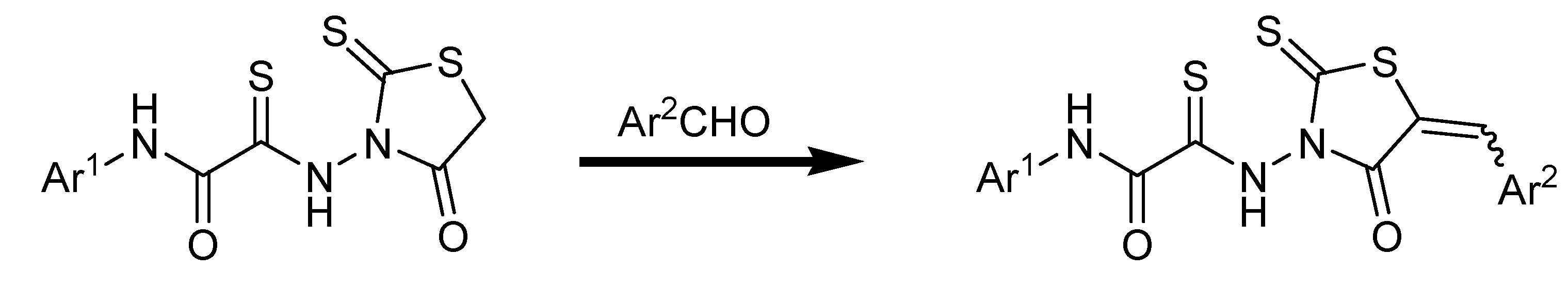
3.2. General Procedures for the Synthesis of 5-Benzylidene-4-oxo-2-thioxothiazolidine (6–54)
A flask containing methanol (10 mL) was charged with the appropriate rhodanine (0.13 mmol, 1.0 equivalents), the appropriate benzaldehyde (0.13 mmol, 1.0 equivalents), and ethylenediammonium diacetate (0.1 equivalents). The solution was stirred at room temperature for 1–2 h. The solvent was evaporated under reduced pressure and the product residue was purified by flash chromatography.
2-{[5-(4-Chlorobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-fluorophenyl)-2-thioxo-acetamide (6). Yield 87%; m.p. 204–205 °C; 1H-NMR: δ 7.09 (d, 2H, Harom, J = 8.7 Hz), 7.55–7.7 (m, 7H, Harom, J = 8.3 Hz), 9.89 (s, 1H, NH); MS (EI) m/z (%): 541 [M+] (78). Anal. Calcd. for C18H11ClFN3O2S3: C, 47.84; H, 2.45; Cl, 7.84; F, 4.20; N, 9.30; O, 7.08; S, 21.28. Found: C, 47.80; H, 2.39; Cl, 7.88; N, 9.38; S, 21.20.
2-{[5-(4-Chlorobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-chlorophenyl)-2-thioxo-acetamide (7). Yield 87%; m.p. 211–212 °C; 1H-NMR: δ 7.51−7.65 (m, 9H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 468 [M+] (85). Anal. Calcd. for C18H11Cl2N3O2S3: C, 46.16; H, 2.37; Cl, 15.14; N, 8.97; O, 6.83; S, 20.54. Found: C, 46.21; H, 2.30; Cl, 15.24; N, 8.91; S, 20.58.
2-{[5-(4-Chlorobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(3-nitrophenyl)-2-thioxo-acetamide (8). Yield 71%; m.p. 221–222 °C; 1H-NMR: δ 7.55–7.65 (m, 5H, Harom), 7.77−7.81 (t, 1H, Harom), 7.94 (d, 1H, Harom, J = 8.2 Hz), 8.17 (d, 1H, Harom, J = 8 Hz), 8.62 (s, 1H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 478 [M+] (62). Anal. Calcd. for C18H11ClN4O4S3: C, 45.14; H, 2.31; Cl, 7.40; N, 11.70; O, 13.36; S, 20.08. Found: C, 45.07; H, 2.28; Cl, 7.40; N, 11.79; S, 20.01.
2-{[5-(4-Chlorobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-methoxyphenyl)-2-thioxoacetamide (9). Yield 90%; m.p. 186–187 °C; 1H-NMR: δ 3.77 (s, 3H, OCH3), 7.09 (d, 2H, Harom, J = 8.7 Hz), 7.51–7.66 (m, 6H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 463 [M+] (93). Anal. Calcd. for C19H14ClN3O3S3: C, 49.18; H, 3.04; Cl, 7.64; N, 9.06; O, 10.34; S, 20.73. Found: C, 49.25; H, 3.14; Cl, 7.56; N, 9.26; S, 20.71.
2-{[5-(1H-indol-3-ylmethylene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-phenyl-2-thioxo-acetamide (10). Yield 83%; m.p. 206–207 °C; 1H-NMR: δ 7.06–7.12 (m, 2H, Harom), 7.26–7.3 (m, 3H, Harom), 7.41 (d, 1H, Harom, J = 7.9 Hz), 7.63 (d, 2H, Harom, J = 7.3 Hz), 7.8 (s, 1H, CH), 8.07 (d, 1H, Harom, J = 7.8 Hz), 8.69 (s, 1H, Harom), 9.8 (s, 1H, NH); MS (EI) m/z (%) 438 [M+] (87). Anal. Calcd. for C20H14N4O2S3: C, 54.78; H, 3.22; N, 12.78; O, 7.30; S, 21.93. Found: C, 54.85; H, 3.29; N, 12.73; S, 21.85.
2-{5-[2-(4-Methylphenyl)-2-oxoethylidene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-N-phenyl-2-thioxoacetamide (11). Yield 65%; m.p. 147–148 °C; 1H-NMR: δ 2.43 (s, 3H, CH3), 7.08–7.13 (t, 1H, Harom, J = 7.3 Hz), 7.18-7.30 (m, 4H, Harom), 7.62 (d, 2H, Harom, J = 7.3 Hz), 7.68 (s, 1H, CH), 7.91 (d, 2H, Harom, J = 8.4 Hz), 9.89 (s, 1H, NH); MS (EI) m/z (%) 441 [M+] (82). Anal. Calcd. for C19H13N3O3S3: C, 53.38; H, 3.06; N, 9.83; O, 11.23; S, 22.50. Found: C, 53.46; H, 3.13; N, 9.76; S, 22.50.
N-(4-fluorophenyl)-2-{5-[(5-nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-2-thioxoacetamide (12). Yield 53%; m.p. 155–156 °C; 1H-NMR: δ 7.09 (d, 2H, Harom, J = 8.6 Hz), 7.59 (d, 1H, Harom, J = 3.6 Hz), 7.67–7.7 (m, 3H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 452 [M+] (35). Anal. Calcd. for C16H9FN4O5S3: C, 42.47; H, 2.00; F, 4.20; N, 12.38; O, 17.68; S, 21.26. Found: C, 42.56; H, 1.89; N, 12.46; S, 21.18.
N-(4-chlorophenyl)-2-{5-[(5-nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-2-thioxoacetamide (13). Yield 58%; m.p. 159–160 °C; 1H-NMR: δ 7.11 (s, 1H, CH), 7.53–7.63 (m, 5H, Harom), 7.68 (d, 1H, Harom, J = 3.6 Hz), 9.89 (s, 1H, NH); MS (EI) m/z (%) 468 [M+] (30). Anal. Calcd. for C16H9ClN4O5S3: C, 40.98; H, 1.93; Cl, 7.56; N, 11.95; O, 17.06; S, 20.51. Found: C, 41.06; H, 1.90; Cl, 7.50; N, 12.04; S, 20.54.
2-{-5-[(5-Nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-N-(3-nitrophenyl)-2-thioxoacetamide (14). Yield 49%; m.p. 152–153 °C; 1H-NMR: δ 7.11 (s, 1H, CH), 7.59 (d, 1H, Harom, J = 3.6 Hz), 7.67 (d, 1H, Harom, J = 3.6 Hz), 7.77–7.81 (t, 1H, Harom, J = 7.9 Hz), 7.94 (d, 1H, Harom, J = 8.2 Hz), 8.17 (d, 1H, Harom, J = 8.0 Hz), 8.62 (s, 1H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 479 [M+] (39). Anal. Calcd. for C16H9N5O7S3: C, 40.08; H, 1.89; N, 14.61; O, 23.36; S, 20.06. Found: C, 40.01; H, 1.83; N, 14.69; S, 20.09.
2-{5-[(5-Nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-2-thioxo-N-[3-(trifluoro-methyl)phenyl]acetamide (15). Yield 53%; m.p. 163–164 °C; 1H-NMR: δ 7.11 (s, 1H, CH), 7.44 (d, 1H, Harom, J = 7.8 Hz), 7.59 (d, 1H, Harom, J = 3.6 Hz), 7.64-7.68 (m, 2H, Harom), 7.87 (d, 1H, Harom, J = 7.7 Hz), 8.18 (s, 1H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 502 [M+] (43). Anal. Calcd. for C17H9F3N4O5S3: C, 40.64; H, 1.81; F, 11.34; N, 11.15; O, 15.92; S, 19.14. Found: C, 40.69; H, 1.74; N, 11.21; S, 19.20.
N-[3,5-bis(trifluoromethyl)phenyl]-2-{5-[(5-nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-2-thioxoacetamide (16). Yield 55%; m.p. 195–196 °C; 1H-NMR: δ 7.11 (s, 1H, CH), 7.59 (d, 1H, Harom, J = 3.6 Hz), 7.67-7.72 (m, 2H, Harom) 8.31 (s, 2H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 570 [M+] (25). Anal. Calcd. for C18H8F6N4O5S3: C, 37.90; H, 1.41; F, 19.98; N, 9.82; O, 14.02; S, 16.86. Found: C, 37.81; H, 1.37; N, 9.87; S, 16.81.
N-(4-methoxyphenyl)-2-{5-[(5-nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-2-thioxoacetamide (17). Yield 73%; m.p. 187–188 °C; 1H-NMR: δ 3.77 (s, 3H, OCH3), 7.09 (d, 2H, Harom, J = 8.8 Hz), 7.11 (s, 1H, CH), 7.51 (d, 2H, Harom, J = 8.8 Hz), 7.59 (d, 1H, Harom, J = 3.6 Hz), 7.68 (d, 1H, Harom, J = 3.6 Hz), 9.89 (s, 1H, NH); MS (EI) m/z (%) 464 [M+] (57). Anal. Calcd. for C17H12N4O6S3: C, 43.96; H, 2.60; N, 12.06; O, 20.67; S, 20.71. Found: C, 44.08; H, 2.67; N, 12.06; S, 20.63.
N-(3,4-dimethoxyphenyl)-2-{5-[(5-nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-2-thioxoacetamide (18). Yield 68%; m.p. 156–157 °C; 1H-NMR: δ 3.71 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 6.88 (s, 1H, Harom), 6.95 (d, 1H, Harom, J = 7.9 Hz), 7.11 (s, 1H, CH), 7.29 (d, 1H, Harom, J = 7.9 Hz), 7.59 (d, 1H, Harom, J = 3.6 Hz), 7.68 (d, 1H, Harom, J = 3.6 Hz), 9.89 (s, 1H, NH); MS (EI) m/z (%) 494 [M+] (67). Anal. Calcd. for C18H14N4O7S3: C, 43.72; H, 2.85; N, 11.33; O, 22.65; S, 19.45. Found: C, 43.81; H, 2.87; N, 11.30; S, 19.47.
2-{5-[(5-Nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-2-thioxo-N-(3,4,5-tri-methoxyphenyl)acetamide (19). Yield 68%; m.p. 201–202 °C; 1H-NMR: δ 3.8 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 6.58 (s, 2H, Harom), 7.11 (s, 1H, CH), 7.59 (d, 1H, Harom, J = 3.6 Hz), 7.68 (d, 1H, Harom, J = 3.6 Hz), 9.9 (s, 1H, NH); MS (EI) m/z (%) 524 [M+] (75). Anal. Calcd. for C19H16N4O8S3: C, 43.51; H, 3.07; N, 10.68; O, 24.40; S, 18.34. Found: C, 43.57; H, 3.12; N, 10.65; S, 18.30.
N-[2,5-diethoxy-4-(1H-tetrazol-1-yl)phenyl]-2-{5-[(5-nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino-2-thioxoacetamide (20). Yield 43%; m.p. 136–138 °C; 1H-NMR: δ 1.4–1.47 (t, 6H, CH3, J = 6.9 Hz), 4.13–4.21 (q, 4H, CH2, J = 6.7 Hz), 7.11 (s, 1H, CH), 7.24 (s, 1H, Harom), 7.32 (s, 1H, Harom),7.59 (d, 1H, Harom, J = 3.6 Hz), 7.68 (d, 1H, Harom, J = 3.6 Hz), 8.96 (s, 1H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 590 [M+] (15). Anal. Calcd. for C21H18N8O7S3: C, 42.71; H, 3.07; N, 18.97; O, 18.96; S, 16.29. Found: C, 42.65; H, 3.01; N, 19.11; S, 16.22.
N-[3-methyl-4-(1H-tetrazol-1-yl)phenyl]-2-{5-[(5-nitro-2-furyl)methylene]-4-oxo-2-thioxo-1,3-thia-zolidin-3-yl}amino-2-thioxoacetamide (21). Yield 40%; m.p. 161–162 °C; 1H-NMR: δ 2.48 (s, 3H, CH3), 7.11 (s, 1H, CH), 7.59 (d, 1H, Harom, J = 3.6 Hz), 7.63-7.68 (m, 2H, Harom), 7.83 (d, 1H, Harom, J = 8.6 Hz), 8.01 (s, 1H, Harom), 8.95 (s, 1H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 516 [M+] (33). Anal. Calcd. for C18H12N8O5S3: C, 41.86; H, 2.34; N, 21.69; O, 15.49; S, 18.62. Found: C, 41.91; H, 2.29; N, 21.77; S, 18.58.
2-{[4-Oxo-2-thioxo-5-(3,4,5-trimethoxybenzylidene)-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-(3,4,5-tri-methoxyphenyl)acetamide (22). Yield 83%; m.p. 196–197 °C; 1H-NMR: δ 3.73 (s, 6H, CH3), 3.80 (s, 6H, CH3), 3.83 (s, 3H, CH3), 3.85 (s, 3H, CH3), 6.58 (s, 2H, Harom), 7.11 (s, 2H, Harom), 7.42 (s, 1H, CH), 9.89 (s, 1H, NH); MS (EI) m/z (%) 579 [M+] (76). Anal. Calcd. for C24H25N3O8S3: C, 49.73; H, 4.35; N, 7.25; O, 22.08; S, 16.59. Found: C, 49.81; H, 4.42; N, 7.19; S, 16.64.
N-[2,5-diethoxy-4-(1H-tetrazol-1-yl)phenyl]-2-{[4-oxo-2-thioxo-5-(3,4,5-trimethoxybenzylidene)-1,3-thiazolidin-3-yl]amino}-2-thioxoacetamide (23). Yield 83%; m.p. 158–159 °C; 1H-NMR: δ 1.4–1.47 (t, 3H, CH3, J = 6.9 Hz), 3.73 (s, 6H, CH3), 3.84 (s, 3H, CH3), 4.12–4.21 (q, 2H, CH2, J = 6.7 Hz), 7.11 (s, 2H, Harom), 7.24 (s, 1H, Harom), 7.32 (s, 1H, Harom), 7.42 (s, 1H, CH), 8.96 (s, 1H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 645 [M+] (12). Anal. Calcd. for C26H27N7O7S3: C, 48.36; H, 4.21; N, 15.18; O, 17.34; S, 14.90. Found: C, 48.38; H, 4.15; N, 15.28; S, 14.85.
N-[3-methyl-4-(1H-tetrazol-1-yl)phenyl]-2-{[4-oxo-2-thioxo-5-(3,4,5-trimethoxybenzylidene)-1,3-thiazolidin-3-yl]amino}-2-thioxoacetamide (24). Yield 77%; m.p. 173–174 °C; 1H-NMR: δ 2.48 (s, 3H, CH3), 3.73 (s, 6H, CH3), 3.84 (s, 3H, CH3), 7.11 (s, 2H, Harom), 7.42 (s, 1H, CH), 7.64 (d, 1H, Harom, J = 8.6 Hz), 7.82 (d, 1H, Harom, J = 8.6 Hz), 8.01 (s, 1H, Harom), 8.95 (s, 1H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 571 [M+] (23). Anal. Calcd. for C23H21N7O5S3: C, 48.33; H, 3.70; N, 17.15; O, 13.99; S, 16.83. Found: C, 48.39; H, 3.74; N, 17.24; S, 16.78.
2-{[4-Oxo-2-thioxo-5-(3,4,5-trimethoxybenzylidene)-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-[3-(tri-fluoromethyl)phenyl]acetamide (25). Yield 89%; m.p. 160–161 °C; 1H-NMR: δ 3.73 (s, 6H, CH3), 3.84 (s, 3H, CH3), 7.11 (s, 2H, Harom), 7.42-7.44 (m, 2H), 7.64-7.68 (t, 1H, Harom, J = 7.8 Hz), 7.88 (d, 1H, Harom, J = 7.7 Hz), 8.17 (s, 1H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 557 [M+] (58). Anal. Calcd. for C22H18F3N3O5S3: C, 47.39; H, 3.25; F, 10.22; N, 7.54; O, 14.35; S, 17.25. Found: C, 47.43; H, 3.19; N, 7.50; S, 17.27.
N-[3,5-bis(trifluoromethyl)phenyl]-2-{[4-oxo-2-thioxo-5-(3,4,5-trimethoxybenzylidene)-1,3-thiazol-idin-3-yl]amino}-2-thioxoacetamide (26). Yield 72%; m.p. 187–188 °C; 1H-NMR: δ 3.73 (s, 6H, CH3), 3.84 (s, 3H, CH3), 7.11 (s, 2H, Harom), 7.42 (s, 1H, CH), 7.7 (s, 1H, Harom), 8.3 (s, 2H, Harom), 9.89 (s, 1H, NH); MS (EI) m/z (%) 625 [M+] (17). Anal. Calcd. for C23H17F6N3O5S3: C, 44.16; H, 2.74; F, 18.22; N, 6.72; O, 12.79; S, 15.38. Found: C, 44.09; H, 2.78; N, 6.67; S, 15.35.
2-{[5-(5-Chloro-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (27). Yield 83%; m.p. 216–217 °C; 1H-NMR: δ 3.80 (s, 6H, CH3), 3.86 (s, 3H, CH3), 6.58 (s, 2H, Harom), 7.04 (d, 1H, Harom, J = 8.9 Hz), 7.21 (d, 1H, Harom, J = 9.0 Hz), 7.73 (s, 1H, Harom), 7.81 (s, 1H, CH), 9.34 (s, 1H, OH), 10.71 (s, 1H, NH); MS (EI) m/z (%) 540 [M+] (89). Anal. Calcd. for C21H18ClN3O6S3: C, 46.71; H, 3.36; Cl, 6.56; N, 7.78; O, 17.78; S, 17.81. Found: C, 46.80; H, 3.41; Cl, 6.60; N, 7.69; S, 17.77.
2-{[5-(5-Bromo-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (28). Yield 87%; m.p. 208–209 °C; 1H-NMR: δ 3.80 (s, 6H, CH3), 3.86 (s, 3H, CH3), 6.58 (s, 2H, Harom), 6.78 (d, 1H, Harom, J = 8.5 Hz), 7.05 (d, 1H, Harom, J = 8.3 Hz), 7.77 (s, 1H, Harom), 7.85 (s, 1H, CH), 9.16 (s, 1H, OH), 10.38 (s, 1H, NH); MS (EI) m/z (%) 584 [M+] (89). Anal. Calcd. for C21H18BrN3O6S3: C, 43.15; H, 3.10; Br, 13.67; N, 7.19; O, 16.42; S, 16.4. Found: C, 43.19; H, 3.13; Br, 13.62; N, 7.11; S, 16.44.
2-{[5-(2-Hydroxy-3-nitrobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (29). Yield 62%; m.p. 193–194 °C; 1H-NMR: δ 3.80 (s, 6H, CH3), 3.86 (s, 3H, CH3), 6.58 (s, 2H, Harom), 7.21–7.25 (t, 1H, Harom, J = 7.9 Hz), 7.87 (d, 1H, Harom, J = 7.7 Hz), 8.01 (s, 1H, CH), 10.26 (s, 1H, OH), 11.83 (s, 1H, NH); MS (EI) m/z (%) 550 [M+] (43). Anal. Calcd. for C21H18N4O8S3: C, 45.81; H, 3.30; N, 10.18; O, 23.25; S, 17.47. Found: C, 45.86; H, 3.32; N, 10.25; S, 17.50.
2-{[5-(2-Hydroxy-5-nitrobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (30). Yield 52%; m.p. 205–206 °C; 1H-NMR: δ 3.80 (s, 6H, CH3), 3.86 (s, 3H, CH3), 6.58 (s, 2H, Harom), 7.23 (d, 1H, Harom, J = 9.3 Hz), 7.91 (d, 1H, Harom, J = 9.0 Hz), 8.02 (s, 1H, CH), 8.19 (s, 1H, Harom), 8.96 (s, 1H, CH), 9.16 (s, 1H, OH), 10.81 (s, 1H, NH); MS (EI) m/z (%) 550 [M+] (43). Anal. Calcd. for C21H18N4O8S3: C, 45.81; H, 3.30; N, 10.18; O, 23.25; S, 17.47. Found: C, 45.78; H, 3.35; N, 10.23; S, 17.42.
2-{[5-(3-Ethoxy-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (31). Yield 90%; m.p. 231–232 °C; 1H-NMR: δ 1.40–1.43 (t, 3H, CH3, J = 7.15 Hz), 3.80 (s, 6H, CH3), 3.86 (s, 3H, CH3), 4.04–4.09 (q, 2H, CH2), 6.58 (s, 2H, Harom), 6.76 (d,1H, Harom, J = 7.8 Hz), 7.17–7.21 (t, 1H, Harom, J = 7.8 Hz), 7.41 (d, 1H, Harom, J = 7.8 Hz), 8.25 (s, 1H, CH), 9.88 (s, 1H, OH), 11.45 (s, 1H, NH); MS (EI) m/z (%) 550 [M+] (43). Anal. Calcd. for C23H23N3O7S3: C, 50.26; H, 4.22; N, 7.64; O, 20.38; S, 17.50. Found: C, 50.36; H, 4.29; N, 7.58; S, 17.52.
2-{[5-(3-Allyl-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (32). Yield 75%; m.p. 174–175 °C; 1H-NMR: δ 3.35–3.38 (m, 2H, Halk), 3.80 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 5.09–5.12 (m, 1H, Halk), 5.20–5.25 (m, 1H, Halk), 6.12–6.22 (m, 1H, Halk), 6.50 (s, 2H, Harom), 7.0 (d, 1H, Harom, J = 7.7 Hz), 7.11–7.15 (t, 1H, Harom, J = 7.7 Hz), 7.52 (d, 1H, Harom, J = 7.7 Hz), 8.25 (s, 1H, CH), 8.81 (s, 1H, OH), 10.21 (s, 1H, NH); MS (EI) m/z (%) 545 [M+] (32). Anal. Calcd. for C24H23N3O6S3: C, 52.83; H, 4.25; N, 7.70; O, 17.59; S, 17.63. Found: C, 52.91; H, 4.29; N, 7.66; S, 17.60.
2-({5-[4-(Diethylamino)-2-hydroxybenzylidene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino)-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (33). Yield 87%; m.p. 210–211 °C; 1H-NMR: δ 1.07–1.12 (t, 6H, CH3, J = 7.2 Hz), 3.23–3.30 (q, 4H, CH2), 3.80 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 6.50 (s, 1H, Harom), 6.58–6.61 (m, 3H, Harom), 7.31 (d, 1H, Harom, J = 9.0 Hz), 8.28 (s, 1H, CH), 9.18 (s, 1H, OH), 10.85 (s, 1H, NH); MS (EI) m/z (%) 576 [M+] (72). Anal. Calcd. for C25H28N4O6S3: C, 52.07; H, 4.89; N, 9.71; O, 16.65; S, 16.68. Found: C, 52.17; H, 4.93; N, 9.76; S, 16.64.
2-[(5-{2-Hydroxy-5-[phenyldiazenyl]benzylidene}-4-oxo-2-thioxo-1,3-thiazolidin-3-yl)amino]-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (34). Yield 52%; m.p. 149–150 °C; 1H-NMR: δ 3.80 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 6.58 (s, 2H, Harom), 6.73 (d, 2H, Harom, J = 7.8 Hz), 7.0−7.04 (t, 1H, Harom, J = 7.3 Hz), 7.11-7.16 (m, 3H, Harom), 7.56 (s, 1H, Harom), 7.68 (d, 1H, Harom, J = 8.7 Hz), 8.31 (s, 1H, CH), 8.98 (s, 1H, OH), 10.05 (s, 1H, NH); MS (EI) m/z (%) 609 [M+] (21). Anal. Calcd. for C27H23N5O6S3: C, 53.19; H, 3.80; N, 11.49; O, 15.74; S, 15.78. Found: C, 53.27; H, 3.84; N, 11.49; S, 15.75.
2-Hydroxy-5-{([4-oxo-3-({2-oxo-2-[(3,4,5-trimethoxyphenyl)amino]-ethanethioyl}amino)-2-thioxo-1,3-thiazolidin-5-ylidene]methyl}benzoic acid (35). Yield 81%; m.p. 239–240 °C; 1H-NMR: δ 3.80 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 6.59 (s, 2H, Harom), 7.19 (d, 1H, Harom, J = 8.6 Hz), 7.82 (d, 1H, Harom, J = 8.5 Hz), 8.01 (s, 1H, CH), 8.17 (s, 1H, Harom), 10.35 (s, 2H, OH), 11.53 (s, 1H, NH); MS (EI) m/z (%) 549 [M+] (77). Anal. Calcd. for C22H19N3O8S3: C, 48.08; H, 3.48; N, 7.65; O, 23.29; S, 17.50. Found: C, 48.13; H, 3.53; N, 7.61; S, 17.48.
2-{[5-(3,5-Dichloro-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (36). Yield 59%; m.p. 215–216 °C; 1H-NMR: δ 3.80 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 6.58 (s, 2H, Harom), 7.37 (s, 1H, Harom), 7.92 (s, 1H, Harom), 8.09 (s, 1H, CH), 10.89 (s, 1H, OH), 11.71 (s, 1H, NH); MS (EI) m/z (%) 574 [M+] (89). Anal. Calcd. for C21H17Cl2N3O6S3: C, 43.91; H, 2.98; Cl, 12.34; N, 7.31; O, 16.71; S, 16.74. Found: C, 43.90; H, 2.95; Cl, 12.41; N, 7.26; S, 16.72.
2-{[5-(3,5-Dibromo-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-2-thioxo-N-(3,4,5-trimethoxyphenyl)acetamide (37). Yield 62%; m.p. 221–222 °C; 1H-NMR: δ 3.80 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 6.58 (s, 2H, Harom), 7.58 (s, 1H, Harom), 7.73 (s, 1H, Harom), 8.17 (s, 1H, CH), 10.26 (s, 1H, OH), 11.11 (s, 1H, NH); MS (EI) m/z (%) 663 [M+] (71). Anal. Calcd. for C21H17Br2N3O6S3: C, 38.02; H, 2.58; Br, 24.09; N, 6.33; S, 14.50.
2-{[5-(5-Bromo-2-hydroxy-3-methoxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(3,4,5-trimethoxyphenyl)acetamide (38). Yield 69%; m.p. 203–204 °C; 1H-NMR: δ 3.76 (s, 3H, OCH3), 3.80 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 6.58 (s, 2H, Harom), 6.88 (s, 1H, Harom), 7.43 (s, 1H, Harom), 8.20 (s, 1H, CH), 9.88 (s, 1H, OH), 10.95 (s, 1H, NH); MS (EI) m/z (%) 614 [M+] (65). Anal. Calcd. for C22H20BrN3O7S3: C, 43.00; H, 3.28; Br, 13.00; N, 6.84; S, 15.65.
2-{[5-(2-Hydroxy-3,5-diiodobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(3,4,5-trimethoxyphenyl)-2-thioxoacetamide (39). Yield 60%; m.p. 235–236 °C; 1H-NMR: δ 3.80 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 6.58 (s, 2H, Harom), 7.42 (s, 1H, Harom), 7.83 (s, 1H, Harom), 8.09 (s, 1H, CH), 10.26 (s, 1H, OH), 11.33 (s, 1H, NH); MS (EI) m/z (%) 757 [M+] (21). Anal. Calcd. for C21H17I2N3O6S3: C, 33.30; H, 2.26; I, 33.51; N, 5.55; S, 12.68.
2-{[5-(2-Hydroxy-3,5-diisopropylbenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(3,4,5-trimethoxyphenyl)-2-thioxoacetamide (40). Yield 71%; m.p. 189–190 °C; 1H-NMR: δ 1.1–1.15 m, 12H, Halk), 2.43-2.54 (m, 1H, Halk), 3.14–3.22 (m, 1H, Halk), 3.80 (s, 6H, OCH3), 3.86 (s, 3H, OCH3), 6.58 (s, 2H, Harom), 6.8 (s, 1H, Harom), 7.16 (s, 1H, Harom), 7.67 (s, 1H, CH), 8.82 (s, 1H, OH), 10.11 (s, 1H, NH); MS (EI) m/z (%) 589 [M+] (45). Anal. Calcd. For C27H31N3O6S3: C, 54.99; H, 5.30; N, 7.12; S, 16.31.
2-{[5-(5-Chloro-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxy-phenyl)-2-thioxoacetamide (41). Yield 86%; m.p. 220–221 °C; 1H-NMR: δ 6.89 (d, 2H, Harom, J = 9 Hz), 7.04 (d, 1H, Harom, J = 8.9 Hz), 7.21 (d, 1H, Harom, J = 8.9 Hz), 7.41 (d, 2H, Harom, J = 9 Hz), 7.73 (s, 1H, CH), 7.79 (s, 1H, Harom), 8.25 (s, 2H, OH), 10.31 (s, 1H, NH); MS (EI) m/z (%) 465 [M+] (99). Anal. Calcd. for C18H12ClN3O4S3: C, 46.40; H, 2.60; Cl, 7.61; N, 9.02; O, 13.73; S, 20.64. Found: C, 46.37; H, 2.64; Cl, 7.60; N, 8.97; S, 20.64.
2-{[5-(5-Bromo-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxy-phenyl)-2-thioxoacetamide (42). Yield 79%; m.p. 190–191 °C; 1H-NMR: δ 6.78 (d, 1H, Harom, J = 8.5 Hz), 6.89 (d, 2H, Harom, J = 9 Hz), 7.06 (d, 1H, Harom, J = 8.5 Hz), 7.43 (d, 2H, Harom, J = 9 Hz), 7.77 (s, 1H, CH), 7.82 (s, 1H, Harom), 8.11 (s, 2H, OH), 10.17 (s, 1H, NH); MS (EI) m/z (%) 510 [M+] (84). Anal. Calcd. for C18H12BrN3O4S3: C, 42.36; H, 2.37; Br, 15.65; N, 8.23; O, 12.54; S, 18.85. Found: C, 42.36; H, 2.45; Br, 15.62; N, 8.20; S, 18.81.
2-{[5-(2-Hydroxy-3-nitrobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxy-phenyl)-2-thioxoacetamide (43). Yield 66%; m.p. 179–180 °C; 1H-NMR: δ 6.88 (d, 2H, Harom, J = 9 Hz), 7.21–7.25 (t, 1H, Harom, J = 7.7 Hz), 7.42 (d, 2H, Harom, J = 9 Hz), 7.87 (d, 2H, Harom, J = 7.7 Hz), 8.01 (s, 1H, CH), 8.94 (s, 2H, OH), 10.76 (s, 1H, NH); MS (EI) m/z (%) 476 [M+] (84). Anal. Calcd. for C18H12N4O6S3: C, 45.37; H, 2.54; N, 11.76; O, 20.15; S, 20.19. Found: C, 45.35; H, 2.60; N, 11.82; S, 20.17.
2-{[5-(2-Hydroxy-5-nitrobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxy-phenyl)-2-thioxoacetamide (44). Yield 69%; m.p. 199–200 °C; 1H-NMR: δ 6.89 (d, 2H, Harom, J = 9 Hz), 7.23 (d, 1H, Harom, J = 9.3 Hz), 7.43 (d, 2H, Harom, J = 8.8 Hz), 7.91 (d, 1H, Harom, J = 9.3 Hz), 8.01 (s, 1H, CH), 8.11 (s, 2H, OH), 8.2 (s, 1H, Harom), 10.05 (s, 1H, NH); MS (EI) m/z (%) 476 [M+] (73). Anal. Calcd. for C18H12N4O6S3: C, 45.37; H, 2.54; N, 11.76; O, 20.15; S, 20.19. Found: C, 45.33; H, 2.60; N, 11.81; S, 20.16.
2-{[5-(3-Ethoxy-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxyphenyl)-2-thioxoacetamide (45). Yield 90%; m.p. 184–185 °C; 1H-NMR: δ 1.4–1.43 (t, 3H, CH3, J = 6.9 Hz), 4.07 (q, 2H, CH2, J = 6.9 Hz), 6.75 (d, 1H, Harom, J = 7.8 Hz), 6.89 (d, 2H, Harom, J = 8.8 Hz), 7.17–7.21 (t, 1H, Harom, J = 7.8 Hz), 7.40-7.43 (m, 3H, Harom), 7.79 (s, 1H, CH), 8.65 (s, 2H, OH), 11.97 (s, 1H, NH); MS (EI) m/z (%) 475 [M+] (95). Anal. Calcd. for C20H17N3O5S3: C, 50.51; H, 3.60; N, 8.84; O, 16.82; S, 20.23. Found: C, 50.65; H, 3.70; N, 8.71; S, 20.22.
2-{[5-(3-Allyl-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxy-phenyl)-2-thioxoacetamide (46). Yield 81%; m.p. 153–154 °C; 1H-NMR: δ 3.26–3.28 (m, 2H, Halk), 5.01–5.05 (m, 1H, Halk), 5.12–5.18 (m, 1H, Halk), 6.02–6.12 (m, 1H, Halk), 6.89 (d, 1H, Harom, J = 9 Hz), 6.99 (d, 1H, Harom, J = 7.8 Hz), 7.11–7.15 (t, 1H, Harom, J = 7.8 Hz), 7.42 (d, 2H, Harom, J = 8.9 Hz), 7.52 (d, 1H, Harom, J = 7.8 Hz), 7.79 (s, 1H, CH), 7.86 (s, 2H, OH), 10.41 (s, 1H, NH); MS (EI) m/z (%) 471 [M+] (48). Anal. Calcd. for C21H17N3O4S3: C, 53.49; H, 3.63; N, 8.91; O, 13.57; S, 20.40. Found: C, 53.55; H, 3.67; N, 8.85; S, 20.43.
2-({5-[4-(Diethylamino)-2-hydroxybenzylidene]-4-oxo-2-thioxo-1,3-thiazolidin-3-yl}amino)-N-(4-hydroxyphenyl)-2-thioxoacetamide (47). Yield 90%; m.p. 206–207 °C; 1H-NMR: δ 1.07–1.12 (t, 6H, CH3, J = 7.2 Hz), 3.24–3.3 (q, 4H, CH2, J = 7.2 Hz), 6.5 (s, 1H, Harom), 6.61 (d, 1H, Harom, J = 9 Hz), 6.89 (d, 2H, Harom, J = 8.9 Hz), 7.32 (d, 1H, Harom, J = 9 Hz), 7.43 (d, 2H, Harom, J = 8.9 Hz), 7.82 (s, 1H, CH), 8.13 (s, 2H, OH), 10.69 (s, 1H, NH); MS (EI) m/z (%) 502 [M+] (84). Anal. Calcd. for C22H22N4O4S3: C, 52.57; H, 4.41; N, 11.15; O, 12.73; S, 19.14. Found: C, 52.61; H, 4.43; N, 11.22; S, 19.13.
N-(4-hydroxyphenyl)-2-[(5-{2-hydroxy-5-[(E)-phenyldiazenyl]benzylidene}-4-oxo-2-thioxo-1,3-thiaz-olidin-3-yl)amino]-2-thioxoacetamide (48). Yield 43%; m.p. 145–146 °C; 1H-NMR: δ 6.89 (d, 2H, Harom, J = 9 Hz), 7.13 (d, 1H, Harom, J = 8.5 Hz), 7.34–7.46 (m, 3H, Harom), 7.67 (d, 1H, Harom, J = 8.5 Hz), 7.84–7.93 (m, 4H, Harom), 7.99 (s, 2H, OH), 9.42 (s, 1H, NH); MS (EI) m/z (%) 535 [M+] (33). Anal. Calcd. for C24H17N5O4S3: C, 53.82; H, 3.20; N, 13.08; O, 11.95; S, 17.96. Found: C, 53.91; H, 3.24; N, 13.15; S, 17.92.
2-Hydroxy-5-{[3-({2-[(4-hydroxyphenyl)amino]-2-oxoethanethioyl}amino)-4-oxo-2-thioxo-1,3-thiazolidin-5-ylidene]methyl}benzoic acid (49). Yield 76%; m.p. 223–224 °C; 1H-NMR: δ 6.89 (d, 2H, Harom, J = 9 Hz), 7.2 (d, 1H, Harom, J = 8.6 Hz), 7.42 (d, 2H, Harom, J = 9 Hz), 7.59 (s, 1H, CH), 7.82 (d, 1H, Harom, J = 8.6Hz), 8.17(s, 1H, Harom), 9.27 (s, 2H, OH), 10.99 (s, 1H, NH); MS (EI) m/z (%) 475 [M+] (82). Anal. Calcd. for C19H13N3O6S3: C, 47.99; H, 2.76; N, 8.84; O, 20.19; S, 20.23. Found: C, 48.05; H, 2.81; N, 8.79; S, 20.22.
2-{[5-(3,5-Dichloro-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxy-phenyl)-2-thioxoacetamide (50). Yield 53%; m.p. 217–218 °C; 1H-NMR: δ 6.89 (d, 2H, Harom, J = 9 Hz), 7.37–7.43 (m, 3H, Harom), 7.63 (s, 1H, CH), 7.91 (s, 1H, Harom), 9.41 (s, 2H, OH), 11.93 (s, 1H, NH); MS (EI) m/z (%) 500 [M+] (77). Anal. Calcd. for C18H11Cl2N3O4S3: C, 43.21; H, 2.22; Cl, 14.17; N, 8.40; O, 12.79; S, 19.22. Found: C, 43.18; H, 2.20; Cl, 14.24; N, 8.39; S, 19.19.
2-{[5-(3,5-Dibromo-2-hydroxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxy-phenyl)-2-thioxoacetamide (51). Yield 87%; m.p. 189–190 °C; 1H-NMR: δ 6.89 (d, 2H, Harom, J = 8.8 Hz), 7.43 (d, 2H, Harom, J = 8.8 Hz), 7.59 (s, 1H, Harom), 7.73 (s, 2H, CH, Harom), 8.93 (s, 2H, OH), 11.02 (s, 1H, NH); MS (EI) m/z (%) 589 [M+] (70). Anal. Calcd. for C18H11Br2N3O4S3: C, 36.69; H, 1.88; Br, 27.12; N, 7.13; O, 10.86; S, 16.32. Found: C, 36.68; H, 1.82; Br, 27.17; N, 7.10; S, 16.29.
2-{[5-(5-Bromo-2-hydroxy-3-methoxybenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxyphenyl)-2-thioxoacetamide (52). Yield 74%; m.p. 176–177 °C; 1H-NMR: δ 3.76 (s, 3H, OCH3), 6.89 (m, 3H, Harom), 7.42 (m, 3H, Harom), 7.74 (s, 1H, CH), 8.65 (s, 2H, OH), 10.12 (s, 1H, NH); MS (EI) m/z (%) 540 [M+] (91). Anal. Calcd. for C19H14BrN3O5S3: C, 42.23; H, 2.61; Br, 14.79; N, 7.78; O, 14.80; S, 17.80. Found: C, 42.29; H, 2.66; Br, 14.82; N, 7.74; S, 17.76.
2-{[5-(2-Hydroxy-3,5-diiodobenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxy-phenyl)-2-thioxoacetamide (53). Yield 56%; m.p. 157–158 °C; 1H-NMR: δ 6.89 (d, 2H, Harom, J = 8.9 Hz), 7.42 (m, 3H, Harom), 7.63 (s, 1H, CH), 7.83 (s, 1H, Harom), 8.94 (s, 2H, OH), 10.86 (s, 1H, NH); MS (EI) m/z (%) 683 [M+] (15). Anal. Calcd. for C18H11I2N3O4S3: C, 31.64; H, 1.62; I, 37.14; N, 6.15; O, 9.37; S, 14.08. Found: C, 31.60; H, 1.58; N, 6.19; S, 14.05.
2-{[5-(2-Hydroxy-3,5-diisopropylbenzylidene)-4-oxo-2-thioxo-1,3-thiazolidin-3-yl]amino}-N-(4-hydroxyphenyl)-2-thioxoacetamide (54). Yield 77%; m.p. 184–185 °C; 1H-NMR: δ 1.1–1.15 (m, 12H, Halk), 2.45–2.54 (m, 1H, Halk), 3.14–3.21 (m, 1H, Halk), 6.8 (s, 1H, Harom), 6.89 (d, 2H, Harom, J = 8.9 Hz), 7.16 (s, 1H, Harom), 7.42 (d, 2H, Harom, J = 8.8 Hz), 7.67 (s, 1H, CH), 7.85 (s, 2H, OH), 10.13 (s, 1H, NH); MS (EI) m/z (%) 515 [M+] (83). Anal. Calcd. for C24H25N3O4S3: C, 55.90; H, 4.89; N, 8.15; O, 12.41; S, 18.65. Found: C, 55.97; H, 4.94; N, 8.10; S, 18.62.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




















































