohex-3-en-1-yl)methyl]-1,2,3-triazol-4-yl}methyl)amine (a9b6). Prepared from 69 mg (0.50 mmol) of a9 and 226 mg (1.65 mmol) of b6. The product was obtained as a mixture of mono, bis (34%) and tris (61%) derivatives. C30H42N10, M = 542.72 g.mol−1. LC-MS: ELSD pur. 61%, UV pur. 62%; Rt = 7.82 min; m/z: 543 ([M+H]+).
Tris ({1-[2-(2-thienyl)ethyl]-1,2,3-triazol-4-yl}methyl)amine (a9b7). Prepared from 69 mg (0.50 mmol) of a9 and 225 mg (1.65 mmol) of b7. The product was obtained as a mixture of mono, bis (51%) and tris (47%) derivatives. C24H24N10S3, M = 590.80 g.mol−1. LC-MS: ELSD pur. 47%, UV pur. 50%; Rt = 8.05min; m/z: 592 ([M+H]+).
Tris {[1-(3,6-anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-1,2,3-triazol-4-yl]methyl}amine (a9b8). Prepared from 76 mg (0.50 mmol) of a9 and 378 mg (1.65 mmol) of b8. The product was obtained as a mixture of mono, bis (44%) and tris (53%) derivatives. C36H54N10O12, M = 818.89 g.mol−1. LC-MS: ELSD pur. 53%, UV pur. 50%; Rt = 1.85min; m/z: 819 ([M+H]+).
1-Benzyl-4-phenyl-1,2,3-triazole (a10b1). Prepared from 55 mg (0.50 mmol) of a10 and 73 mg (0.55 mmol)of b1. The product was obtained as a beige solid (95 mg, 81%). C15H13N3, M = 235.29 g.mol−1. m.p. 128 ºC. FTIR: ν 3144, 3037, 2975, 1496, 1451, 1044 cm−1. 1H-NMR (CDCl3): δ 3.90 (s, 3H), 5.55 (s, 2H), 7.27–7.36 (m, 5H), 7.99 (s, 1H) ppm. 13C-NMR (CDCl3): δ 52.2, 54.5, 127.3, 128.3, 129.2, 129.3, 133.6, 140.3, 161.1 ppm. LC-MS: ELSD pur. 93%, UV pur. 100%, Rt = 8.86 min, m/z: 236 ([M+H]+, 100%).
1-Ethoxycarbonylmethyl-4-phenyl-1,2,3-triazole (a10b2). Prepared from 55 mg (0.50 mmol) of a10 and 71 mg (0.55 mmol) of b2. The product was obtained as a white solid (92 mg, 80%). C12H13N3O2, M = 231.26 g.mol−1. m.p. 104 ºC. FTIR: ν 3302, 3140, 3079, 3004, 2950, 1758, 1464, 1082, 1044 cm−1. 1H-NMR (CDCl3): δ 1.30 (t, J = 7.2 Hz, 3H), 3.95 (s, 3H), 4.28 (q, J = 7.2 Hz, 2H), 5.25 (s, 2H), 8.30 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.4, 51.4, 52.7, 63.2, 129.4, 161.4, 166.0 ppm. LC-MS: ELSD pur. 91%, UV pur. 100%, Rt = 8.10 min, m/z: 232 ([M+H]+, 100%).
1-(3-Hydroxypropyl)-4-phenyl-1,2,3-triazole (a10b3). Prepared from 55 mg (0.50 mmol) of a10 and 56 mg (0.55 mmol) of b3. The product was obtained as a white solid (88 mg, 87%). C11H13N3O, M = 203.25 g.mol−1. m.p. 90.5 ºC. FTIR: ν 3315, 3120, 2950, 2875, 1086 cm−1. 1H-NMR (CDCl3): δ 2.18 (q, J = 6.0 Hz, 2H), 3.67 (t, J = 6.0 Hz, 2H), 3.93 (s, 3H), 4.62 (t, J = 6.0 Hz, 2H), 8.22 (s, 1H) ppm. 13C-NMR (CDCl3): δ 32.4, 47.5, 52.2, 58.3, 128.2, 139.7, 161.2 ppm. LC-MS: ELSD pur. 98%, UV pur. 100%, Rt = 2.95 min, m/z: 204 ([M+H]+, 100%).
1-(3-Trifluoroacetamidopropyl)-4-phenyl-1,2,3-triazole (a10b4). Prepared from 55 mg (0.50 mmol) of a10 and 108 mg (0.55 mmol) of b4. The product was obtained as a white solid (132 mg, 88%). C13H13F3N4O, M = 298.27 g.mol−1. m.p. 159 ºC. FTIR: ν 3211, 3045, 2953, 2896, 1721, 1193, 1144 cm−1. 1H-NMR (CDCl3): δ 2.30 (t, J = 7.2 Hz, 2H), 3.27 (q, J = 6.9 Hz, 2H), 4.44 (t, J = 7.0 Hz, 2H), 7,33–7,85 (m, 5H), 8.57 (s, 1H) ppm. 13C-NMR (CDCl3): δ 28.7, 36.6, 47.2, 117.7, 121.4, 125.1, 127.8, 128.8, 130.7, 146.2, 156.1 ppm. LC-MS: ELSD pur. 95%, UV pur. 100%, Rt = 7.88 min, m/z: 299 ([M+H]+, 100%).
1-(2,2-Dimethyl-1,3-dioxolan-4-yl)methyl-4-phenyl-1,2,3-triazole (a10b5). Prepared from 55 mg (0.50 mmol) of a10 and 86 mg (0.55 mmol) of b5. The product was obtained as a beige solid (96 mg, 74%). C14H17N3O2, M = 259.31 g.mol−1. m.p. 110 ºC. FTIR: ν 3144, 2991, 2925, 1260, 1069, 1048 cm−1. 1H-NMR (CDCl3): δ 1.32 (s, 3H), 1.37 (s, 3H), 3.73 (dd, J = 5.3, 8.7 Hz, 1H), 4.10 (dd, J = 6.1, 8.7 Hz, 1H), 4.36–4.56 (m, 3H), 4.74 (s, 2H), 7.73 (s, 1H) ppm. 13C-NMR (CDCl3): δ 25.2, 26.7, 52.3, 66.4, 110.2, 120.9, 125.7, 128.2, 128.8, 130.6, 147.8 ppm. LC-MS: ELSD pur. 97%, UV pur. 100%, Rt = 7.96 min, m/z: 260 ([M+H]+, 100%).
1-[(Cyclohex-3-en-1-yl)methyl]-4-phenyl-1,2,3-triazole (a10b6). Prepared from 55 mg (0.50 mmol) of a10 and 75 mg (0.55 mmol) of b6. The product was obtained as a brown solid (98 mg, 82%). C15H17N3, M = 239.32 g.mol−1. m.p. 93 ºC. FTIR: ν 3140, 3074, 3020, 2921, 2842, 1222, 1044 cm−1. 1H-NMR (CDCl3): δ 1.32 (m, 2H), 1.75 (m, 2H), 2.02 (m, 2H), 2.20 (m, 1H), 4.27 (d, J = 7.3 Hz, 2H), 4.78 (s, 2H), 5.60–5.72 (m, 2H), 7.58 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.6, 26.3, 29.3, 35.2, 55.8, 120.3, 125.2, 126.1, 127.5, 128.5, 129.2, 131.1, 148.0 ppm. LC-MS: ELSD pur. 94%, UV pur. 100%, Rt = 9.37 min, m/z: 240 ([M+H]+, 100%).
4-Phenyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a10b7). Prepared from 55 mg (0.50 mmol) of a10 and 84 mg (0.55 mmol) of b7. The product was obtained as a beige solid (96 mg, 76%). C14H13N3S, M = 255.34 g.mol−1. m.p. 126 ºC. FTIR: ν 3086, 2962, 2924, 1222, 1085 cm−1. 1H-NMR (CDCl3): δ 3.42 (t, J = 7.1 Hz, 2H), 4.59 (t, J = 7.1 Hz, 2H), 4.73 (s, 2H), 6.74 (d, J = 3.3 Hz, 1H), 6.91 (dd, J = 3.3, 5.0 Hz, 1H), 7.16 (d, J = 5.1 Hz, 1H), 7.47 (s, 1H) ppm. 13C-NMR (CDCl3): δ 31.3, 52.2, 120.4, 125.0, 126.1, 126.7, 128.5, 129.2, 131.0, 139.3, 148.0 ppm. LC-MS: ELSD pur. 100%, UV pur. 100%, Rt = 8.86 min, m/z: 256 ([M+H]+, 100%).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-phenyl-1,2,3-triazole (a10b8). Prepared from 55 mg (0.50 mmol) of a10 and 126 mg (0.55 mmol) of b8. The product was obtained as a orange solid (134 mg, 81%). C17H21N3O4, M = 331.37 g.mol−1. m.p. 89.5 ºC. FTIR: ν 3452, 3137, 2987, 2929, 1381, 1277, 1210, 1081 cm−1. 1H-NMR (CDCl3): δ 1.35 (s, 3H), 1.52 (s, 3H), 3.19 (dd, J = 3.7, 5.8 Hz, 1H), 3.46 (dd, J = 3.5, 10.8 Hz, 1H), 4.08 (d, J = 10.8 Hz, 1H), 4.47 (dd, J = 5.8, 9.3 Hz, 1H), 4.62 (dd, J = 3.5, 14.3 Hz, 1H), 4.76 (dd, J = 5.7, 14.4 Hz, 1H), 4.80 (dd, J = 3.5, 6.2 Hz, 1H), 4.87 (dd, J = 3.5, 6.2 Hz, 1H), 7.32–7.85 (m, 5H), 8.01 (s, 1H) ppm. 13C-NMR (CDCl3): δ 24.7, 26.3, 52.9, 69.6, 73.0, 81.0, 81.8, 84.7, 113.0 122.1, 126.1, 128.5, 129.2, 131.0, 148.0 ppm. LC-MS: ELSD pur. 70%, UV pur. 100%, Rt = 5.17 min, m/z: 332 ([M+H]+, 66%), 354 ([M+Na]+, 34%).
1-Benzyl-4-octyl-1,2,3-triazole (a11b1). Prepared from 69 mg (0.50 mmol) of a11 and 73 mg (0.55 mmol) of b1. The product was obtained as a beige solid (108 mg, 80%). C17H25N3, M = 271.41 g.mol−1. m.p. 64–66 ºC. FTIR: ν 3102, 2958, 2924, 2854, 1483, 1451, 1256, 1197, 1098, 1052, 844 cm−1. 1H-NMR (CDCl3): δ 0.86 (t, J = 6 Hz, 3H), 1.24–1.28 (m, 12H), 1.58 (t, J = 2 Hz, 2H) 5.29 (s, 2H) 7.24–7.34 (m, 5H), 7.36 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.7, 21.0, 23.1, 29.8, 29.9, 30.0, 30.1, 32.4, 55.4, 128.5, 129.4, 129.6, 135.9 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 10.02 min; m/z: 273 ([M+H]+).
1-Ethoxycarbonylmethyl-4-octyl-1,2,3-triazole (a11b2). Prepared from 69 mg (0.50 mmol) of a11 and 71 mg (0.55 mmol) of b2. The product was obtained as a brown oil (115 mg, 86%). C14H25N3O2, M = 267.37 g.mol−1. FTIR: ν 3124, 2983, 2958, 1746, 1256, 1206 cm−1. 1H-NMR (CDCl3): δ 0.87 (t, J = 6 Hz, 3H), 1.28–1.31 (m, 10H), 1.62–1.64 (m, 3H), 1.70 (m, 5H), 4.2 (q, J = 7 Hz, 2H), 4.93 (s, 1H), 7.26 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.4(8), 14.5, 23.1, 26.0, 29.5, 29.5, 29.6, 32.2, 50.6, 63.9, 167.0 ppm. LC-MS: ELSD pur. 90%, UV pur. 100%; Rt = 9.54 min; m/z: 268 ([M+H]+).
1-(3-Hydroxypropyl)-4-octyl-1,2,3-triazole (a11b3). Prepared from 69 mg (0.50 mmol) of a11 and 56 mg (0.55 mmol) of b3. The product was obtained as a yellow oil (77 mg, 65%). C13H25N3O, M = 239.36 g.mol−1. FTIR: ν 3347, 3120, 2953, 2937, 1247, 1197, 1118, 1056, 844 cm−1. 1H-NMR (CDCl3): δ 0.86 (t, J = 6 Hz, 3H), 1.26–1.29 (m, 10H), 1.67 (q, J = 3 Hz, 2H) 1.98 (q, J = 3 Hz, 2H), 2.52 (t, J = 6 Hz, 2H), 3.23 (t, J = 9 Hz, 2H), 3.45 (t, J = 9 Hz, 2H), 7.36 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.5, 18.8, 23.07, 29.6, 29.6(8), 29.7, 29.8, 32.3, 33.1, 43.1, 59.3, 84.5, 131.3 ppm. LC-MS: ELSD pur. 97%, UV pur. 100%; Rt = 7.82 min; m/z: 240 ([M+H]+).
1-(3-Trifluoroacetamidopropyl)-4-octyl-1,2,3-triazole (a11b4). Prepared from 78 mg (0.50 mmol) of a11 and 108 mg (0.55 mmol) of b4. The product was obtained as a pale green solid (165 mg, 99%). C14H25F3N4O, M = 334.39 g.mol−1. m.p.104–106 ºC. FTIR: ν 3207, 3048, 2924, 2854, 1712, 1571, 1463, 1197, 1139 cm−1. 1H-NMR (CD3CN): δ 0.87 (q, J = 3 Hz, 3H), 1.27–1.32 (m, 10H), 1.60–1.62 (m, 2H), 2.63 (t, J = 3 Hz, 2H), 3.27 (q, J = 2 Hz, 2H), 3.34–3.37 (m, 2H), 4.34 (t, J = 6 Hz, 2H), 7.55 (s, H), 7.67 (s, H) ppm. 13C-NMR (CD3CN): δ 14.3, 21.7, 23.3, 28.6, 29.8, 29.9, 29.9, 30.0, 30.2, 37.7, 48.1 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 9.32 min; m/z: 352 ([M+NH4]+).
1-(2,2-Dimethyl-1,3-dioxolan-4-yl)methyl-4-octyl-1,2,3-triazole (a11b5). Prepared from 78 mg (0.50 mmol) of a11 and 86 mg (0.55 mmol) of b5. The product was obtained as a pale green solid (146 mg, 99%). C16H27N3O2, M = 295.43 g.mol−1. m.p. 70–72 ºC. FTIR: ν 3122, 3064, 2953, 2920, 1461, 1378, 1267, 1057 cm−1. 1H-NMR (CDCl3): δ 0.67 (t, J = 6 Hz, 3H), 1.06–1.12 (m, 8H), 1.15 (s, 3H), 1.18 (s, 3H), 1.47–1.49 (m, 2H), 2.54 (t, J = 6 Hz, 2H), 3.50–3.55 (m, 2H), 3.88–3.92 (m, 2H), 4.19–4.34 (m, 3H), 7.22 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.2, 22.8, 23.4, 25.4, 26.8, 29.3, 29.4, 29.5, 29.6, 32.0, 52.9, 66.4, 110.2 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 9.06 min; m/z: 296 ([M+H]+).
1-[(Cyclohex-3-en-1-yl)methyl]-4-octyl-1,2,3-triazole (a11b6). Prepared from 69 mg (0.50 mmol) of a11 and 75 mg (0.55 mmol) of b6. The product was obtained as a brown oil (136 mg, 99%). C15H27N3O2, M = 275.44 g.mol−1. FTIR: ν 3127, 3019, 2933, 2854, 1463, 1375, 1052 cm−1. 1H-NMR (CDCl3): δ 0.69 (t, J = 4.5 Hz, 3H), 1.08–1.13 (m, 8H), 1.58 (s, 3H), 2.01 (s, 3H), 2.06–2.66 (m, 7H), 4.16 (d, J = 9 Hz, 2H), 5.53–5.58 (m, 2H), 7.19 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.5, 23.0, 24.6, 26.1, 26.2, 29.3, 29.6, 29.7, 29.9, 32.2, 35.1, 55.7, 125.3, 155.9 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 10.41 min; m/z: 276 ([M+H]+).
4-Octyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a11b7). Prepared from 69 mg (0.50 mmol) of a11 and 75 mg (0.55 mmol) of b7. The product was obtained as a pale green solid (106 mg, 73%). C16H25N3S, M = 291.46 g.mol−1. m.p. 72–74 ºC. FTIR: ν 3102, 3057, 2962, 2929, 2854, 1455, 1060 cm−1. 1H-NMR (CDCl3): δ 0.65 (t, J = 13 Hz, 3H), 1.08–1.13 (m, 8H), 1.42 (s, 3H) 2.49 (t, J = 6 Hz, 2H) 3.21 (t, J = 7.5 Hz, 2H), 4.37 (t, J = 7.5 Hz, 3H), 6. 52 (d, J = 3 Hz, 1H), 6.72 (t, J = 15 Hz, 1H), 6.88 (s, 1H), 6.98 (d, J = 6 Hz, 1H) ppm. 13C-NMR (CDCl3): δ 14.5, 23.1, 26.0, 29.6, 29.6, 29.7, 29.9, 31.3, 52.0, 124.9, 126.5, 127.6, 130.2 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 9.64 min; m/z: 293 ([M+H]+).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-octyl-1,2,3-triazole (a11b8). Prepared from 76 mg (0.50 mmol) of a11 and 126 mg (0.55 mmol) of b8. The product was obtained as a pale green solid (128 mg, 70%). C19H33N3O4, M = 367.49g g.mol−1. m.p.120–122 ºC. FTIR: ν 3269, 2994, 1721, 1555,1438, 1197,1185, 1159, 1102, 1056 cm−1. 1H-NMR (CDCl3): δ 0.64 (t, J = 13 Hz, 3H), 1.19–1.25 (m, 21H), 1.41–1.43 (m, 5H), 2.63 (t, J = 3 Hz, 2H), 3.41 (dd, J = 7, 13.5 Hz, 2H), 4.01 (dd, J = 3, 6 Hz, 2H), 4.33–4.79 (m, 8H), 7.44 (s, 1H) ppm. 13C-NMR (CDCl3): δ 14.5, 23.1, 24.7, 24.8, 29.6, 29.7, 29.7, 29.8, 32.2, 52.7, 69.7, 73.0, 81.0, 81.7, 82.2, 130.5 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 9.10 min; m/z: 368 ([M+H]+).
1-Benzyl-4-trimethylsilyl-1,2,3-triazole (a12b1). Prepared from 246 mg (0.50 mmol) of a12 and 67 mg (0.50 mmol) of b1. The product was obtained as a pale green solid (82 mg, 71%). C12H17N3Si, M = 231.38 g.mol−1. m.p. 74–76 ºC. FTIR: ν 3286, 3115, 3061, 2953, 2920, 1674, 1542, 1438, 1318, 1280, 1168 cm−1. 1H-NMR (CDCl3): δ 0.11 (s, 9H), 5.56 (s, 2H), 7.24–7.34 (m, 5 H), 7.26 (s, 1H) ppm. 13C-NMR (CDCl3): δ 0.02, 54.6, 129.2, 129.4, 130.2, 136.1 ppm. LC-MS: ELSD pur. 96%, UV pur. 100%; Rt = 11.23 min; m/z: 232 ([M+H]+).
1-Ethoxycarbonylmethyl-4-trimethylsilyl-1,2,3-triazole (a12b2). Prepared from 246 mg (2.50 mmol) of a12 and 62 mg (0.50 mmol) of b2. The product was obtained as a brown oil (107 mg, 86%). C9H17N3O2Si, M = 227.34 g.mol−1. FTIR: ν 2928, 2854, 1746, 1455, 1372, 1213, 1023 cm−1. 1H-NMR (CDCl3): δ 0.12 (s, 9H), 1.12 (t, J = 8 Hz, 3H), 4.08 (q, J = 1 Hz, 2H), 5.01 (s, 1H), 7.49 (s, 1H) ppm. 13C-NMR (CDCl3): δ 0.02, 15.2, 51.4, 63.5, 131.5, 167.7 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 8.50 min; m/z: 228 ([M+H]+).
1-(3-Hydroxypropyl)-4-trimethylsilyl-1,2,3-triazole (a12b3). Prepared from 246 mg (0.50 mmol) of a12 and 51 mg (0.50 mmol) of b3. The product was obtained as a pale green solid (80 mg, 73%). C8H17N3OSi, M = 199.33 g.mol−1. m.p. 58–60 ºC. FTIR: ν 3290, 2920, 2850, 1650, 1461, 1213, 1172, 1049 cm−1. 1H-NMR (CDCl3): δ 0.12 (s, 9H), 1.94 (q, J = 2 Hz, 2H), 3.44–3.47 (m, 2H), 3.56 (t, J = 4.5 Hz, 1H), 4.36 (t, J = 6 Hz, 2H), 7.41 (s, 1H) ppm. 13C-NMR (CDCl3): δ 0.02, 34.1, 47.6, 59.7, 130.7 ppm. LC-MS: ELSD pur. 90%, UV pur. 100%; Rt = 2.74 min; m/z: 200 ([M+H]+).
1-(3-Trifluoroacetamidopropyl)-4-trimethylsilyl-1,2,3-triazole (a12b4). Prepared from 246 mg (2.50 mmol) of a12 and 79 mg (0.50 mmol) of b4. The product was obtained as a pale green solid (160 mg, 99%). C10H17F3N4OSi, M = 294.35 g.mol−1. m.p. 120–122 ºC. FTIR: ν 3186, 3124, 3073, 2962, 1721, 1571, 1185, 1156 cm−1. 1H-NMR (CD3CN): δ 0.15 (s, 9H), 1.81 (t, J = 3 Hz, 2H), 3.15–3.17 (m, 2H), 4.28 (q, J = 3 Hz, 2H), 7.49–7.50 (m, 1H), 7.68 (s, 1H) ppm. 13C-NMR (CD3CN): δ 0.02, 28.7, 36.5, 47.7, 132.6, 157.7 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 10.15 min; m/z: 335 ([M+MeCN]+).
1-(2,2-Dimethyl-1,3-dioxolan-4-yl)methyl-4-trimethylsilyl-1,2,3-triazole (a12b5). Prepared from 246 mg (2.50 mmol) of a12 and 79 mg (0.50 mmol) of b5. The product was obtained as a yellow oil (92 mg, 66%). C10H19N3O2Si, M = 255.39 g.mol−1. FTIR: ν 2987, 2962, 2895, 1488, 1375, 1247, 1069, 840 cm−1. 1H-NMR (CDCl3): δ 0.32 (s, 9H), 1.16 (s, 3H), 1.18 (s, 3H), 3.12–3.20 (m, 2H), 3.57–3.61 (m, 1H), 3.85–3.88 (m, 2H), 4.28–4.31 (m, 3H), 7.63 (s, 1H) ppm. 13C-NMR (CDCl3): δ 0.02, 26.4, 27.4, 54.0, 128.2, 128.2 ppm. LC-MS: ELSD pur. 92 %, UV pur. 100%; Rt = 9.50 min; m/z: 296 ([M+MeCN]+).
1-[(Cyclohex-3-en-1-yl)methyl]-4-trimethylsilyl-1,2,3-triazole (a12b6). Prepared from 246 mg (2.50 mmol) of a12 and 52 mg (0.50 mmol) of b6. The product was obtained as a brown oil (128 mg, 99%). C12H21N3Si, M = 235.41 g.mol−1. FTIR: ν 2924, 2850, 1455, 1251, 840 cm−1. 1H-NMR (CDCl3): δ 0.32 (s, 9H), 1.30–1.33 (m, 2H), 1.99–2.00 (m, 2H), 2.06–2.09 (m,2H), 4.29 (d, J = 3 Hz, 2H), 5.63–5.65 (m, 1H), 5.67–5.69 (m, 1H), 7.49 (s, 1H) ppm. 13C-NMR (CDCl3): δ 0.02, 25.3, 27.0, 35.9, 55.7, 128.0, 128.2 ppm. LC-MS: ELSD pur. 90%, UV pur. 100%; Rt = 10.15 min; m/z: 276 ([M+MeCN]+).
4-Trimethylsilyl-1-[2-(2-thienyl)ethyl]-1,2,3-triazole (a12b7). Prepared from 246 mg (2.50 mmol) of a12 and 77 mg (0.50 mmol) of b7. The product was obtained as a beige solid (87 mg, 63%). C11H17N3SSi, M = 251.43 g.mol−1. m.p. 76–78 ºC. FTIR: ν 3095, 2962, 2904, 2857, 1251, 1197, 840 cm−1. 1H-NMR (CDCl3): δ 0.28 (s, 9H), 3.42 (t, J = 6 Hz, 2H), 4.63 (t, J = 6 Hz, 2H), 6.71 (d, J = 3 Hz, 1H), 6.91 (t, J = 6 Hz, 1H), 7.18 (d, J = 6 Hz, 1H), 7.32 (s, 1H) ppm. 13C-NMR (CDCl3): δ 0.02, 32.2, 52.3, 125.6, 127.3, 128.7, 130.4, 140.1 ppm. LC-MS: ELSD pur. 95%, UV pur.100%; Rt = 9.02 min; m/z: 292 ([M+MeCN]+).
1-(3,6-Anhydro-1-deoxy-4,5-O-isopropylidene-D-glucitol-1-yl)-4-trimethylsilyl-1,2,3-triazole (a12b8). Prepared from 246 mg (2.50 mmol) of a12 and 115 mg (0.55 mmol) of b8. The product was obtained as a yellow oil (118 mg, 66%). C14H25N3O4Si, M = 327.46 g.mol−1. FTIR: ν 3451, 2924, 2857, 1375, 1272, 1210, 1098, 804 cm−1. 1H-NMR (CDCl3): δ 0.21 (s, 9H), 1.12 (s, 1H), 1.25 (s, 1H), 2.95 (m, 1H), 3.25–3.31 (m, 3H), 3.86–3.89 (m, 3H), 3.99 (t, J = 6 Hz, 1H), 4.41 (m, 4H), 4.66–4.68 (m, 2H), 7.54 (s, 1H) ppm. 13C-NMR (CDCl3): δ 0.02, 25.4, 25.5, 53.0, 70.3, 73.7, 81.7, 82.4, 83.0, 113.6 ppm. LC-MS: ELSD pur. 99%, UV pur. 100%; Rt = 4.94 min; m/z: 328 ([M+H]+).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

