Lead Generation and Optimization Based on Protein-Ligand Complementarity

Abstract
:1. Introduction
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein names | PDB entry | Name of ligands | PDB entry | MW |
|---|---|---|---|---|
| Map kinase P38 | 1oz1 | 3-(4-FLUOROPHENYL)-2-PYRIDIN-4-YL-1 H-PYRROLO- [3,2- B]PYRIDIN-1-OL | FPH 1 | 305.306 |
| Map kinase ERK2 | 2oji | N-BENZYL-4-[4-(3-CHLOROPHENYL)-1H-PYRAZOL-3-YL]- 1H-PYRROLE-2-CARBOXAMIDE | 33A | 376.839 |
| c-Jun N-terminal kinase 3 (JNK3) | 2ok1 | N-BENZYL-4-[4-(3-CHLOROPHENYL)-1H-PYRAZOL-3- YL]-1H-PYRROLE-2-CARBOXAMIDE | 33A | 376.839 |
| c-Jun N-terminal kinase 3 (JNK3) | NA | N-(3,4-DICHLOROPHENYL)-4-HYDROXY-1-METHYL-2,2-DIOXO-1,2-DIHYDRO-2LAMBDA~6~-THIENO[3,2-c][1,2]THIAZINE-3-CARBOXAMIDE | Z1208 2 | 405.278 |
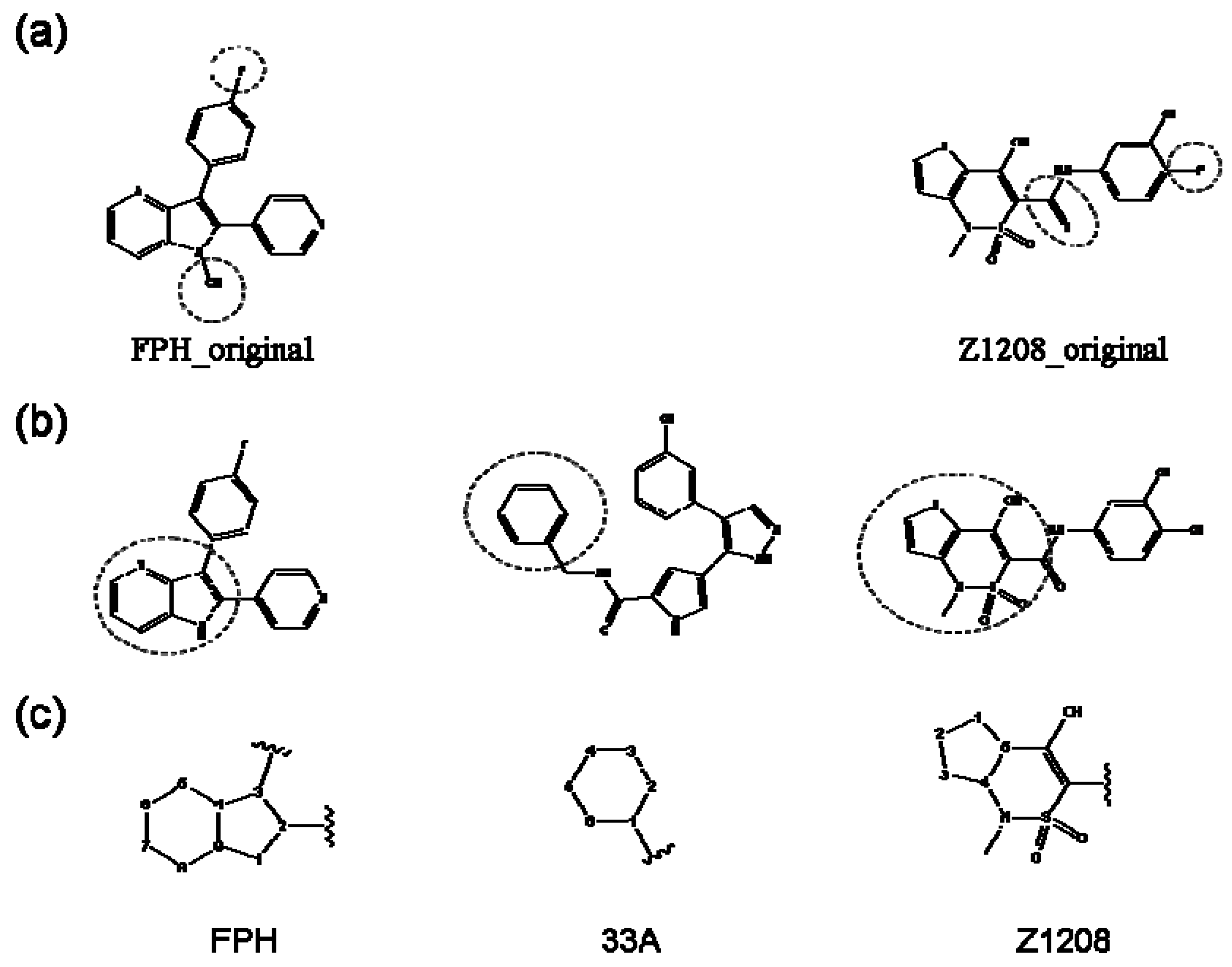
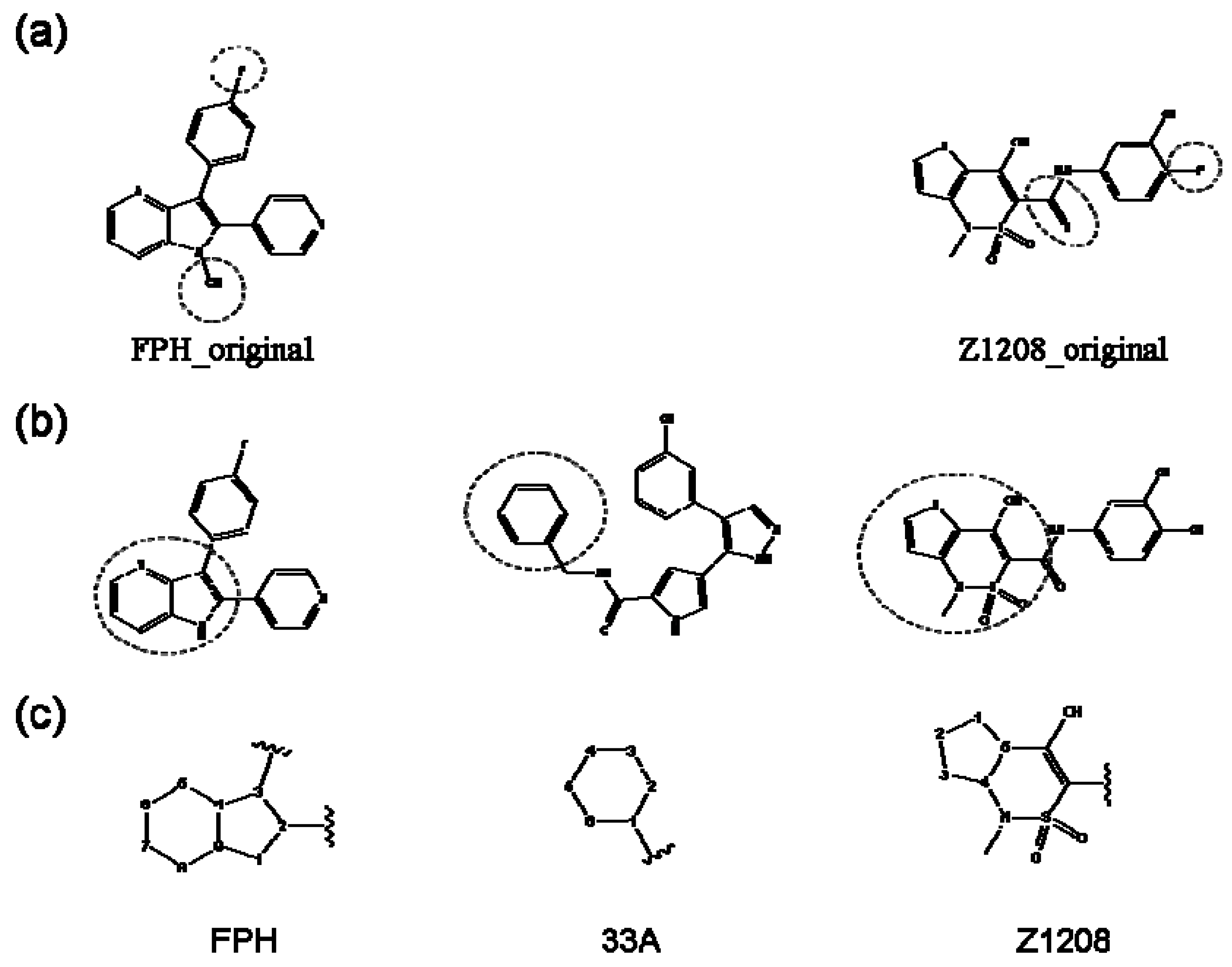
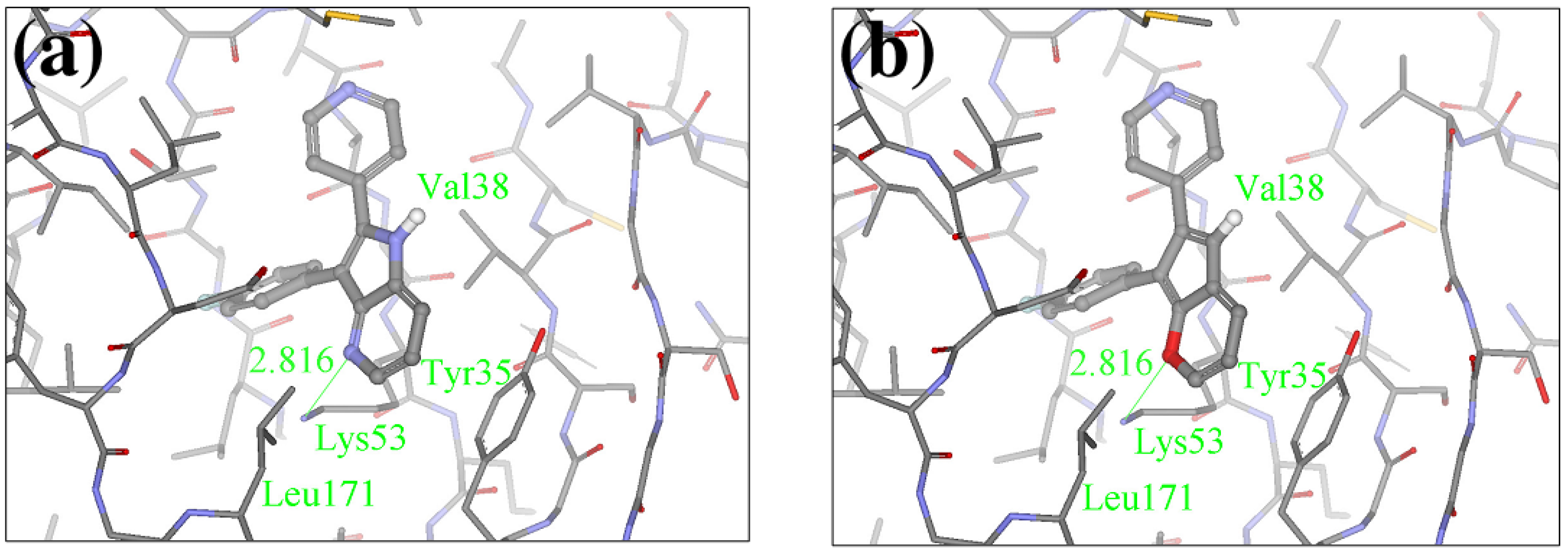
2.1. Redesign of the FPH ligand
| Ranking | Compounds | Score (Kcal/mol) | Ranking | Compounds | Score (Kcal/mol) |
|---|---|---|---|---|---|
| 1 |  | -38.536 | 6 | 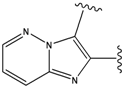 | -37.916 |
| 2 | 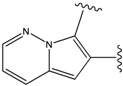 | -38.51 | 7 | 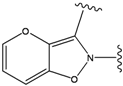 | -37.834 |
| 3 | 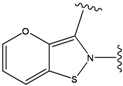 | -38.474 | 8 |  | -37.602 |
| 4 | 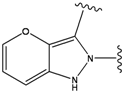 | -38.036 | 9 | 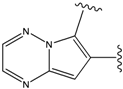 | -37.577 |
| 5 |  | -37.941 | 10 | 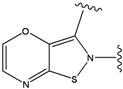 | -37.541 |

| Compounds | Substitution | Ranking | Score (Kcal/mol) | IC50 (nM)1 |
|---|---|---|---|---|
| 9 |  | 29 | -36.501 | 6.5 |
| 15 |  | 153 | 40.118 | 3100 |
| 19a | 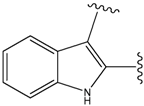 | 156 | 40.214 | 1800 |
| 22 | 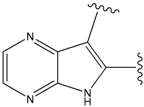 | 56 | -35.568 | 53 |
| 24 | 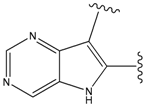 | 66 | -34.414 | 895 |
| 35 |  | 34 | -36.299 | 86 |
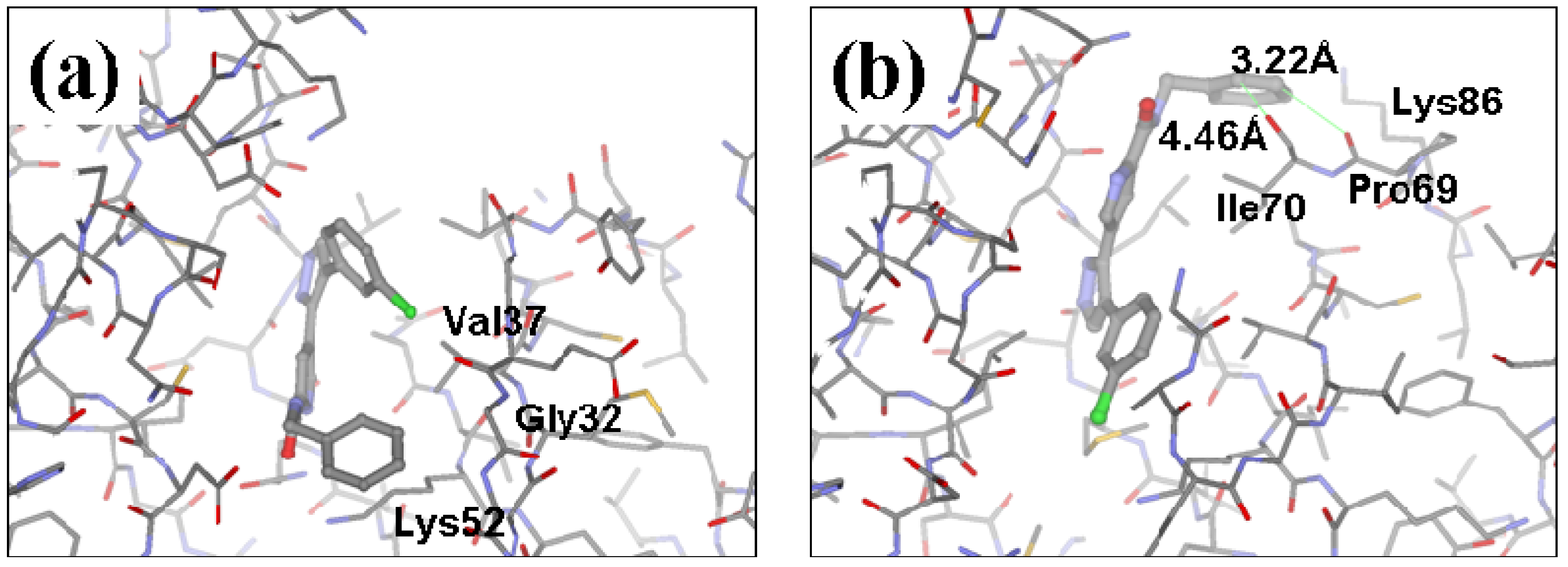
2.2. Redesign of the 33A scaffold to optimize ERK2 binding

| ERK2 | JNK3 | ||||||
|---|---|---|---|---|---|---|---|
| Ranking | Substitutions | Score | Ki, uM1 | Ranking | Substitutions | Score | Ki, uM1 |
| 1 | 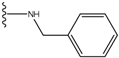 | -28.016 | 0.086 | 1 |  | -43.488 | 0.55 |
| 2 | 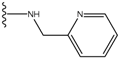 | -27.643 | --- | 2 | 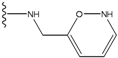 | -42.738 | --- |
| 3 |  | -27.342 | 0.23 | 3 | 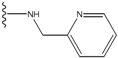 | -42.557 | --- |
| 4 |  | -26.969 | --- | 4 | 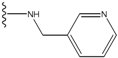 | -42.117 | ND2 |
| 5 | 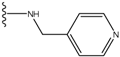 | -25.62 | 0.16 | 5 |  | -41.903 | ND2 |
2.3. Redesign of the 33A scaffold to optimize JNK3 binding
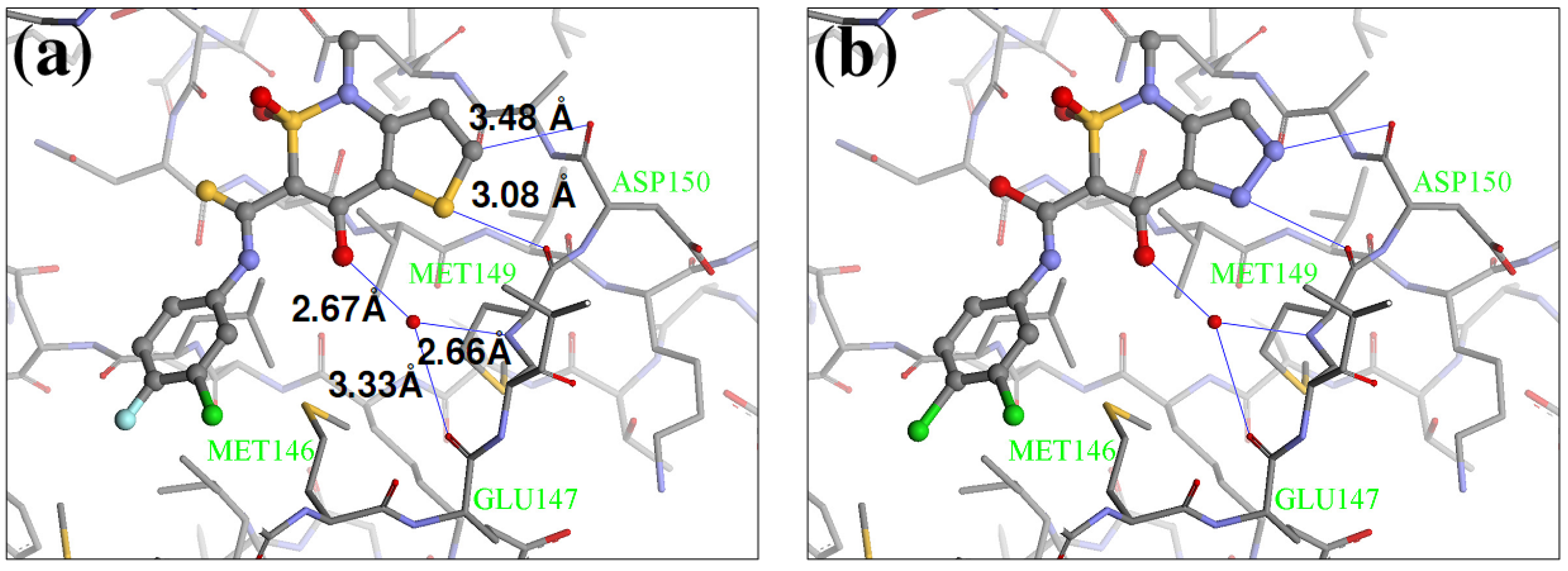
2.4. Redesign of the Z1208 scaffold bound to JNK3.
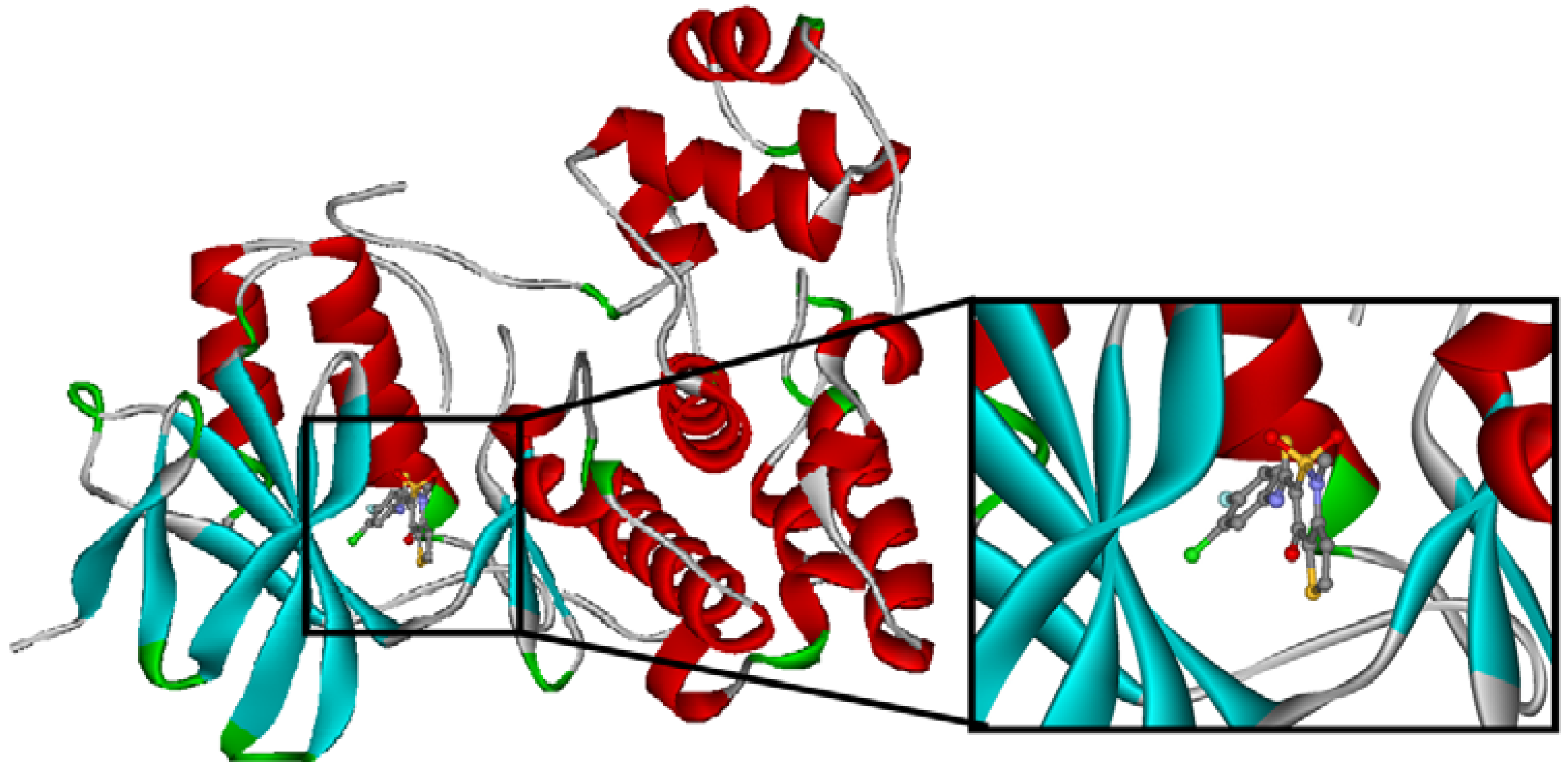

| Ranking | Substitutions | Score | IC50 (uM) | Ranking | Substitutions | Score | IC50 (uM) |
|---|---|---|---|---|---|---|---|
| 1 |  | -37.376 | NA | 6 |  | -35.815 | NA |
| 2 |  | -36.783 | NA | 7 | 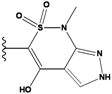 | -35.783 | NA |
| 3 | 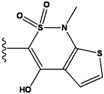 | -36.6 | NA | 8 |  | -35.569 | |
| 4 |  | -36.285 | 9 | 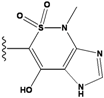 | -35.189 | NA | |
| 5 | 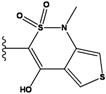 | -36.145 | NA | 10 | 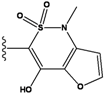 | -35.175 | NA |
2.5. Discussion
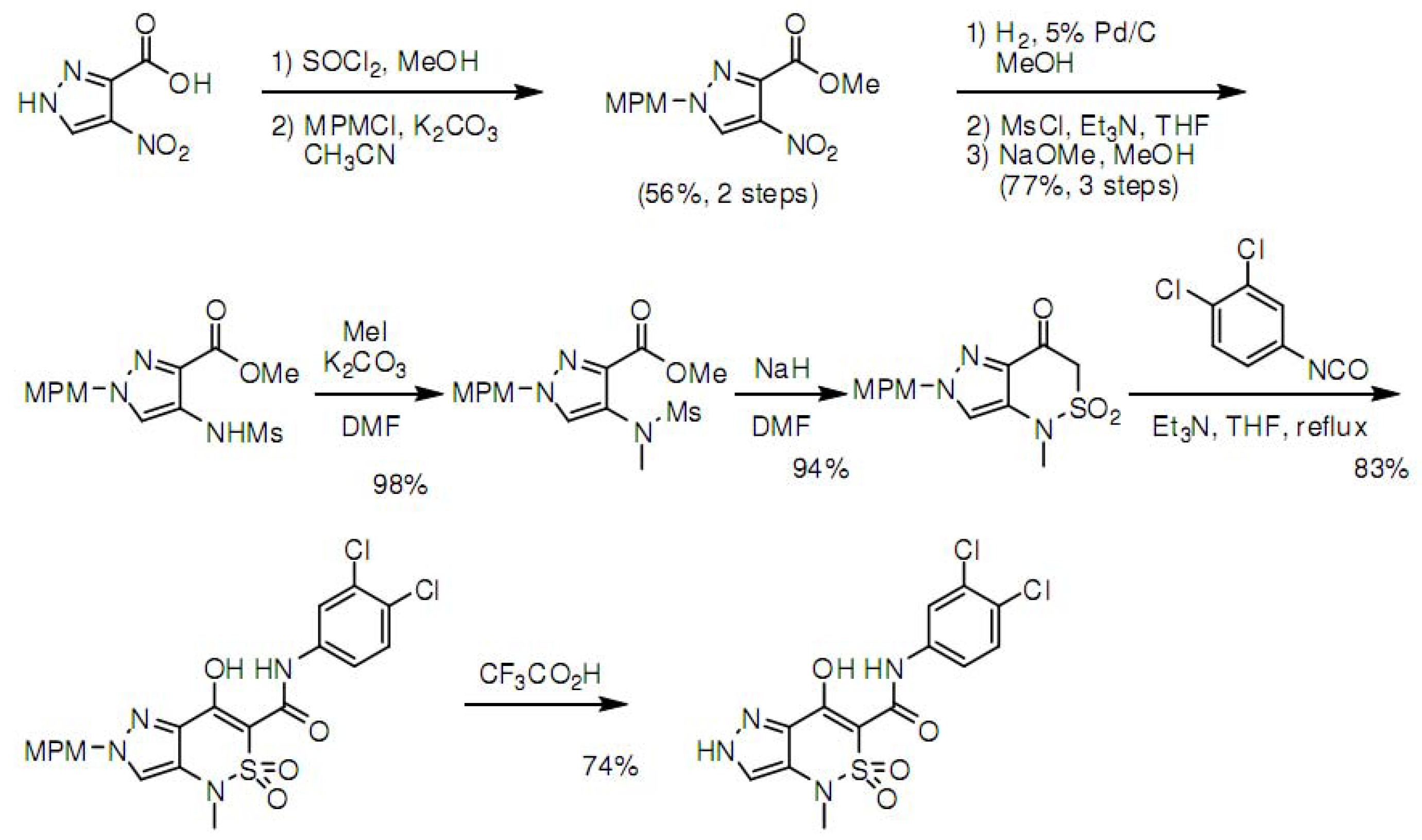
Additional validation of our method by synthesis of one molecule
3. Experimental
3.1. Lead optimization procedure
| Atom groups | Bond type | No. of bonds | No. of hydrogens | Atom groups | Bond type | No. of bonds | No. of hydrogens |
|---|---|---|---|---|---|---|---|
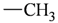 | sp3 | 4 | 3 |  | sp3 | 2 | 1 |
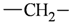 | sp3 | 4 | 2 |  | all | 2 | 0 |
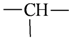 | sp3 | 4 | 1 | 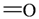 | all | 2 | 0 |
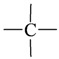 | sp3 | 4 | 0 | ||||
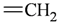 | sp2 | 4 | 2 | 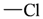 | sp3 | 1 | 0 |
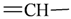 | sp2 | 4 | 1 | ||||
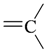 | sp2 | 4 | 0 | ||||
 | sp3 | 3 | 2 | ||||
 | all | 3 | 1 | 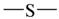 | all | 2 | 0 |
 | sp2 | 3 | 0 | 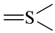 | all | 4 | 0 |
 | all | 3 | 0 | 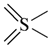 | all | 6 | 0 |

 ,
,
 ,
,
 and
and  were calculated in the same manner. This method has been designed to provide a ranked list of compounds with better “drug-type” properties (more stable, druglikeness and synthesizable compounds) than other approaches.
were calculated in the same manner. This method has been designed to provide a ranked list of compounds with better “drug-type” properties (more stable, druglikeness and synthesizable compounds) than other approaches.
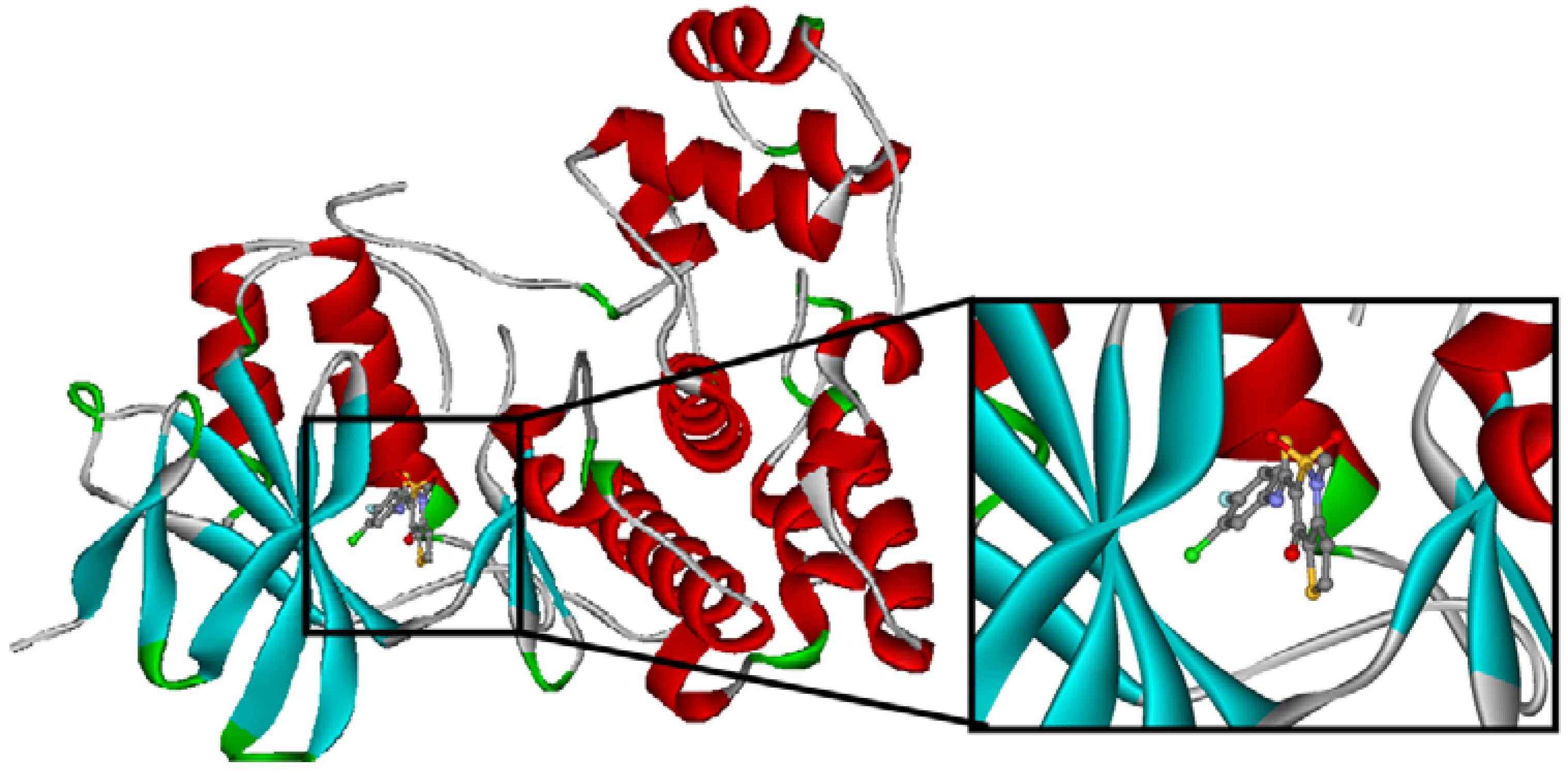
3.2. Application to serine/threonine protein kinases
3.3. Inhibition assay
3.4. Synthesis of Z1208-8

Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References
- Yamazaki, K.; Kusunose, N.; Fujita, K.; Sato, H.; Asano, S.; Dan, A.; Kanaoka, M. Identification of phosphodiesterase-1 and 5 dual inhibitors by a ligand-based virtual screening optimized for lead evolution. Bioorg. Med. Chem. Lett. 2006, 16, 1371–1379. [Google Scholar] [CrossRef]
- Yan, S.; Appleby, T.; Larson, G.; Wu, J.Z.; Hamatake, R.K.; Hong, Z.; Yao, N. Thiazolone-acylsulfonamides as novel HCV NS5B polymerase allosteric inhibitors: convergence of structure-based drug design and X-ray crystallographic study. Bioorg. Med. Chem. Lett. 2007, 17, 1991–1995. [Google Scholar]
- Carosati, E.; Mannhold, R.; Wahl, P.; Hansen, J.B.; Fremming, T.; Zamora, I.; Cianchetta, G.; Baroni, M. Virtual screening for novel openers of pancreatic K(ATP) channels. J. Med. Chem. 2007, 50, 2117–2126. [Google Scholar] [CrossRef]
- Meng, E.C.; Gschwend, D.A.; Blaney, J.M.; Kuntz, I.D. Orientational sampling and rigid-body minimization in molecular docking. Proteins 1993, 17, 266–278. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P. Docking small-molecule ligands into active sites. Curr. Opin. Biotechnol. 1995, 6, 652–656. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. eHiTS: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef]
- Zhao, H. Scaffold selection and scaffold hopping in lead generation: a medicinal chemistry perspective. Drug Discov. Today 2007, 12, 149–155. [Google Scholar] [CrossRef]
- Schnecke, V.; Bostrom, J. Computational chemistry-driven decision making in lead generation. Drug Discov. Today 2006, 11, 43–50. [Google Scholar] [CrossRef]
- Bohm, H.J. LUDI: rule-based automatic design of new substituents for enzyme inhibitor leads. J. Comput. Aided Mol. Des. 1992, 6, 593–606. [Google Scholar] [CrossRef]
- DeWitte, R.S.; Ishchenko, A.V.; Shakhnovich, E.I. SMoG: de Novo design method based on simple, fast, and accurate free energy estimates. 2. Case studies in molecular design. J. Am. Chem. Soc. 1997, 119, 4608–4617. [Google Scholar]
- Gehlhaar, D.K.; Moerder, K.E.; Zichi, D.; Sherman, C.J.; Ogden, R.C.; Freer, S.T. De novo design of enzyme inhibitors by Monte Carlo ligand generation. J. Med. Chem. 1995, 38, 466–472. [Google Scholar] [CrossRef]
- Nishibata, Y.; Itai, A. Confirmation of usefulness of a structure construction program based on three-dimensional receptor structure for rational lead generation. J. Med. Chem. 1993, 36, 2921–2928. [Google Scholar] [CrossRef]
- Ahlstrom, M.M.; Ridderstrom, M.; Luthman, K.; Zamora, I. Virtual screening and scaffold hopping based on GRID molecular interaction fields. J. Chem. Inf. Model 2005, 45, 1313–1323. [Google Scholar] [CrossRef]
- Bergmann, R.; Linusson, A.; Zamora, I. SHOP: Scaffold HOPping by GRID-Based Similarity Searches. J. Med. Chem. 2007, 50, 2708–2717. [Google Scholar] [CrossRef]
- Schneider, G.; Neidhart, W.; Giller, T.; Schmid, G. "Scaffold-Hopping" by Topological Pharmacophore Search: A Contribution to Virtual Screening. Angew. Chem. Int. Ed. Engl. 1999, 38, 2894–2896. [Google Scholar] [CrossRef]
- Abolmaali, S.F.; Ostermann, C.; Zell, A. The Compressed Feature Matrix--a novel descriptor for adaptive similarity search. J. Mol. Model. 2003, 9, 66–75. [Google Scholar] [CrossRef]
- Renner, S.; Schneider, G. Scaffold-hopping potential of ligand-based similarity concepts. ChemMedChem 2006, 1, 181–185. [Google Scholar] [CrossRef]
- Barker, E.J.; Buttar, D.; Cosgrove, D.A.; Gardiner, E.J.; Kitts, P.; Willett, P.; Gillet, V.J. Scaffold hopping using clique detection applied to reduced graphs. J. Chem. Inf. Model. 2006, 46, 503–511. [Google Scholar] [CrossRef]
- Naerum, L.; Norskov-Lauritsen, L.; Olesen, P.H. Scaffold hopping and optimization towards libraries of glycogen synthase kinase-3 inhibitors. Bioorg. Med. Chem. Lett. 2002, 12, 1525–1528. [Google Scholar] [CrossRef]
- Lloyd, D.G.; Buenemann, C.L.; Todorov, N.P.; Manallack, D.T.; Dean, P.M. Scaffold hopping in de novo design. Ligand generation in the absence of receptor information. J. Med. Chem. 2004, 47, 493–496. [Google Scholar] [CrossRef]
- Nair, P.C.; Sobhia, M.E. Fingerprint Directed Scaffold Hopping for Identification of CCR2 Antagonists. J. Chem. Inf. Model. 2008, 48, 1891–1902. [Google Scholar] [CrossRef]
- Mason, J.S.; Morize, I.; Menard, P.R.; Cheney, D.L.; Hulme, C.; Labaudiniere, R.F. New 4-point pharmacophore method for molecular similarity and diversity applications: overview of the method and applications, including a novel approach to the design of combinatorial libraries containing privileged substructures. J. Med. Chem. 1999, 42, 3251–64. [Google Scholar] [CrossRef]
- Andrews, K.M.; Cramer, R.D. Toward general methods of targeted library design: topomer shape similarity searching with diverse structures as queries. J. Med. Chem. 2000, 43, 1723–1740. [Google Scholar] [CrossRef]
- Jenkins, J.L.; Glick, M.; Davies, J.W. A 3D similarity method for scaffold hopping from known drugs or natural ligands to new chemotypes. J. Med. Chem. 2004, 47, 6144–6159. [Google Scholar] [CrossRef]
- Bohl, M.; Dunbar, J.; Gifford, E.M.; Heritage, T.; Wild, D.J.; Willett, P.; Wilton, D.J. Scaffold Searching: Automated Identification of Similar Ring Systems for the Design of Combinatorial Libraries. Quant. Struct.-Act. Relat. 2002, 21, 590–597. [Google Scholar] [CrossRef]
- Jorgensen, W.L. Rusting of the lock and key model for protein-ligand binding. Science 1991, 254, 954–955. [Google Scholar]
- Grant, M.A. Protein structure prediction in structure-based ligand design and virtual screening. Comb. Chem. High Throughput Screening 2009, 12, 940–960. [Google Scholar] [CrossRef]
- Aronov, A.M.; Baker, C.; Bemis, G.W.; Cao, J.; Chen, G.; Ford, P.J.; Germann, U.A.; Green, J.; Hale, M.R.; Jacobs, M.; Janetka, J.W.; Maltais, F.; Martinez-Botella, G.; Namchuk, M.N.; Straub, J.; Tang, Q.; Xie, X. Flipped out: structure-guided design of selective pyrazolylpyrrole ERK inhibitors. J. Med. Chem. 2007, 50, 1280–1287. [Google Scholar]
- Trejo, A.; Arzeno, H.; Browner, M.; Chanda, S.; Cheng, S.; Comer, D.D.; Dalrymple, S.A.; Dunten, P.; Lafargue, J.; Lovejoy, B.; Freire-Moar, J.; Lim, J.; McIntosh, J.; Miller, J.; Papp, E.; Reuter, D.; Roberts, R.; Sanpablo, F.; Saunders, J.; Song, K.; Villasenor, A.; Warren, S.D.; Welch, M.; Weller, P.; Whiteley, P.E.; Zeng, L.; Goldstein, D.M. Design and synthesis of 4-azaindoles as inhibitors of p38 MAP kinase. J. Med. Chem. 2003, 46, 4702–13. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–42. [Google Scholar]
- Scapin, G.; Patel, S.B.; Lisnock, J.; Becker, J.W.; LoGrasso, P.V. The structure of JNK3 in complex with small molecule inhibitors: structural basis for potency and selectivity. Chem. Biol. 2003, 10, 705–712. [Google Scholar] [CrossRef]
- Ogata, K.; Isomura, T.; Yamashita, H.; Kubodera, H. A Quantitative Approach to the Estimation of Chemical Space from a Given Geometry by the Combination of Atomic Species. QSAR Comb. Sci. 2007, 26, 596–607. [Google Scholar] [CrossRef]
- Rishton, G.M. Nonleadlikeness and leadlikeness in biochemical screening. Drug Discov. Today 2003, 8, 86–96. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Ooi, T.; Oobatake, M.; Nemethy, G.; Scheraga, H.A. Accessible surface areas as a measure of the thermodynamic parameters of hydration of peptides. Proc. Natl. Acad. Sci. USA 1987, 84, 3086–3090. [Google Scholar] [CrossRef]
- Peifer, C.; Kinkel, K.; Abadleh, M.; Schollmeyer, D.; Laufer, S. From five- to six-membered rings: 3,4-diarylquinolinone as lead for novel p38MAP kinase inhibitors. J. Med. Chem. 2007, 50, 1213–21. [Google Scholar] [CrossRef]
- Kulkarni, R.G.; Srivani, P.; Achaiah, G.; Sastry, G.N. Strategies to design pyrazolyl urea derivatives for p38 kinase inhibition: a molecular modeling study. J. Comput. Aided Mol. Des. 2007, 21, 155–66. [Google Scholar] [CrossRef]
- Gaillard, P.; Jeanclaude-Etter, I.; Ardissone, V.; Arkinstall, S.; Cambet, Y.; Camps, M.; Chabert, C.; Church, D.; Cirillo, R.; Gretener, D.; Halazy, S.; Nichols, A.; Szyndralewiez, C.; Vitte, P.A.; Gotteland, J.P. Design and synthesis of the first generation of novel potent, selective, and in vivo active (benzothiazol-2-yl)acetonitrile inhibitors of the c-Jun N-terminal kinase. J. Med. Chem. 2005, 48, 4596–4607. [Google Scholar]
- Xie, X.; Gu, Y.; Fox, T.; Coll, J.T.; Fleming, M.A.; Markland, W.; Caron, P.R.; Wilson, K.P.; Su, M.S. Crystal structure of JNK3: a kinase implicated in neuronal apoptosis. Structure 1998, 6, 983–991. [Google Scholar] [CrossRef]
- Lombardino, J.G. Preparation of Some 4-Hydroxyl-1-Methyl-1h-2.1-Benzothiazine-3-Carboxanilide 2,2-Dioxides. J. Heterocycl. Chem. 1972, 9, 315–317. [Google Scholar] [CrossRef]
- Coppo, F.T.; Fawzi, M.M. Novel heterocycles. Synthesis of 2,3-dihydro-6-methyl-2-phenyl-4H,6H-pyrano[3,2-c][2,1]benzothiazin-4-one 5,5-dioxide and related compounds. J. Heterocycl. Chem. 1998, 35, 983–987. [Google Scholar]
- Coppo, F.T.; Fawzi, M.M. Synthesis of 1-methyl-7-(trifluoromethyl)-1H-pyrido[2,3-c][1,2]thiazin-4(3H)-one 2,2-dioxide. J.Heterocycl. Chem. 1998, 35, 499–501. [Google Scholar] [CrossRef]
- Coppola, G.M.; Hardtmann, G.E. Novel Heterocycles .4. Synthesis of the Pyrido[2,3-C]-1,2-Thiazine Ring-System. J. Heterocycl.Chem. 1979, 16, 1361–1363. [Google Scholar] [CrossRef]
© 2010 by the authors;
Share and Cite
Ogata, K.; Isomura, T.; Kawata, S.; Yamashita, H.; Kubodera, H.; Wodak, S.J. Lead Generation and Optimization Based on Protein-Ligand Complementarity. Molecules 2010, 15, 4382-4400. https://doi.org/10.3390/molecules15064382
Ogata K, Isomura T, Kawata S, Yamashita H, Kubodera H, Wodak SJ. Lead Generation and Optimization Based on Protein-Ligand Complementarity. Molecules. 2010; 15(6):4382-4400. https://doi.org/10.3390/molecules15064382
Chicago/Turabian StyleOgata, Koji, Tetsu Isomura, Shinji Kawata, Hiroshi Yamashita, Hideo Kubodera, and Shoshana J. Wodak. 2010. "Lead Generation and Optimization Based on Protein-Ligand Complementarity" Molecules 15, no. 6: 4382-4400. https://doi.org/10.3390/molecules15064382
APA StyleOgata, K., Isomura, T., Kawata, S., Yamashita, H., Kubodera, H., & Wodak, S. J. (2010). Lead Generation and Optimization Based on Protein-Ligand Complementarity. Molecules, 15(6), 4382-4400. https://doi.org/10.3390/molecules15064382