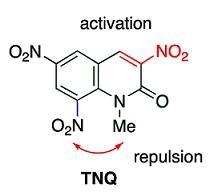
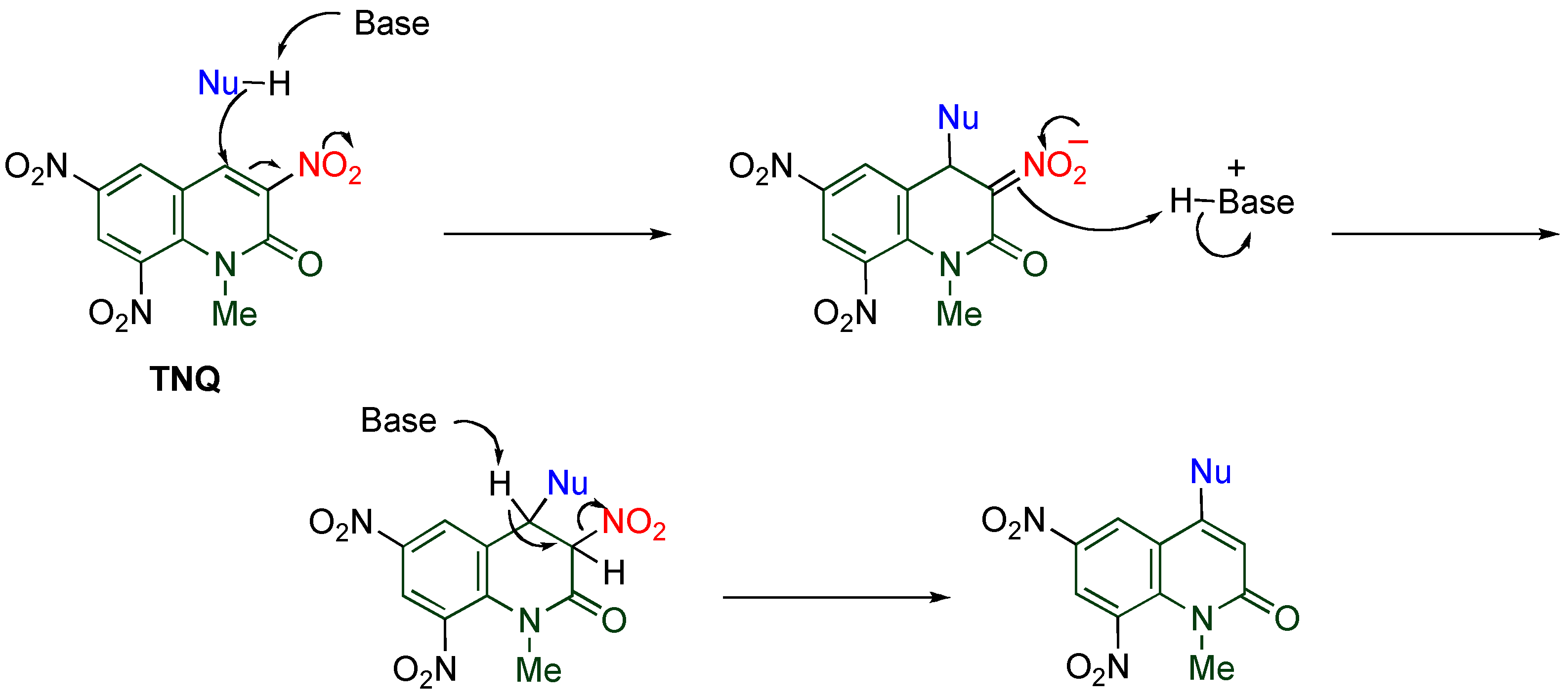
At a glance polynitrated quinolones would appear to be explosive, however, even
TNQ is quite stable under usual conditions, which enables the storage and the use of nitroquinolones without special care. Despite this stability,
TNQ exhibits significantly high reactivity, which is unusual compared with other nitroquinolones. Indeed,
TNQ reacts with versatile nucleophiles to undergo
cine-substitution, in which the nucleophile substitutes at the 4-position accompanied by elimination of the vicinal nitro group leading to 4-substituted 6,8-dinitro-1-methyl-2-quinolones (
Scheme 12). This reaction enables the regioselective functionalization at the 4-position with forming a C-N or a C-C bond. Furthermore,
TNQ undergoes cycloaddition efficiently under mild conditions to afford polycyclic MeQone derivatives. The high reactivity of
TNQ is caused by the presence of a nitro group at the 8-position, which is also mentioned in the present section.
Scheme 12.
cine-Substitution of TNQ.
4.1. Reactions with Amines
The conjugate addition of primary amines readily occurs at the 4-position of
TNQ, and the resulting adduct is converted to an ammonium salt by another molecule of amine. When the salt is heated, a small amounts of
cine-substituted product is formed, together with recovery of a large amount of
TNQ. In this reaction, 3,4-dihydroquinolone is formed under equilibrium at reflux temperature, from which the former product is afforded by deprotonation at the 4-position via
route a, and the latter product is a result of deprotonation at the 3-position via
route b (
Table 6) [
42].
Table 6.
Reaction of TNQ with primary amines.
Table 6.
Reaction of TNQ with primary amines.
![Molecules 15 05174 i006]() |
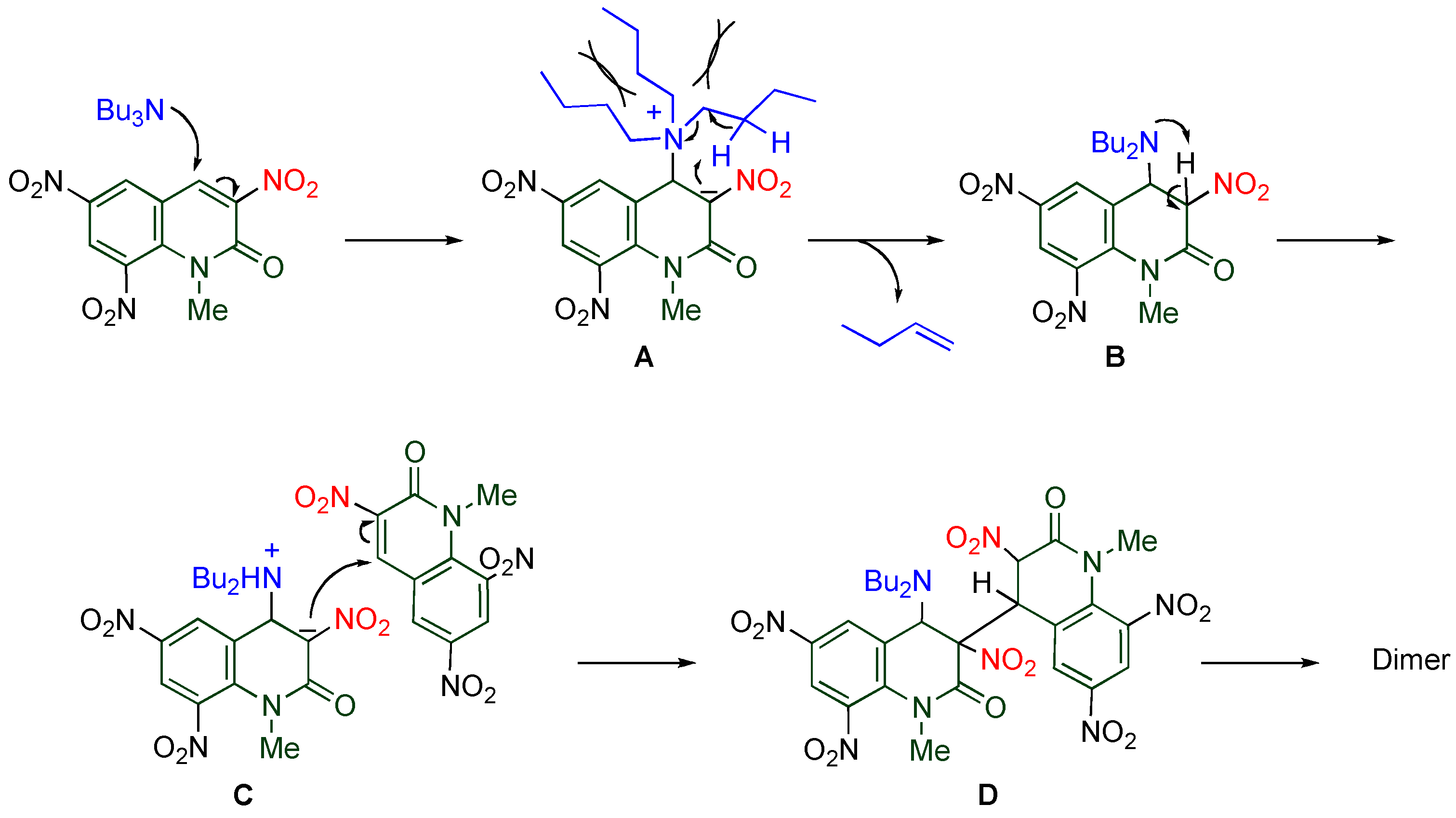
On the other hand, dimerization proceeds in the reaction of
TNQ with less nucleophilic tertiary amines, by which the dimer connected between the 3- and the 4’-positions is formed. The reaction rate is significantly affected by the length and the number of alkyl chains of the amine (
Table 7). Namely, tributylamine causes the reaction faster than tripropylamine and triethylamine, and no reaction proceeds in the cases of trimethylamine and tribenzylamine. In addition, more than two long alkyl chains are necessary for the reaction to occur efficiently [
20].
Table 7.
Dimerization of
TNQ depending on amine structure.
Table 7.
Dimerization of TNQ depending on amine structure.
| R | R’ | Yield/% |
|---|
| Bu | Bu | 93 |
| Pr | Pr | 76 |
| Et | Et | 34 |
| Bu | Me | 79 |
| Me | Bu | 18 |
A plausible mechanism is illustrated in
Scheme 13. The reaction is initiated by addition of tributylamine to the electron-deficient 4-position of
TNQ yielding zwitterion
A, from which β-elimination releasing 1-butene proceeds to afford dihydroquinolone
B. The successive proton transfer from the 3-position to the adjacent dibutylamino group leads to zwitterion
C which then reacts with another
TNQ to cause
cine-substitution, giving the dimer via dihydroquinolone adduct
D. The key step of the present reaction is the intramolecular prototropy of zwitterion
A accompanying elimination of an alkyl chain as an alkene. Since trimethylamine and tribenzylamine have no β-hydrogens, only elimination of tertiary amine from the adduct proceeds to give
TNQ again. In the case of alkyldimethylamines, the alkyl chain avoids steric repulsion with the adjacent nitro group, and the suitable conformation for β-elimination barely occurs. On the other hand, one of the alkyl chains surely locates nearby the 3-nitro group when the amino group has more than two alkyl groups. Hence, tertiary amines should have proper steric bulk for a smooth reaction [
20].
Scheme 13.
A plausible mechanism for the dimerization of TNQ.
Scheme 13.
A plausible mechanism for the dimerization of TNQ.
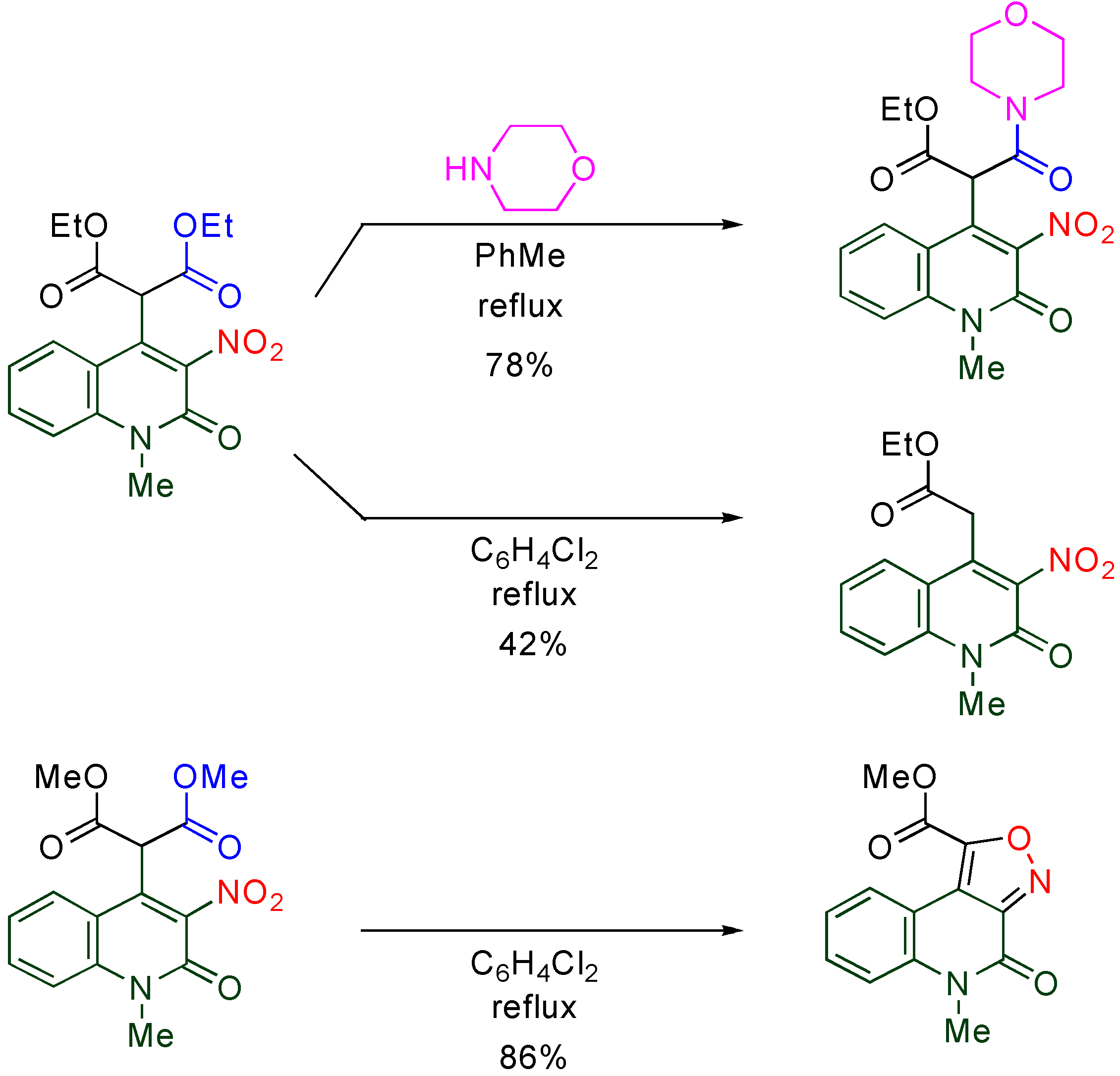
4.2. Cine-Substitution by 1,3-Dicarbonyl Compounds
The
cine-substitution of
TNQ by C-H acids is a useful protocol for regioselectively forming a C-C bond at the 4-position leading to versatile skeletons. 1,3-Dicarbonyl compounds such as β-diketones, β-keto esters and β-diesters easily react with
TNQ in the presence of triethylamine at room temperature to afford 4-functionalized 6,8-dinitro-2-quinolones (
Table 8) [
18].
Table 8.
Reactions of nitroquinolones with 1,3-dicarbonyl compounds.
Table 8.
Reactions of nitroquinolones with 1,3-dicarbonyl compounds.
| X | R | | R1 | R2 | Yield/% |
|---|
| NO2 | Me | (TNQ) | Me | Me | 88 |
| NO2 | Me | (TNQ) | -(CH2)3- | 60 |
| NO2 | Me | (TNQ) | Me | OEt | 93 |
| NO2 | Me | (TNQ) | CH2COOEt | OEt | 26 |
| NO2 | Me | (TNQ) | OEt | OEt | 99 |
| NO2 | H | (TNQ-H) | Me | Me | 0 |
| H | Me | (3,6-DNQ) | Me | Me | 0 |
On the other hand, neither demethylated trinitroquinolone (
TNQ-H) nor 3,6-dinitroquinolone (
3,6-DNQ) causes
cine-substitution, even at elevated temperature. These facts suggest that both 1-methyl and 8-nitro groups are necessary for the high reactivity of
TNQ [
43].
MOPAC (PM3) molecular orbital calculations for
3,6-DNQ, and
TNQ-H reveal that both the benzene and pyridone rings present are almost coplanar. In contrast, the 8-nitro group of
TNQ-Me has no coplanarity with the quinolone ring, which turns through 67.7°. Furthermore, the 2-quinolone ring is torsionally strained by the steric compression of the 1-methyl and the 8-nitro groups. The dihedralangle between C8-NO2 bond and N1-Me one is 30.0° (
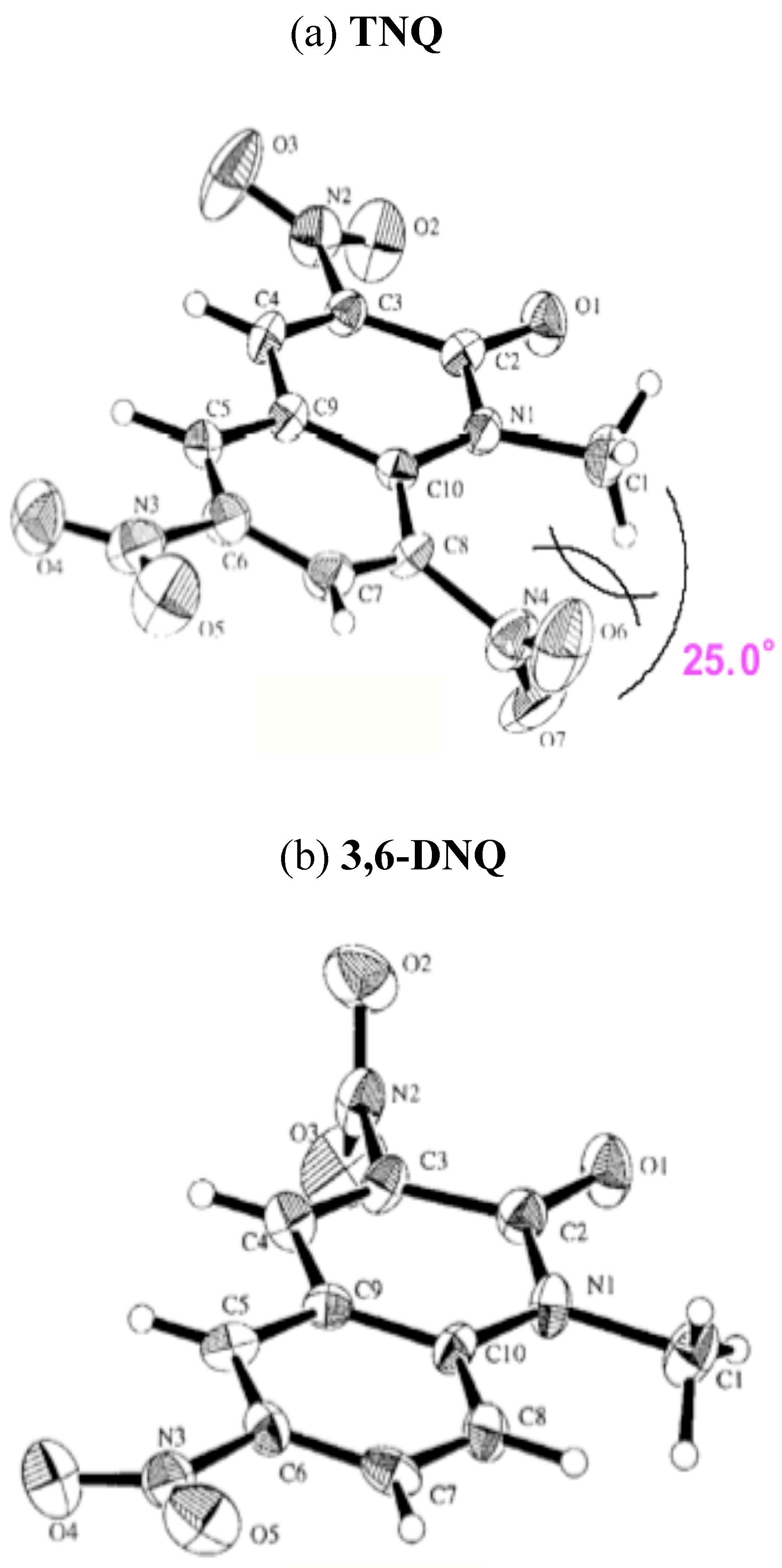
Table 9). Indeed, X-ray analyses for
3,6-DNQ and
TNQ agree well with the calculated results; the 8-NO2 group of
TNQ was turned through 55.8° and the dihedral angle is 25.0° (
Figure 2). From the agreed results of X-ray analyses to calculated ones for
TNQ and
3,6-DNQ, the actual dihedral angle between the pyridone ring and the benzene ring of
TNQ-H is also presumed to be a small [
43].
Table 9.
Dihedral angles between
![Molecules 15 05174 i013]()
and
![Molecules 15 05174 i014]()
bonds.
Table 9.
Dihedral angles between ![Molecules 15 05174 i013]() and
and ![Molecules 15 05174 i014]() bonds.
bonds.
| | R | | Calculated | Actual |
|---|
| NO2 | Me | TNQ | 30.0° | 25.0° |
| H | Me | 3,6-DNQ | 0.7° | 0.9° |
| NO2 | H | TNQ-H | 0.04° | −− |
Figure 2.
(a) An ORTEP view of TNQ. (b) An ORTEP view of 3,6-DNQ. (30% probability ellipsoids).
Figure 2.
(a) An ORTEP view of TNQ. (b) An ORTEP view of 3,6-DNQ. (30% probability ellipsoids).
The 8-nitro group cannot diminish the electron density of the reaction site (the 4-position) because of the large distance and the fact its torsion interferes with the conjugation, thus, the MeQone framework is activated sterically rather than electronically. Namely, the nitro group significantly distorts the quinolone ring by steric repulsion with the 1-methyl group. As a result, the pyridone moiety of the MeQone cannot be coplanar with the benzene ring, which prevents the π-orbitals from overlapping effectively. Consequently, the pyridone moiety exhibits nitroalkene property rather than aromaticity, and which should be a major reason for the extremely high reactivity of TNQ.
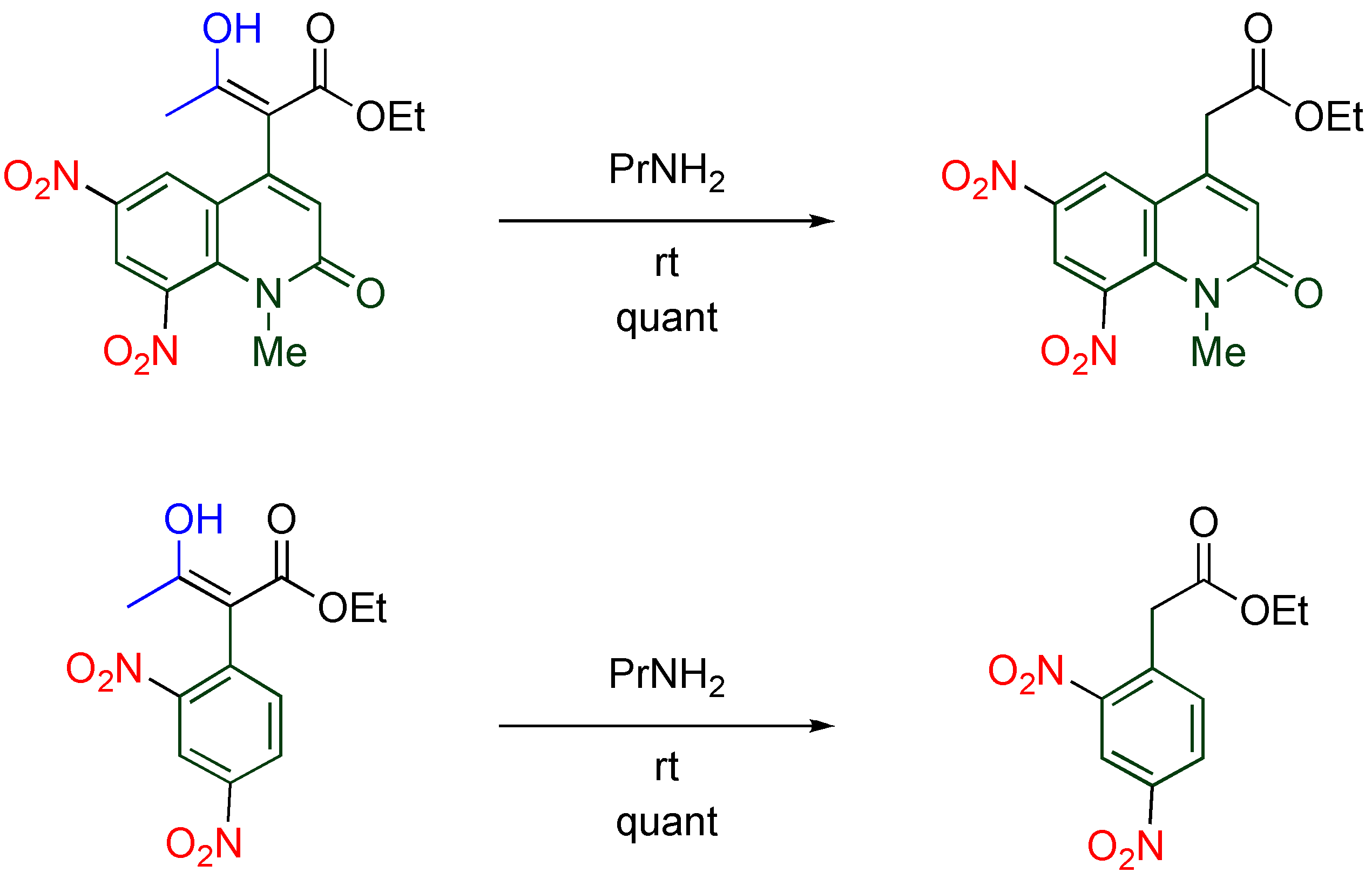
4.4. Cine-Substitution by Enamines, Ketones, and Nitroalkanes
The
cine-substitution similarly proceeds when other carbon nucleophiles such as enamines and ketones are employed [
44]. In the reaction of
TNQ with enamines, morpholinium salts of 3,4-dihydro-quinolones are formed, in which intermediate iminium ions are hydrolyzed and deprotonation at the 3-position by the liberated morpholine proceeds (
Table 10).
Table 10.
Reaction of
TNQ with enamines.
Table 10.
Reaction of TNQ with enamines.
| R1 | R2 | R3 | Yield/% |
|---|
| Ph | Me | H | 43 |
| Ph | Ph | H | 98 |
| -(CH2)3- | H | 40 |
| H | Me | Me | 98 |
Table 11.
Acylmethylation of the MeQone framework.
Table 11.
Acylmethylation of the MeQone framework.
| R1 | R2 | R3 | Yield/% |
|---|
| Ph | H | H | 83 |
| Ph | Me | H | 77 |
| Ph | Ph | H | 69 |
| Ph | Me | Me | 0 |
| -(CH2)4- | H | 82 |
| Me | H | H | 83 |
| Et | Me | H | 18 |
| H | Me | Me | 41 |
| 2-Pyridyl | H | H | 74 |
| 2-Furyl | H | H | 45 |
Acylmethylation of the MeQone framework is also performed directly by conducting reactions of
TNQ with ketones in the presence of triethylamine. This reaction is applicable to aliphatic, alicyclic, aromatic, and heteroaromatic ketones (
Table 11) [
44]. Since the acylmethyl group is expected to serve as scaffold for further chemical transformations, this method would be useful for construction of a new libraries of compounds having a MeQone framework.
Table 12.
Nitroalkylation of the MeQone framework.
Table 12.
Nitroalkylation of the MeQone framework.
![Molecules 15 05174 i012]() |
Nitroalkylation is also performed upon treatment of
TNQ with nitroalkane as a C-H acid (
Table 12) [
45]. While primary nitroalkanes undergo the
cine-substitution efficiently, secondary nitroalkanes are less reactive, requiring longer reaction time,. A tautomeric mixture of 3,4-dihydroquinolones is obtained when the reaction is conducted at room temperature.
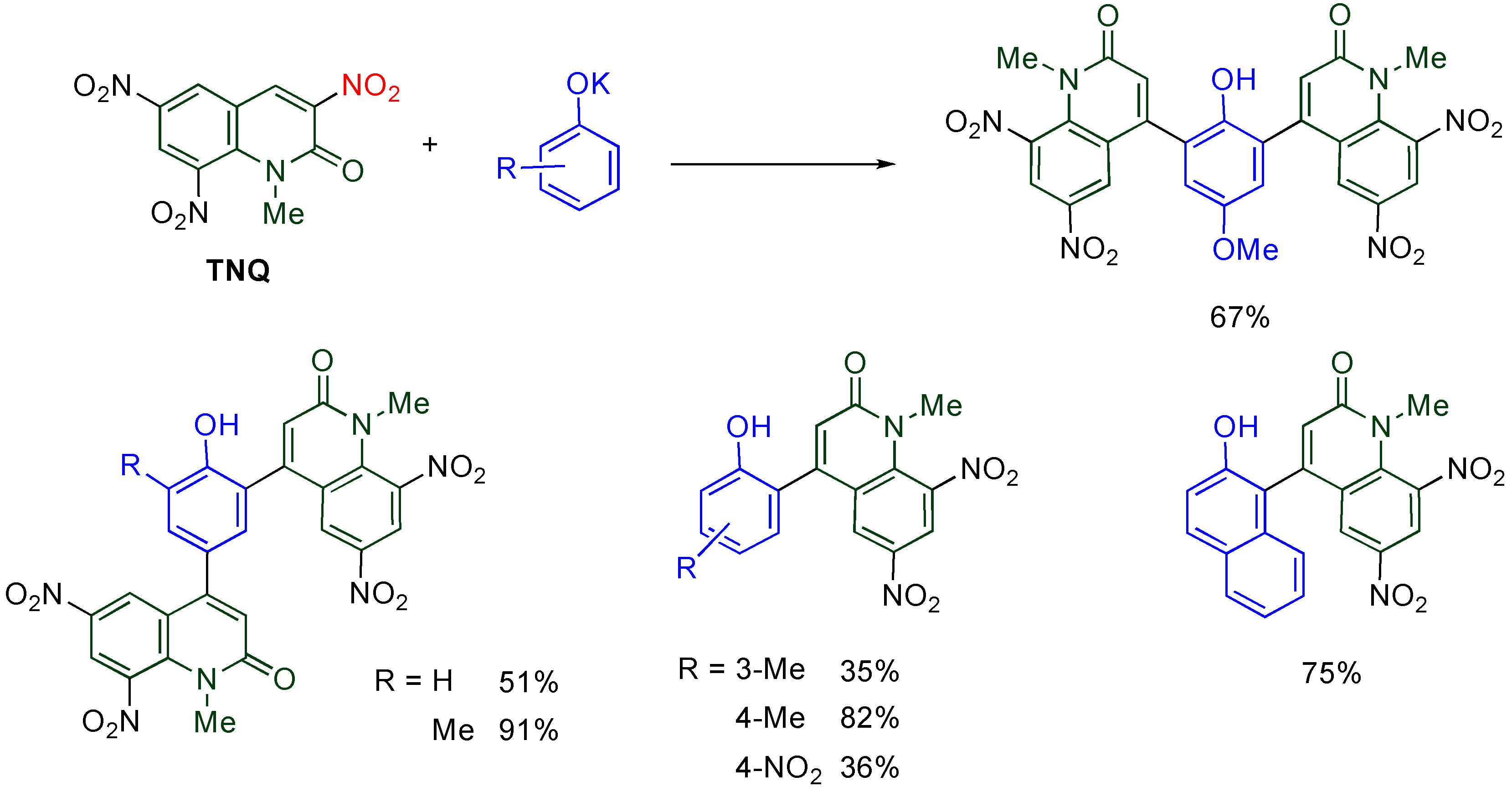
4.5. Cine-Substitution by Phenoxides
Direct arylation of MeQone is one of the more useful modifications from a viewpoint of further functionalization, however, it is quite difficult because the accompanying destruction of the aromaticity of both quinolone and benzene rings. This difficulty is overcome when a combination of electrophilic
TNQ and nucleophilic phenoxide ions is employed [
46].
While phenol, 2-methylphenol, and 4-methoxyphenol undergo double substitution to afford bis(quinolyl)phenols, bulky or electron-poor phenoxides give monoquinolylphenols as a sole product (
Scheme 15). In the reaction of
TNQ with 2-methylphenoxide, a single substituted product is not detected, even when the molar ratio of reagents is changed from 1/1 to 2/1. These experimental facts indicate that the second substitution proceeds much faster than the first one and half of the amount of phenoxide was consumed as the base.
Scheme 15.
Arylation of the MeQone framework.
Scheme 15.
Arylation of the MeQone framework.
Scheme 16.
A plausible mechanism for the present arylation
Scheme 16.
A plausible mechanism for the present arylation
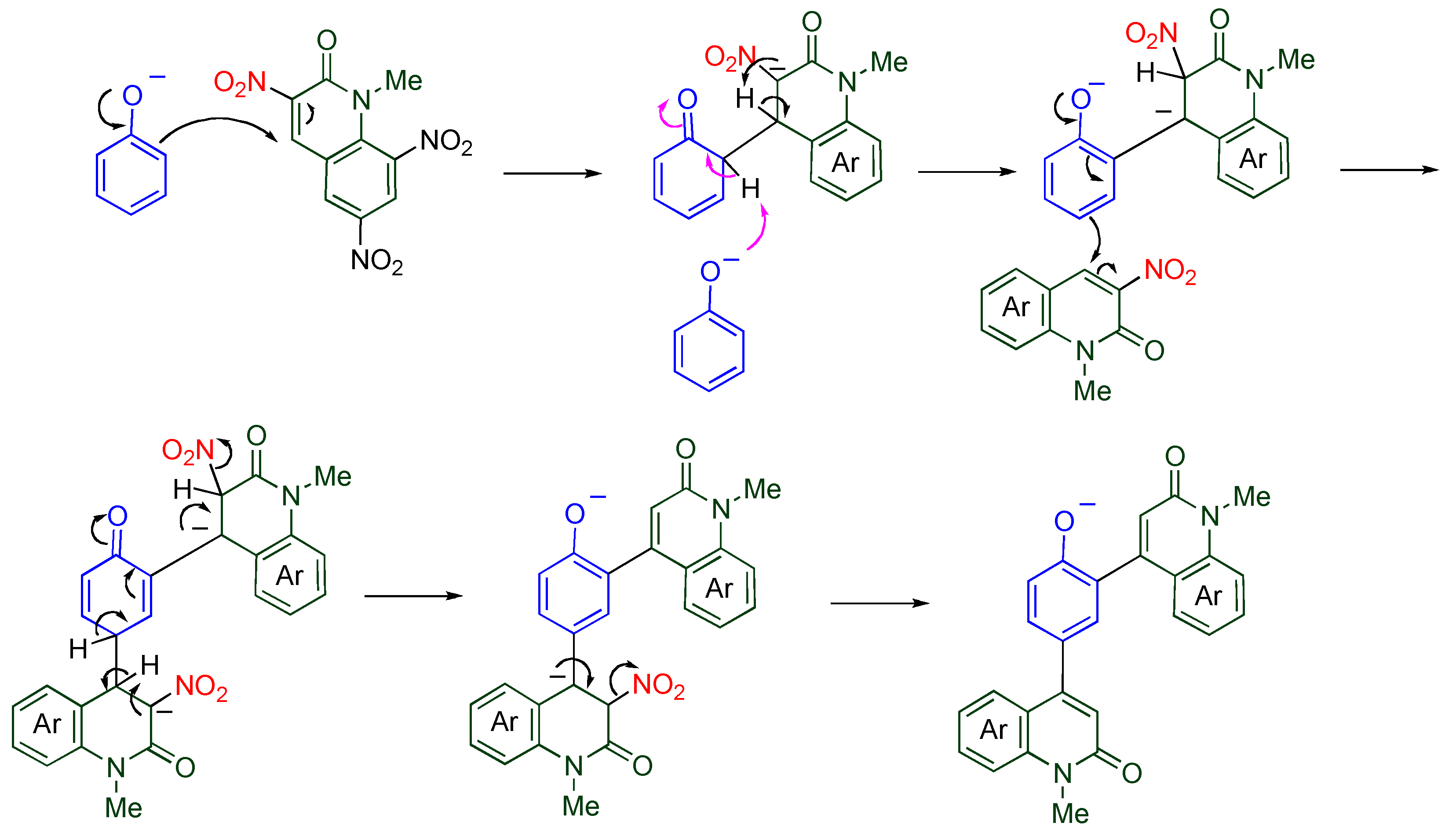
A plausible mechanism is illustrated in
Scheme 16. The phenoxide adds to the 4-position of
TNQ, and another phenoxide assists aromatization of the benzene ring. In the quinolone ring, the proton transfer also occurs from the 4-position to the 3-position. Since the resultant dianionic phenoxide is more reactive than the unsubstituted phenoxide, the second substitution proceeds much faster than the first one. The final product is formed by aromatization of the quinolone ring with loss of nitrite ion [
46]. From the standpoint of the benzene ring, the present reaction can be regarded as an electrophilic arylation, which is well-known to be quite difficult, because MeQone is also an aromatic compound.
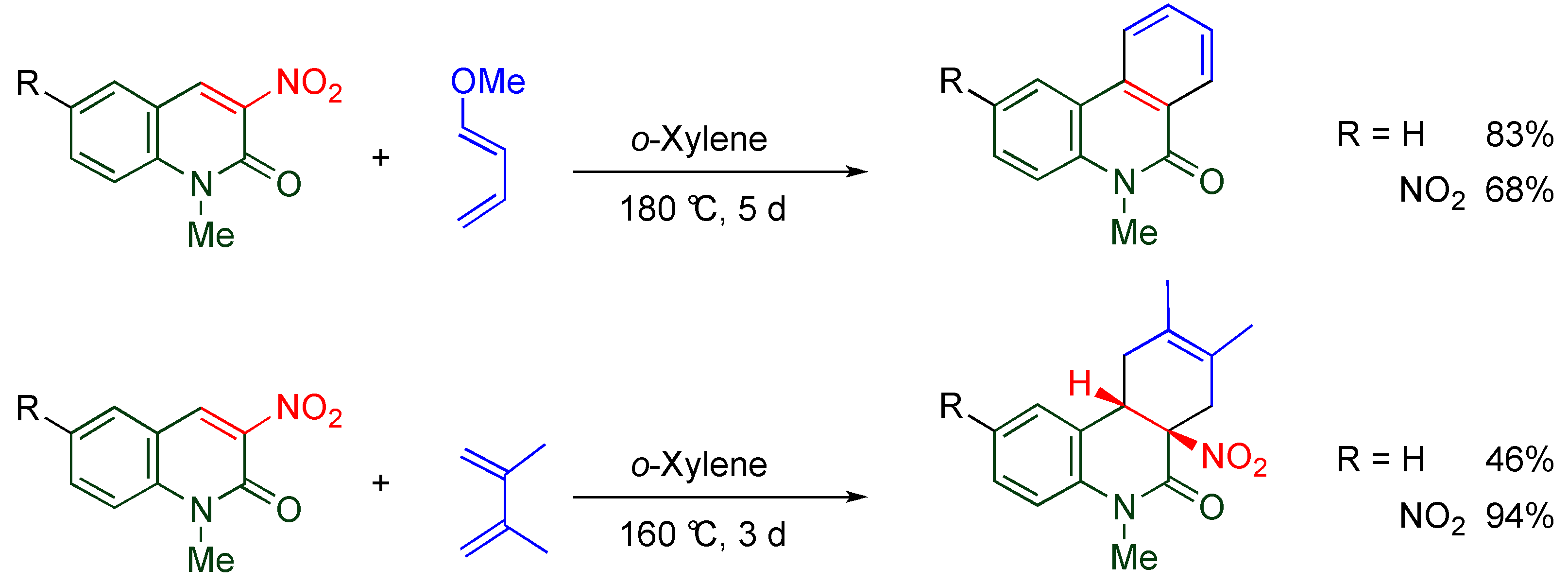
4.6. Cycloaddition of TNQ
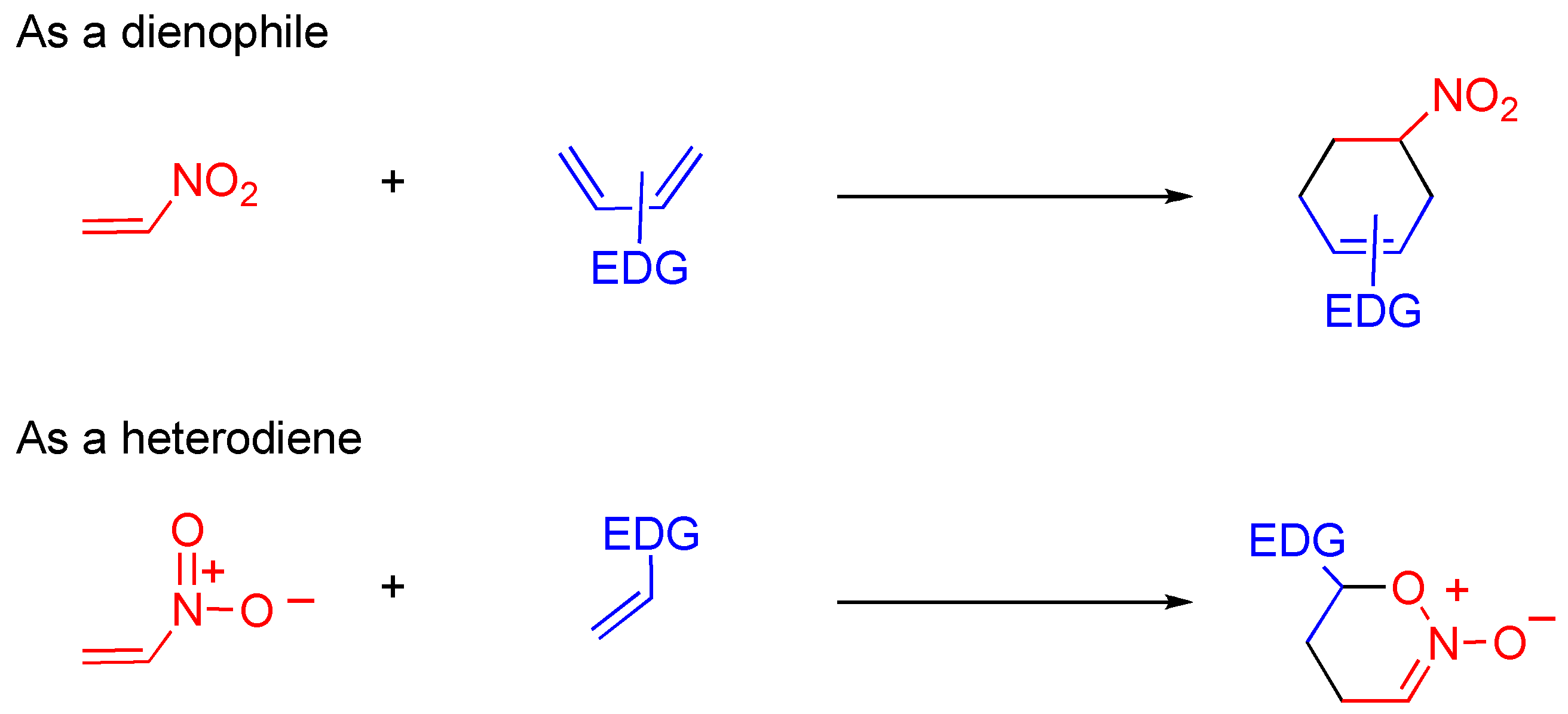
Nitroalkenes are often used for cycloaddition in which they show dual reactivity as dienophile and heterodiene, under different conditions (
Scheme 17) [
47,
48,
49]. As mentioned in
Section 4.2, the pyridone moiety reveals nitroalkene properties rather than aromaticity, which means that the C3-C4 moiety of
TNQ causes cycloaddition under mild conditions while Diels-Alder reactions of 3-nitroquinlone and 3,6-dinitroquinolone with electron-rich dienes require quite severe conditions;
i.e., at 180 °C for 5 days in
o-xylene (
Scheme 11) [
40,
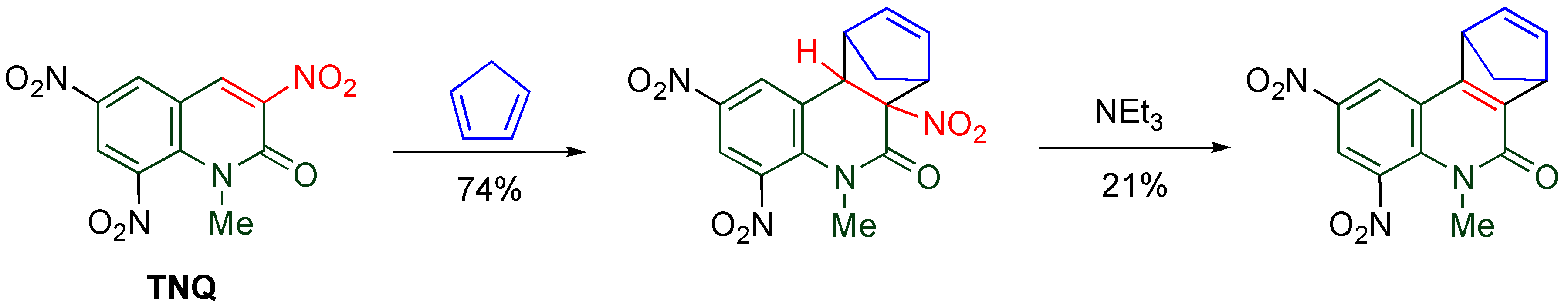
41]. Indeed, efficient cycloaddition proceeds leading to tetracyclic compounds when
TNQ is allowed to react with cyclopentadiene at 80 °C (
Scheme 18) [
50]. The cycloadduct aromatizes with elimination of nitrous acid upon treatment with triethylamine.
Scheme 17.
Dual reactivity of nitroalkene in the cycloaddition.
Scheme 17.
Dual reactivity of nitroalkene in the cycloaddition.
Scheme 18.
Diels-Alder reaction of TNQ with cyclopentadiene.
Scheme 18.
Diels-Alder reaction of TNQ with cyclopentadiene.
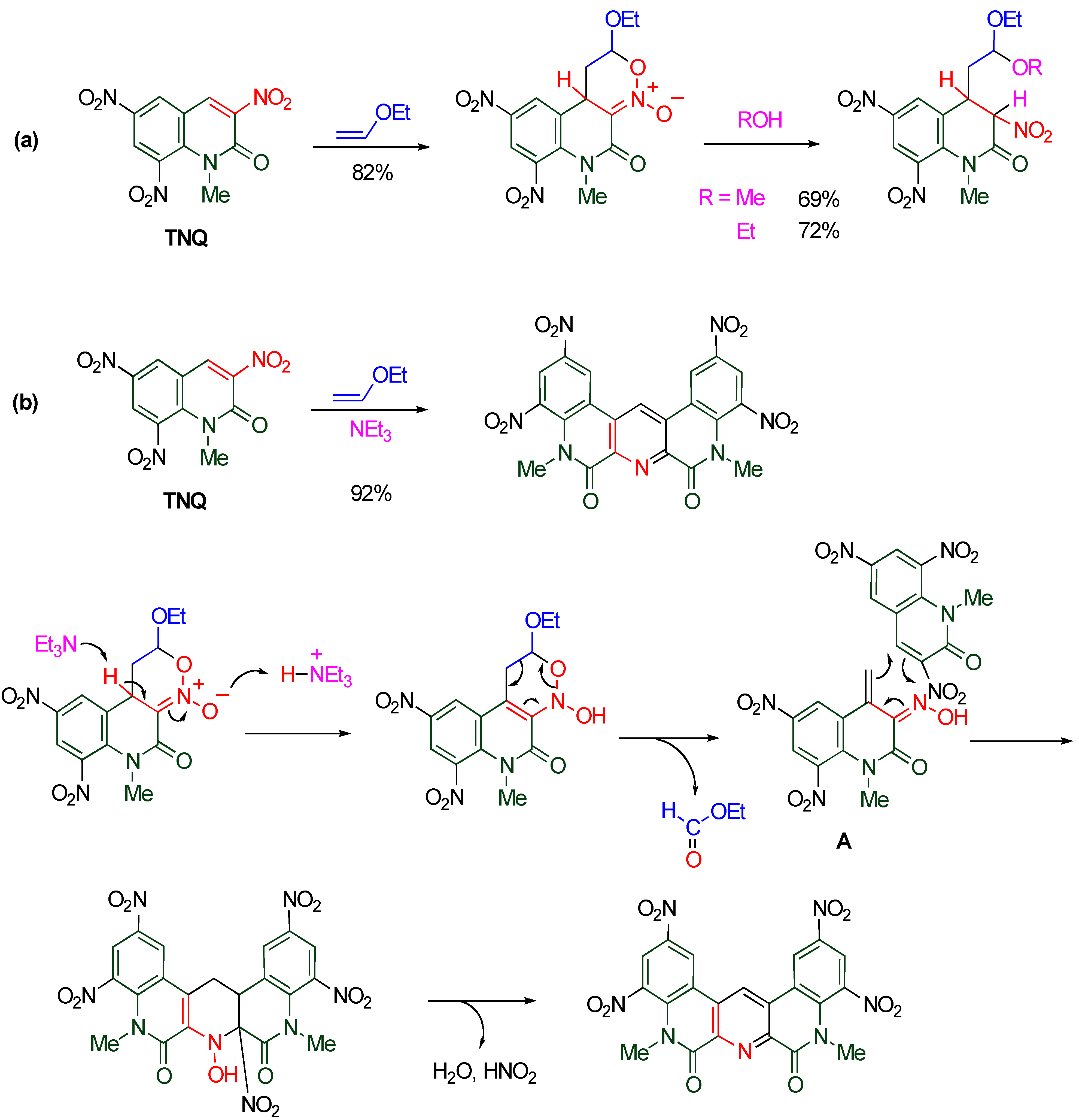
The nitroalkene moiety of
TNQ also serves as a heterodiene in the reaction with ethoxyethene to construct a fused oxazine ring. The cycloadduct is easily converted to an acetal by the ring opening reaction upon heating in alcohol. On the other hand, a quinolino[3,4-
b][1,9]diazaphenanthrene derivative is formed when the same substrates are treated in the presence of triethylamine (
Scheme 19) [
51].
Scheme 19.
(a) Cycloaddition with ethoxyethene in the absence of NEt3. (b) Cycloaddition with ethoxyethene in the presence of NEt3.
Scheme 19.
(a) Cycloaddition with ethoxyethene in the absence of NEt3. (b) Cycloaddition with ethoxyethene in the presence of NEt3.
This reaction is initiated by cycloaddition of
TNQ with ethoxyethene affording a cyclic nitronate. Triethylamine accelerates the prototropy from the pyridine ring to the oxygen atom of the nitronate, and then, a retro Diels-Alder reaction occurs to give the α,β-unsaturated oxime
A with a loss of ethyl formate. The cycloaddition of intermediate
A with another molecule of
TNQ constructs a new pyridine ring, and the subsequent aromatization furnishes the polycyclic product together with elimination of nitrous acid and water. In the present process, the former oxime
A behaves as an electron-rich heterodiene and the latter
TNQ behaves as an electron-poor dienophile [
51]. This mechanism is supported by the experimental fact that polycyclic diazaphenanthrene is isolated in a moderate yield as a result of cycloaddition of
TNQ with α,β-unsaturated oxime as the electron-rich heterodiene (
Scheme 20).
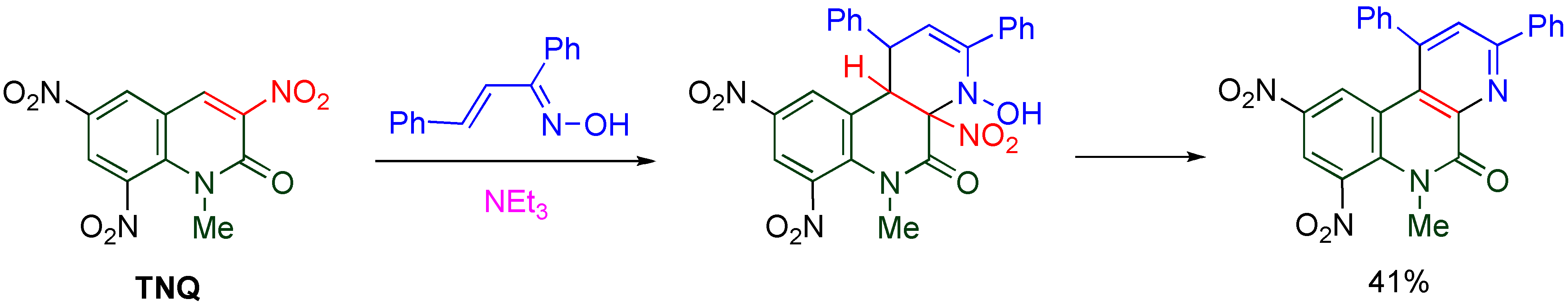
Scheme 20.
Cycloaddition of TNQ with α,β-unsaturated oxime.
Scheme 20.
Cycloaddition of TNQ with α,β-unsaturated oxime.
In general, dual reactivity is not observed at the same time except for a single example [
52] which yields both products under same conditions, however, nitro substituted bicyclo[2.2.2]octane is formed from cyclic nitronate via [
3,
3]-sigmatropic rearrangement. Thus, this is the first example showing dual reactivity in the same reaction system under mild conditions.































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






