Pre-Ischemic Treadmill Training Induces Tolerance to Brain Ischemia: Involvement of Glutamate and ERK1/2

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Physiological parameters
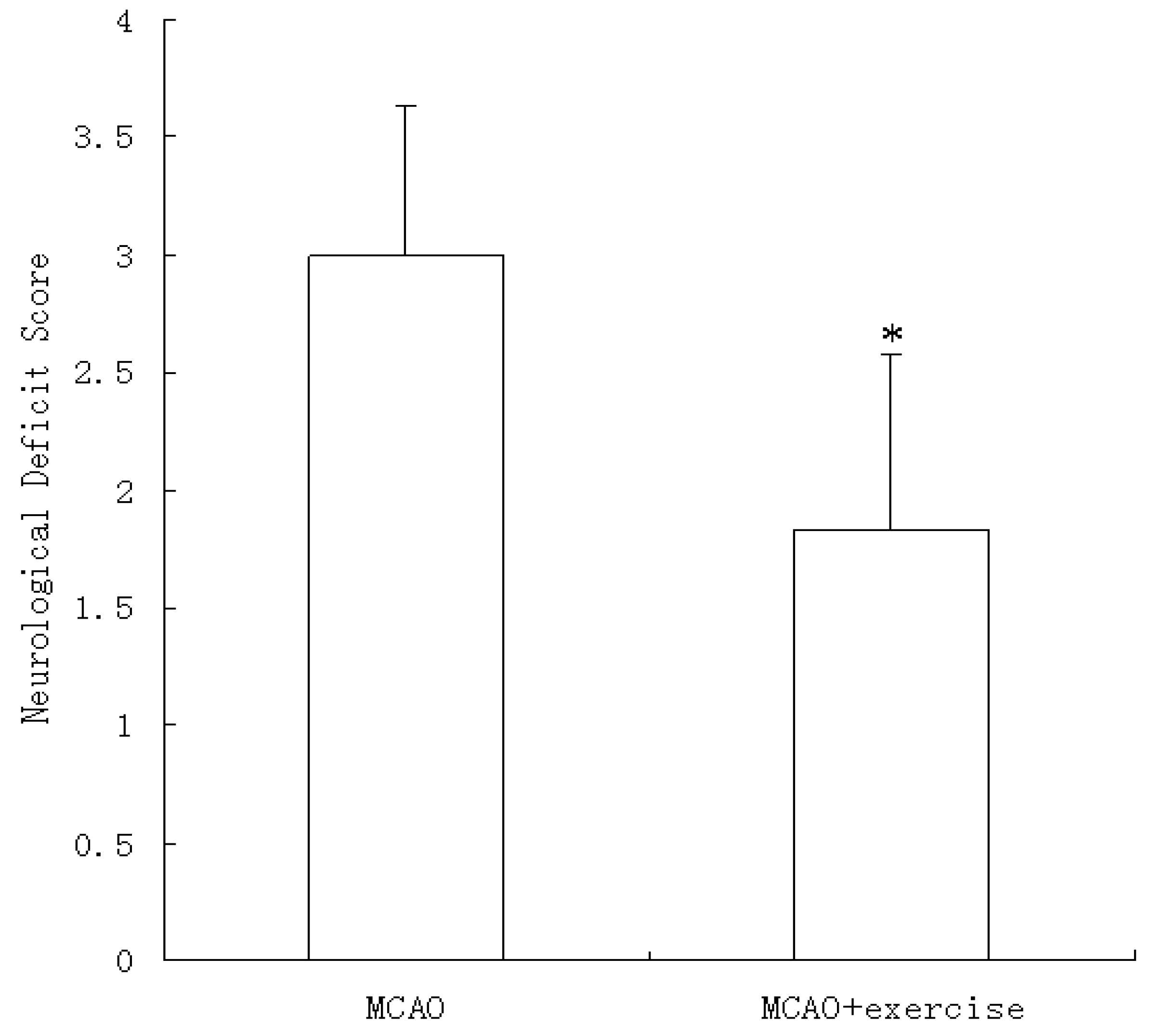
2.2. Behavioral scores

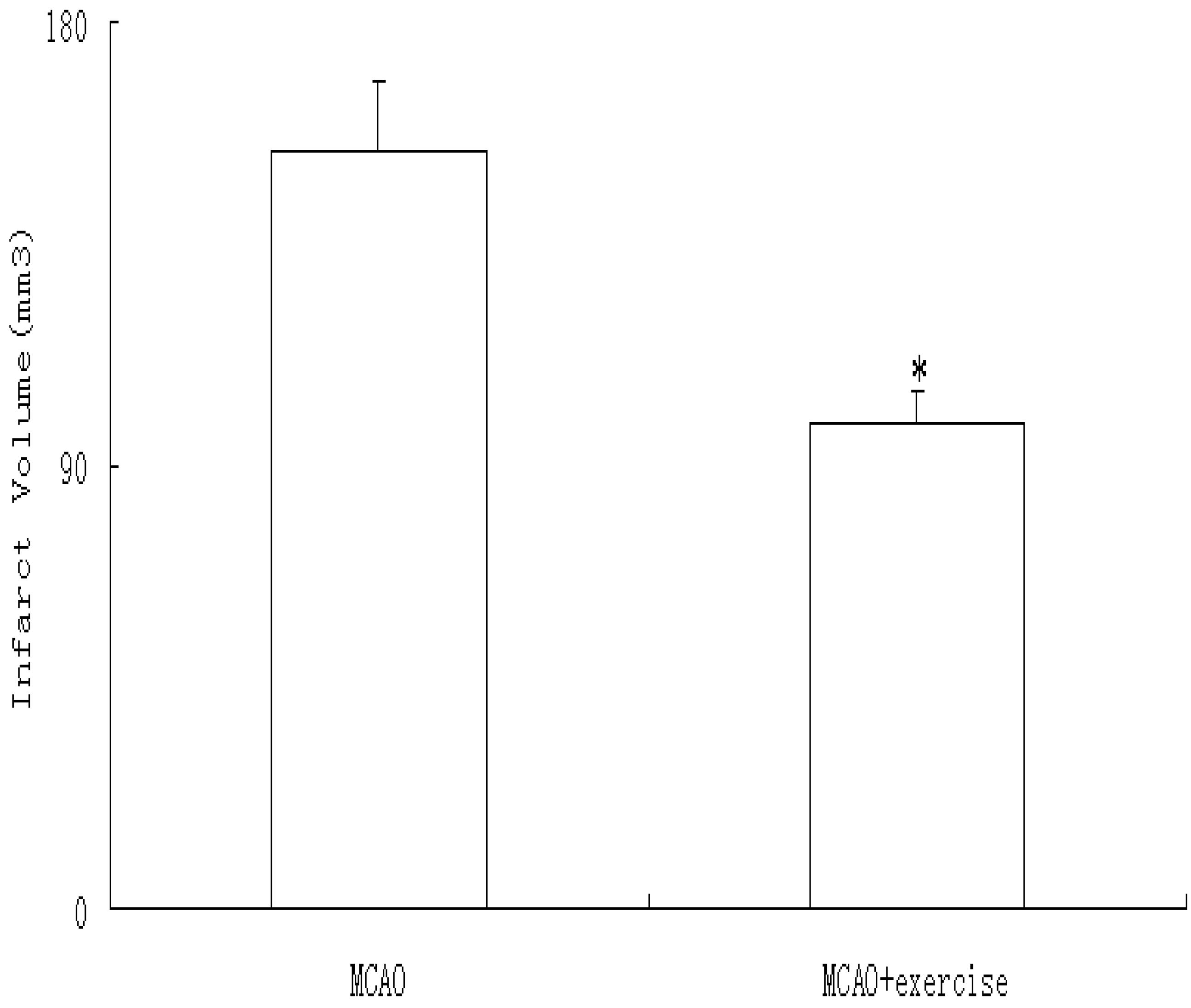
2.3. Infarct volume
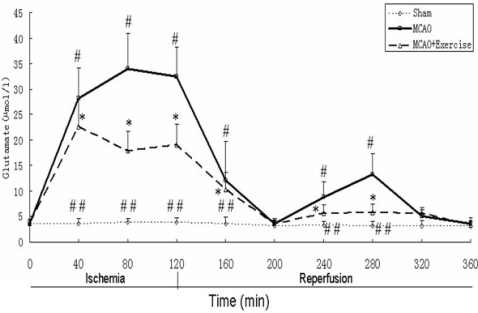
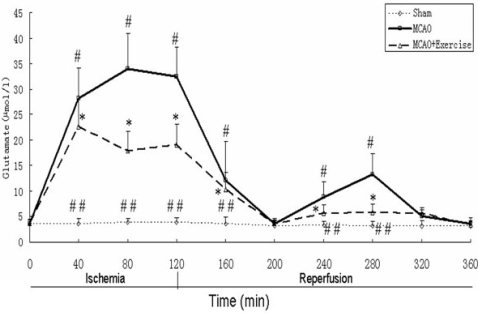
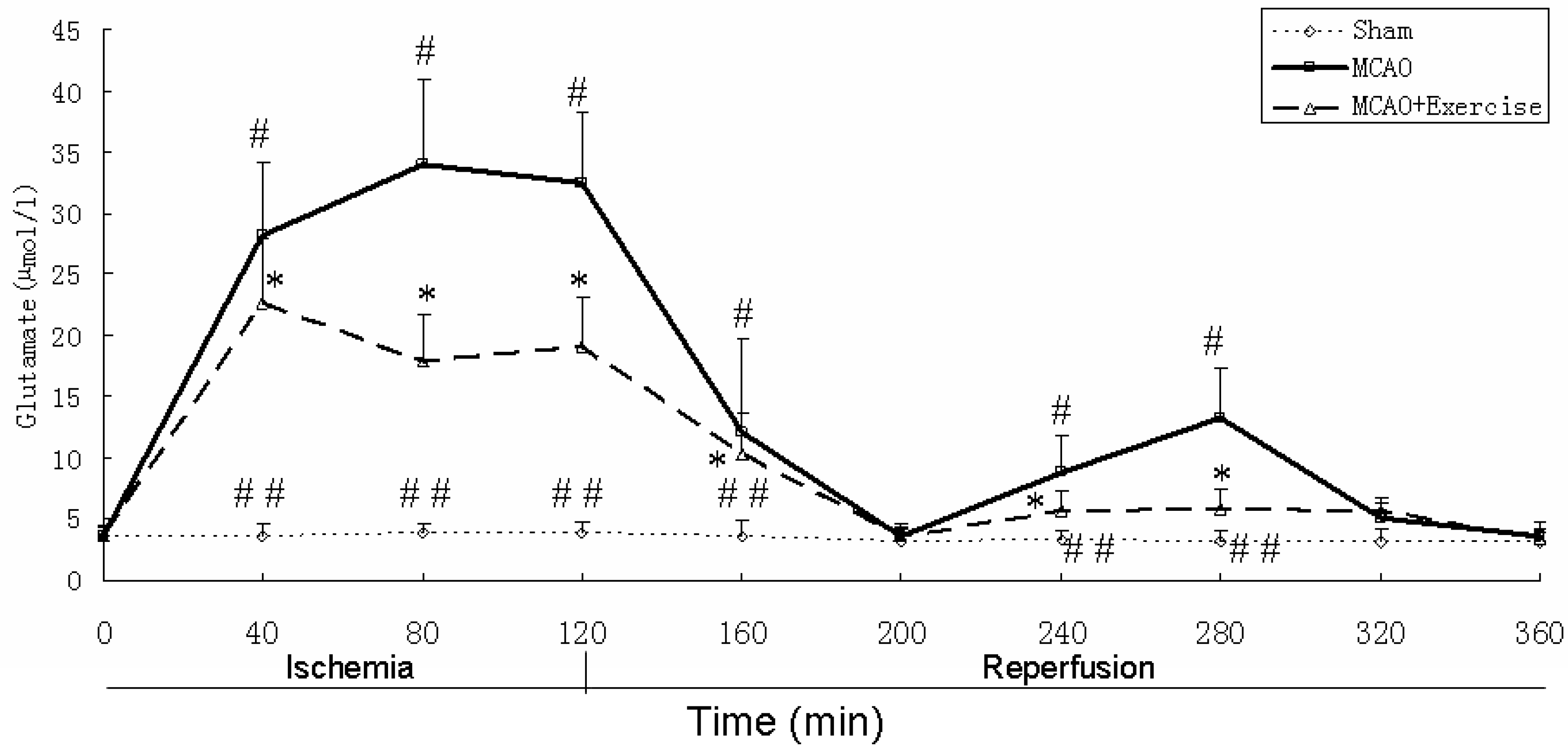
2.4. Glutamate levels


2.5. ERK1/2 and Phospho-ERK1/2

2.6. Discussion
3. Experimental
3.1. Subjects
3.2. Treadmill training
3.3. MCAO model
3.4. In vivo microdialysis
3.5. Amino acid measurement by HPLC
3.6. Determination of brain infarct volume
3.7. Western blot
3.8. Statistical analysis
4. Conclusions
Acknowledgements
References and Notes
- Kitagawa, K.; Matsumoto, M.; Tagaya, M.; Hata, R.; Ueda, H.; Niinobe, M.; Handa, N.; Fukunaga, R.; Kimura, K.; Mikoshiba, K.; Kamada, T. Ischemic tolerance phenomenon found in the brain. Brain Res. 1990, 528, 21–24. [Google Scholar] [CrossRef]
- Gidday, J.M.; Fitzgibbons, J.C.; Shah, A.R.; Park, T.S. Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci. Lett. 1994, 168, 221–224. [Google Scholar] [CrossRef]
- PerezPinzon, M.A; Mumford, P.L.; Rosenthal, M.; Sick, T.J. Anoxic preconditioning in hippocampal slices: Role of adenosine. Neuroscience 1996, 75, 687–694. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Matsumoto, M.; Kuwabara, K.; Kitagawa, K.; Suzuki, K.; Taniguchi, N.; Kamada, T. Influence of oxidative stress on induced tolerance to ischemia in gerbil hippocampal neurons. Brain Res. 1992, 599, 246–252. [Google Scholar]
- Riepe, M.W.; Esclaire, F.; Kasischke, K.; Schreiber, S.; Nakase, H.; Kempski, O.; Ludolph, A.C.; Dirnagl, U.; Hugon, J. Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: ''Chemical preconditioning''. J. Cereb. Blood Flow Metabol. 1997, 17, 257–264. [Google Scholar]
- Bigdeli, M.R.; Hajizadeh, S.; Froozandeh, M.; Rasulian, B.; Heidarianpour, A.; Khoshbaten, A. Prolonged and intermittent normobaric hyperoxia induce different degrees of ischemic tolerance in rat brain tissue. Brain Res. 2007, 1152, 228–233. [Google Scholar] [CrossRef]
- Wang, Q.; Peng, Y.; Chen, S.Y.; Gou, XC.; Hu, B.; Du, J.; Lu, Y.; Xiong, L. Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke 2009, 40, 2157–2164. [Google Scholar] [CrossRef]
- Liebelt, B.; Papapetrou, P.; Ali, A.; Guo, M.; Ji, X.; Peng, C.; Rogers, R.; Curry, A.; Jimenez, D.; Ding, Y. Eexercise preconditioning reduces neuronal apoptosis in stroke by up-regulating heat shock protein-70(heat shock protein-72) and extracellular-signal-regulated-kinase 1/2. Neuroscience 2010, 166, 1091–1100. [Google Scholar] [CrossRef]
- Ang, E.T.; Wong, P.T.H.; Moochhala, S.; Ng, Y.K. Neuroprotection associated with running: Is it a result of increased endogenous neurotrophic factors? Neuroscience 2003, 118, 335–345. [Google Scholar]
- Ding, Y.H.; Ding, Y.C.; Li, J.; Besser, D.A.; Rafols, J.A. Exercise pre-conditioning strengthens brain microvascular integrity in a rat stroke model . Neurol. Res. 2005, 28, 184–189. [Google Scholar]
- Ding, Y.H.; Young, C.N.; Luan, X.D.; Li, J.; Rafols, J.A.; Clark, J.C.; McAllister, J.P.; Ding, Y.C. Exercise preconditioning ameliorates inflammatory injury in ischemic rats during reperfusion. Acta Neuropathol. 2005, 109, 237–246. [Google Scholar] [CrossRef]
- Endres, M.; Gertz, K.; Lindauer, U.; Katchanov, J.; Schultze, J.; Schrock, H.; Nickenig, G.; Kuschinsky, W.; Dirnagl, U.; Laufs, U. Mechanisms of stroke protection by physical activity. Ann. Neurol. 2003, 54, 582–590. [Google Scholar] [CrossRef]
- Li, J.; Luan, X.D.; Clark, J.C.; Rafols, J.A.; Ding, Y.C. Neuroprotection against transient cerebral ischemia by exercise pre-conditioning in rats. Neurol. Res. 2004, 26, 404–408. [Google Scholar] [CrossRef]
- Stummer, W.; Baethmann, A.; Murr, R.; Schurer, L.; Kempski, O.S. Cerebral protection against ischemia by locomotor activity in gerbils - underlying mechanisms. Stroke 1995, 26, 1423–1429. [Google Scholar]
- Wang, R.Y.; Yang, Y.R.; Yu, S.M. Protective effects of treadmill training on infarction in rats. Brain Res. 2001, 922, 140–143. [Google Scholar]
- Guyot, L.L.; Diaz, F.G.; O'Regan, M.H.; McLeod, S.; Park, H.; Phillis, J.W. Real-time measurement of glutamate release from the ischemic penumbra of the rat cerebral cortex using a focal middle cerebral artery occlusion model. Neurosci. Lett. 2001, 299, 37–40. [Google Scholar] [CrossRef]
- Smith, W.S. Pathophysiology of focal cerebral ischemia: A therapeutic perspective. J. Vasc. Interven. Radiol. 2004, 15, S3–12. [Google Scholar] [CrossRef]
- Pradillo, J.M.; Hurtado, O.; Romera, C.; Cardenas, A.; Fernandez-Tome, P.; Alonso-Escolano, D.; Lorenzo, P.; Moro, MA.; Lizasoain, I. TNFR1 mediates increased neuronal membrane EAAT3 expression after in vivo cerebral ischemic preconditioning. Neuroscience 2006, 138, 1171–1178. [Google Scholar] [CrossRef]
- Romera, C.; Hurtado, O.; Botella, S.H.; Lizasoain, I.; Cardenas, A.; Fernandez-Tome, P.; Leza, J.C.; Lorenzo, P.; Moro, M.A. In vitro ischemic tolerance involves upregulation of glutamate transport partly mediated by the TACE/ADAM17-tumor necrosis factor-alpha pathway. J. Neurosci. 2004, 24, 1350–1357. [Google Scholar] [CrossRef]
- Jia, J.; Hu, Y.S.; Wu, Y.; Liu, G.; Yu, H.X.; Zheng, Q.P.; Zhu, D.N.; Xia, C.M.; Cao, Z.J. Pre-ischemic treadmill training affects glutamate and gamma aminobutyric acid levels in the striatal dialysate of a rat model of cerebral ischemia. Life Sci. 2009, 84, 505–511. [Google Scholar]
- Stummer, W.; Weber, K.; Tranmer, B.; Baethmann, A.; Kempski, O. Reduced mortality and brain damage after locomotor activity in gerbil forebrain ischemia. Stroke 1994, 25, 1862–1869. [Google Scholar] [CrossRef]
- Guo, M.; Cox, B.; Mahale, S.; Davis, W.; Carranza, A.; Hayes, K.; Sprague, S.; Jimenez, D.; Ding, Y. Pre-ischemic exercise reduces matrix metalloproteinase-9 expression and ameliorates blood-brain barrier dysfunction in stroke. Neuroscience 2008, 151, 340–351. [Google Scholar]
- Lu, Z.M.; Xu, S.C. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 2006, 58, 621–631. [Google Scholar] [CrossRef]
- Alessandrini, A.; Namura, S.; Moskowitz, M.A.; Bonventre, J.V. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 12866–12869. [Google Scholar] [CrossRef]
- Namura, S.; Iihara, K.; Takami, S.; Nagata, I.; Kikuchi, H.; Matsushita, K.; Moskowitz, M.A.; Bonventre, J.V.; Alessandrini, A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2001, 98, 11569–11574. [Google Scholar]
- Ozawa, S.; Kamiya, H.; Tsuzuki, K. Glutamate receptors in the mammalian central nervous system. Prog. Neurobiol. 1998, 54, 581–618. [Google Scholar] [CrossRef]
- Izquierdo, I.; Medina, J.H. Memory formation: The sequence of biochemical events in the hippocampus and its connection to activity in other brain structures. Neurobiol. Learn. Memory 1997, 68, 285–316. [Google Scholar] [CrossRef]
- Benveniste, H.; Drejer, J.; Schousboe, A.; Diemer, N.H. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral-ischemia monitored by intracerebral microdialysis. J. Neurochem. 1984, 43, 1369–1374. [Google Scholar] [CrossRef]
- Matsumoto, K.; Graf, R.; Rosner, G.; Taguchi, J.; Heiss, W.D. Elevation of neuroactive substances in the cortex of in the cortex of cats during prolonged focal ischemia. J. Cereb. Blood Flow Metabol. 1993, 13, 586–594. [Google Scholar]
- Melani, A.; Pantoni, L.; Corsi, C.; Bianchi, L.; Monopoli, A.; Bertorelli, R.; Pepeu, G.; Pedata, F. Striatal outflow of adenosine, excitatory amino acids, gamma-aminobutyric acid, and taurine in awake freely moving rats after middle cerebral artery occlusion - Correlations with neurological deficit and histopathological damage. Stroke 1999, 30, 2448–2454. [Google Scholar] [CrossRef]
- Saransaari, P.; Oja, S.S. Characteristics of GABA release induced by free radicals in mouse hippocampal slices. Neurochem. Res. 2008, 33, 384–393. [Google Scholar]
- Shimizu-Sasamata, M.; Kano, T.; Rogowska, J.; Wolf, G.L.; Moskowitz, M.A.; Lo, E.H. YM872, a highly water-soluble AMPA receptor antagonist, preserves the hemodynamic penumbra and reduces brain injury after permanent focal ischemia in rats. Stroke 1998, 29, 2141–2147. [Google Scholar] [CrossRef]
- Grasshoff, C.; Gillessen, T. Effects of propofol on N-methyl-D-aspartate receptor-mediated calcium increase in cultured rat cerebrocortical neurons. Eur. J. Anaesth. 2005, 22, 467–470. [Google Scholar]
- JacksonFriedman, C.; Lyden, P.D.; Nunez, S.; Jin, A.; Zweifler, R. High dose baclofen is neuroprotective but also causes intracerebral hemorrhage: A quantal bioassay study using the intraluminal suture occlusion method. Exp. Neurol. 1997, 147, 346–352. [Google Scholar] [CrossRef]
- Stanciu, M.; Wang, Y.; Kentor, R.; Burke, N.; Watkins, S.; Kress, G.; Reynolds, I.; Klann, E.; Angiolieri, M.R.; Johnson, J.W.; DeFranco, D.B. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000, 275, 12200–12206. [Google Scholar]
- Zea, L.E.; Weinstein, P.R.; Carlson, S. Reversible middle cerebral artery occlusion without craniectomy in rat. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef]
- Jung, K.H.; Chu, K.; Ko, S.Y. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke 2006, 37, 2744–2750. [Google Scholar] [CrossRef]
- Sample Availability: Not Available.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, F.; Wu, Y.; Jia, J.; Hu, Y.-S. Pre-Ischemic Treadmill Training Induces Tolerance to Brain Ischemia: Involvement of Glutamate and ERK1/2. Molecules 2010, 15, 5246-5257. https://doi.org/10.3390/molecules15085246
Zhang F, Wu Y, Jia J, Hu Y-S. Pre-Ischemic Treadmill Training Induces Tolerance to Brain Ischemia: Involvement of Glutamate and ERK1/2. Molecules. 2010; 15(8):5246-5257. https://doi.org/10.3390/molecules15085246
Chicago/Turabian StyleZhang, Feng, Yi Wu, Jie Jia, and Yong-Shan Hu. 2010. "Pre-Ischemic Treadmill Training Induces Tolerance to Brain Ischemia: Involvement of Glutamate and ERK1/2" Molecules 15, no. 8: 5246-5257. https://doi.org/10.3390/molecules15085246