4D-QSAR: Perspectives in Drug Design

Abstract
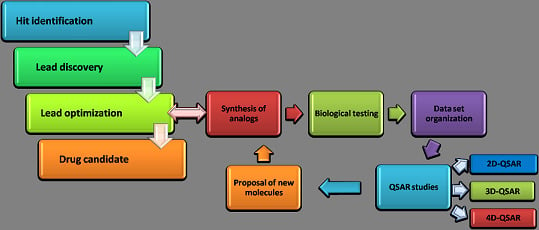
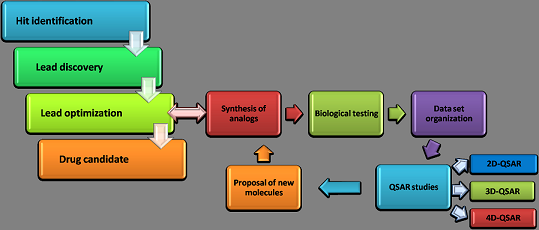
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

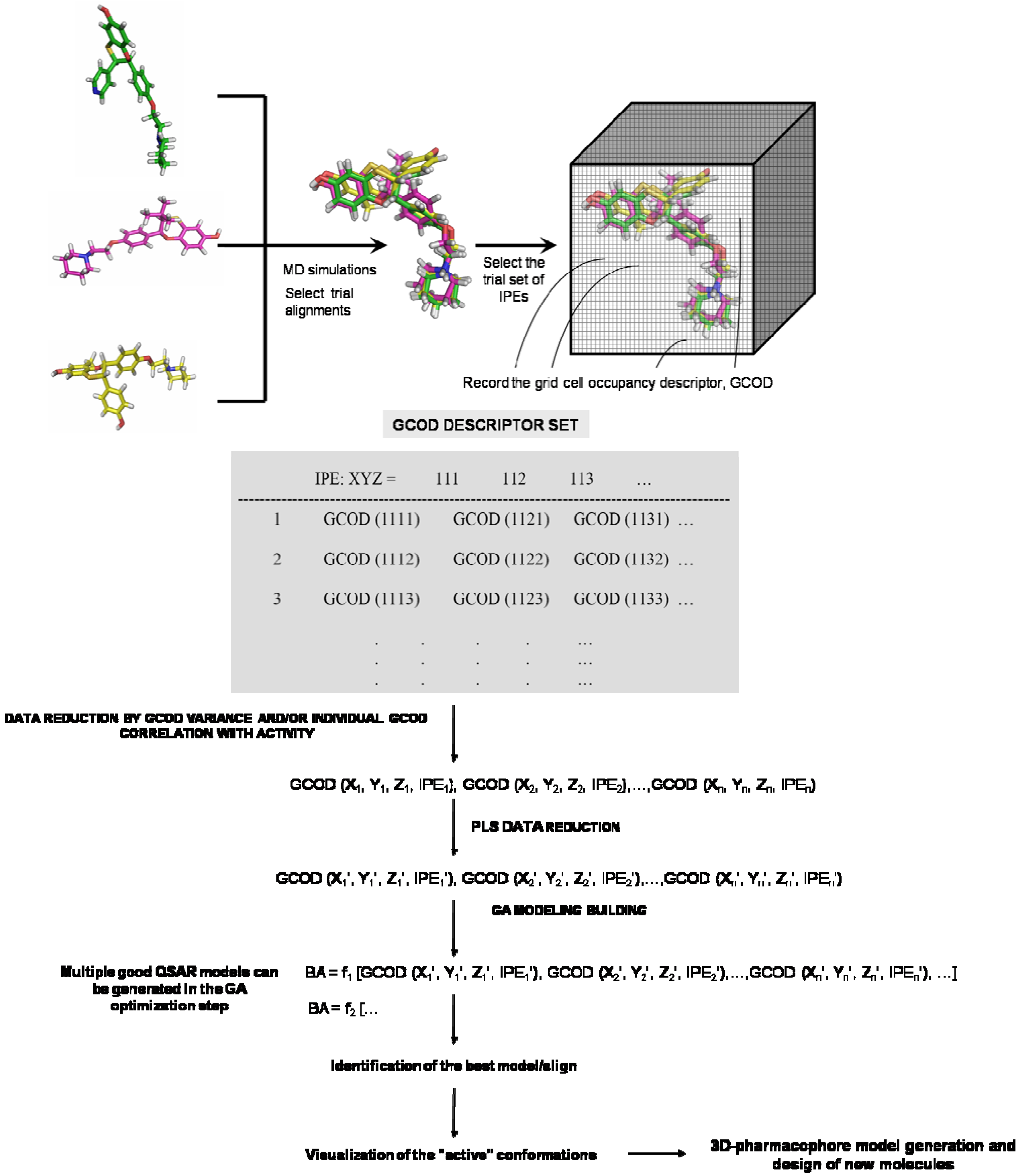
2. 4D-QSAR

3. Successful Applications of 4D-QSAR


4. 4D-QSAR: Pharmacokinetic Studies and ADMET Prediction
5. 4D-Formalism: ‘Practical’ Application in Drug Design
6. Conclusions
Acknowledgements
References and Notes
- Bleicher, K.H.; Böhm, H.-J.; Müller, K.; Alanine, A.I. Hit and lead generation: beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar]
- Lombardino, J.G.; Lowe, J.A. The role of the medicinal chemist in drug discovery--then and now. Nat. Rev. Drug Discov. 2004, 3, 853–862. [Google Scholar] [CrossRef]
- Zhao, H. Scaffold selection and scaffold hopping in lead generation: a medicinal chemistry perspective. Drug Discov. Today 2007, 12, 149–155. [Google Scholar] [CrossRef]
- Andricopulo, A.D.; Salum, L.B.; Abraham, D.J. Structure-Based Drug Design Strategies in Medicinal Chemistry. Curr. Top. Med. Chem. 2009, 9, 771–790. [Google Scholar] [CrossRef]
- Salum, L.B.; Andricopulo, A.D. Fragment-based QSAR: perspectives in drug design. Mol. Divers. 2009, 13, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Guido, R.V.C.; Oliva, G.; Andricopulo, A.D. Virtual screening and its integration with modern drug design technologies. Curr. Med. Chem. 2008, 15, 37–46. [Google Scholar] [CrossRef]
- Santos-Filho, O.A.; Hopfinger, A.J.; Cherkasov, A.; de Alencastro, R.B. The receptor-dependent QSAR paradigm: an overview of the current state of the art. Med. Chem. (Shāriqah (United Arab Emirates)) 2009, 5, 359–366. [Google Scholar] [CrossRef]
- Hansch, C.; Fujita, T. Rho-sigma-pi analysis . Method for correlation of biological activity + chemical structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Free, S.M.; Wilson, J.W. A mathematical contribution to structure-activity studies. J. Med. Chem. 1964, 7, 395–399. [Google Scholar] [CrossRef]
- Kubinyi, H. QSAR: Hansch Analysis and Related Approaches. In Methods and Principles in Medicinal Chemistry; Mannhold, R., Kroogsgard-Larsen, P., Timmerman, H., Eds.; Wiley-VCH: Weinheim, Germany, 1993; Volume 1, p. 240. [Google Scholar]
- Taylor, J.B.; Triggle, D.J. Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherland, 2007. [Google Scholar]
- Terfloth, L. Chemoinformatics: A Textbook; Gasteiger, J., Engel, T., Eds.; Wiley-VCH: Weinheim, Germany, 2003; pp. 401–437. [Google Scholar]
- Cohen, N. Guidebook on Molecular Modeling in Drug Design; Academic Press: London, UK, 1996; Volume 1, p. 361. [Google Scholar]
- Esposito, E.X.; Hopfinger, A.J.; Madura, J.D. Chemoinformatics: concepts, methods, and tools for drug discovery. In Chemoinformatics: Concepts, Methods, and Tools for Drug Discovery; Bajorath, J., Ed.; Humana Press: Totowa, NJ, USA, 2004; Volume 275, pp. 131–213. [Google Scholar]
- Kubinyi, H. 3D QSAR in Drug Design. Theory, Methods and Applications. Escom: Leiden, The Netherlands, 1993; p. 759. [Google Scholar]
- Ooms, F. Molecular modeling and computer aided drug design. Examples of their applications in medicinal chemistry. Curr. Med. Chem. 2000, 7, 141–158. [Google Scholar] [CrossRef]
- Hopfinger, A. A QSAR investigation of dihydrofolate-reductase inhibition by baker triazines based upon molecular shape-analysis. J. Am. Chem. Soc. 1980, 102, 7196–7206. [Google Scholar] [CrossRef]
- Hopfinger, A.J. Inhibition of dihydrofolate reductase: structure-activity correlations of 2,4-diamino-5-benzylpyrimidines based upon molecular shape analysis. J. Med. Chem. 1981, 24, 818–22. [Google Scholar] [CrossRef]
- Hopfinger, A.; Wang, S.; Tokarski, J.; Jin, B.; Albuquerque, M.; Madhav, P.; Duraiswami, C. Construction of 3D-QSAR models using the 4D-QSAR analysis formalism. J. Am. Chem. Soc. 1997, 119, 10509–10524. [Google Scholar] [CrossRef]
- Albuquerque, M.; Brito, M.; Cunha, E.; Alencastro, R.; Antunes, O.; Castro, H.; Rodrigues, C. Multidimensional-QSAR: Beyond the third-dimension in drug design. Curr. Methods Med. Chem. Biol. Phys. 2007, 1, 91–100. [Google Scholar]
- Albuquerque, M.G.; Hopfinger, A.J.; Barreiro, E.J.; de Alencastro, R.B. Four-dimensional quantitative structure-activity relationship analysis of a series of interphenylene 7-oxabicycloheptane oxazole thromboxane A2 receptor antagonists. J. Chem. Inf. Comput. Sci. 1998, 38, 925–938. [Google Scholar] [CrossRef]
- Hopfinger, A. 4D-QSAR Package User’s Manual, 3.0; The Chem21 Group Inc.: Lake Forest, IL, USA, 2001. [Google Scholar]
- Rogers, D.G.; Hopfinger, A.J. Applications of genetic function approximation to quantitative-structure-activity relationships and quantitative structure-property relationships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Cramer III, R.D.; Patterson, D.E.; Bunce, J.D. Comparative Molecular Field Analyses (CoMFA). 1. Effect of Shape on Binding of Steroids to Carrier Proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar]
- Goodford, P. Multivariate characterisation of molecules for QSAR. J. Chemometrics 1996, 10, 107–117. [Google Scholar] [CrossRef]
- Goodford, P. GRID. Molecular Discovery Ltd: University of Oxford, England, UK, 2000. [Google Scholar]
- Clementi, S. GOLPE 3.0; Multivariate Infometric Analyses (MIA): Perugia, Italy, 1995. [Google Scholar]
- Kearsley, S.K.; Smith, G.M. An alternative method for the alignment of molecular structures: Maximizing electrostatic and steric overlap. Tetrahedron Comp. Methodol. 1990, 3, 615–633. [Google Scholar] [CrossRef]
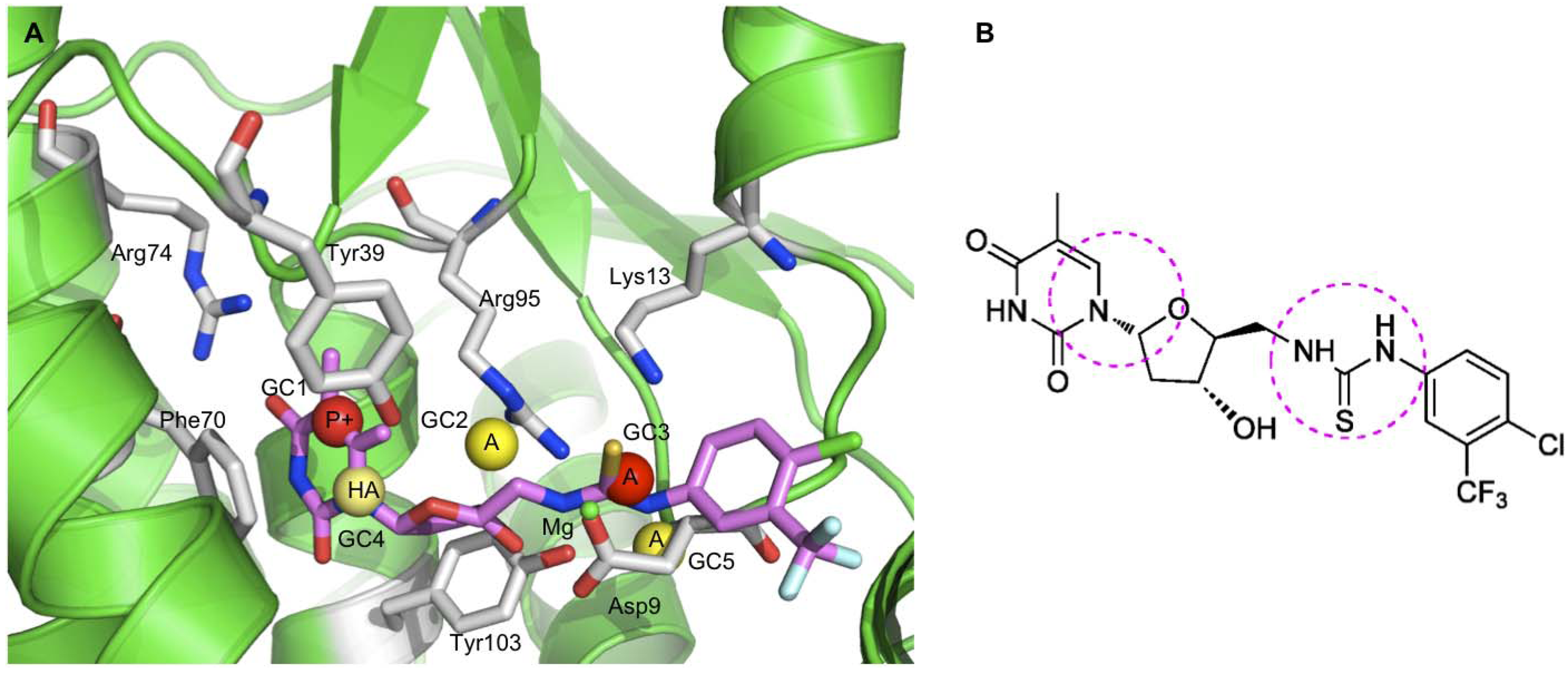
- Andrade, C.H.; Pasqualoto, K.F.M.; Ferreira, E.I.; Hopfinger, A.J. Rational design and 3D-pharmacophore mapping of 5'-thiourea-substituted alpha-thymidine analogues as mycobacterial TMPK inhibitors. J. Chem. Inf. Model. 2009, 49, 1070–1078. [Google Scholar] [CrossRef]
- Hong, X.; Hopfinger, A.J. 3D-pharmacophores of flavonoid binding at the benzodiazepine GABA(A) receptor site using 4D-QSAR analysis. J. Chem. Inf. Comput. Sci. 2003, 43, 324–36. [Google Scholar] [CrossRef]
- Krasowski, M.D.; Hong, X.; Hopfinger, A.J.; Harrison, N.L. 4D-QSAR analysis of a set of propofol analogues: mapping binding sites for an anesthetic phenol on the GABA(A) receptor. J. Med. Chem. 2002, 45, 3210–3221. [Google Scholar] [CrossRef]
- Liu, J.; Pan, D.; Tseng, Y.; Hopfinger, A.J. 4D-QSAR analysis of a series of antifungal p450 inhibitors and 3D-pharmacophore comparisons as a function of alignment. J. Chem. Inf. Comput. Sci. 2003, 43, 2170–2179. [Google Scholar] [CrossRef]
- Thipnate, P.; Liu, J.; Hannongbua, S.; Hopfinger, A.J. 3D pharmacophore mapping using 4D QSAR analysis for the cytotoxicity of lamellarins against human hormone-dependent T47D breast cancer cells. J. Chem. Inf. Model. 2009, 49, 2312–2322. [Google Scholar] [CrossRef]
- Senese, C.L.; Duca, J.; Pan, D.; Hopfinger, A.J.; Tseng, Y.J. 4D-fingerprints, universal QSAR and QSPR descriptors. J. Chem. Inf. Comput. Sci. 2004, 44, 1526–1539. [Google Scholar] [CrossRef]
- Iyer, M.; Hopfinger, A.J. Treating chemical diversity in QSAR analysis: modeling diverse HIV-1 integrase inhibitors using 4D fingerprints. J. Chem. Inf. Model. 2007, 47, 1945–1960. [Google Scholar] [CrossRef]
- Romeiro, N.C.; Albuquerque, M.G.; de Alencastro, R.B.; Ravi, M.; Hopfinger, A.J. Construction of 4D-QSAR models for use in the design of novel p38-MAPK inhibitors. J. Comput. Aided Mol. Des. 2005, 19, 385–400. [Google Scholar] [CrossRef]
- Pasqualoto, K.F.M.; Ferreira, E.I.; Santos-Filho, O.A.; Hopfinger, A.J. Rational design of new antituberculosis agents: receptor-independent four-dimensional quantitative structure-activity relationship analysis of a set of isoniazid derivatives. J. Med. Chem. 2004, 47, 3755–3764. [Google Scholar]
- Ravi, M.; Hopfinger, A. J.; Hormann, R. E.; Dinan, L. 4D-QSAR analysis of a set of ecdysteroids and a comparison to CoMFA modeling. J. Chem. Inf. Comput. Sci. 2001, 41, 1587–1604. [Google Scholar] [CrossRef]
- Van Daele, I.; Munier-Lehmann, H.; Froeyen, M.; Balzarini, J.; Van Calenbergh, S. Rational design of 5'-thiourea-substituted alpha-thymidine analogues as thymidine monophosphate kinase inhibitors capable of inhibiting mycobacterial growth. J. Med. Chem. 2007, 50, 5281–5292. [Google Scholar] [CrossRef] [Green Version]
- de la Sierra, I.L.; Munier-Lehmann, H.; Gilles, A.M.; Barzu, O.; Delarue, M. X-ray structure of TMP kinase from Mycobacterium tuberculosis complexed with TMP at 1.95 angstrom resolution. J. Mol. Biol. 2001, 311, 87–100. [Google Scholar] [CrossRef]
- Santos-Filho, O.A.; Hopfinger, A.J. Structure-based QSAR analysis of a set of 4-hydroxy-5,6-dihydropyrones as inhibitors of HIV-1 protease: an application of the receptor-dependent (RD) 4D-QSAR formalism. J. Chem. Inf. Model. 2006, 46, 345–354. [Google Scholar] [CrossRef]
- Pan, D.; Tseng, Y.; Hopfinger, A.J. Quantitative structure-based design: formalism and application of receptor-dependent RD-4D-QSAR analysis to a set of glucose analogue inhibitors of glycogen phosphorylase. J. Chem. Inf. Comput. Sci. 2003, 43, 1591–1607. [Google Scholar] [CrossRef]
- Hopfinger, A.; Reaka, A.; Venkatarangan, P.; Duca, J.; Wang, S. Construction of a virtual nigh throughput screen by 4D-QSAR analysis: Application to a combinatorial library of glucose inhibitors of glycogen phosphorylase b. J. Chem. Inf. Comput. Sci. 1999, 39, 1151–1160. [Google Scholar] [CrossRef]
- Duca, J.S.; Tseng, Y.F.; Hopfinger, A.J. 4D-QSPR analysis and virtual screening in materials science. Adv. Mater. (Weinheim, Ger.) 2001, 13, 1713–1717. [Google Scholar] [CrossRef]
- Lombardo, F.; Gifford, E.; Shalaeva, M.Y. In silico ADME prediction: data, models, facts and myths. Mini Rev. Med. Chem. 2003, 3, 861–875. [Google Scholar] [CrossRef]
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discovery 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Iyer, M.; Mishra, R.; Han, Y.; Hopfinger, A.J. Predicting blood-brain barrier partitioning of organic molecules using membrane-interaction QSAR analysis. Pharm. Res. 2002, 19, 1611–1621. [Google Scholar] [CrossRef]
- Keseru, G.M.; Molnar, L. High-throughput prediction of blood-brain partitioning: A thermodynamic approach. J. Chem. Inf. Comput. Sci. 2001, 41, 120–128. [Google Scholar] [CrossRef]
- Pan, D.; Iyer, M.; Liu, J.; Li, Y.; Hopfinger, A.J. Constructing optimum blood brain barrier QSAR models using a combination of 4D-molecular similarity measures and cluster analysis. J. Chem. Inf. Comput. Sci. 2004, 44, 2083–2098. [Google Scholar] [CrossRef]
- Platts, J.A.; Abraham, M.H.; Zhao, Y.H.; Hersey, A.; Ijaz, L.; Butina, D. Correlation and prediction of a large blood-brain distribution data set - an LFER study. Eur. J. Med. Chem. 2001, 36, 719–730. [Google Scholar] [CrossRef]
- Subramaniam, S.; Mehrotra, M.; Gupta, D. Virtual high throughput screening (vHTS) - A perspective. Bioinformation 2008, 3, 14–17. [Google Scholar]
- Hopfinger, A.J.; Duca, J.S. Extraction of pharmacophore information from high-throughput screens. Curr. Opin. Biotechnol. 2000, 11, 97–103. [Google Scholar] [CrossRef]
- Duca, J.S.; Hopfinger, A.J. Estimation of molecular similarity based on 4D-QSAR analysis: formalism and validation. J. Chem. Inf. Comput. Sci. 2001, 41, 1367–1387. [Google Scholar] [CrossRef]
- Duca, J.S.; Hopfinger, A.J. 4D-QSPR analysis and virtual screening of calcite growth inhibitor libraries. Chem. Mater. 2000, 12, 3821–3829. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Andrade, C.H.; Pasqualoto, K.F.M.; Ferreira, E.I.; Hopfinger, A.J. 4D-QSAR: Perspectives in Drug Design. Molecules 2010, 15, 3281-3294. https://doi.org/10.3390/molecules15053281
Andrade CH, Pasqualoto KFM, Ferreira EI, Hopfinger AJ. 4D-QSAR: Perspectives in Drug Design. Molecules. 2010; 15(5):3281-3294. https://doi.org/10.3390/molecules15053281
Chicago/Turabian StyleAndrade, Carolina H., Kerly F. M. Pasqualoto, Elizabeth I. Ferreira, and Anton J. Hopfinger. 2010. "4D-QSAR: Perspectives in Drug Design" Molecules 15, no. 5: 3281-3294. https://doi.org/10.3390/molecules15053281
APA StyleAndrade, C. H., Pasqualoto, K. F. M., Ferreira, E. I., & Hopfinger, A. J. (2010). 4D-QSAR: Perspectives in Drug Design. Molecules, 15(5), 3281-3294. https://doi.org/10.3390/molecules15053281