Effects of Time Point Measurement on the Reconstruction of Gene Regulatory Networks

Abstract
:
1. Introduction
2. Experimental
2.1. Data and software
2.2. Method
2.2.1. Dynamic Bayesian network method

 is the vector composed by variables at time i and
is the vector composed by variables at time i and  is the vector composed by jth variable at all times.
is the vector composed by jth variable at all times.
 denotes the random variables that correspond to the parents of node i.
denotes the random variables that correspond to the parents of node i.
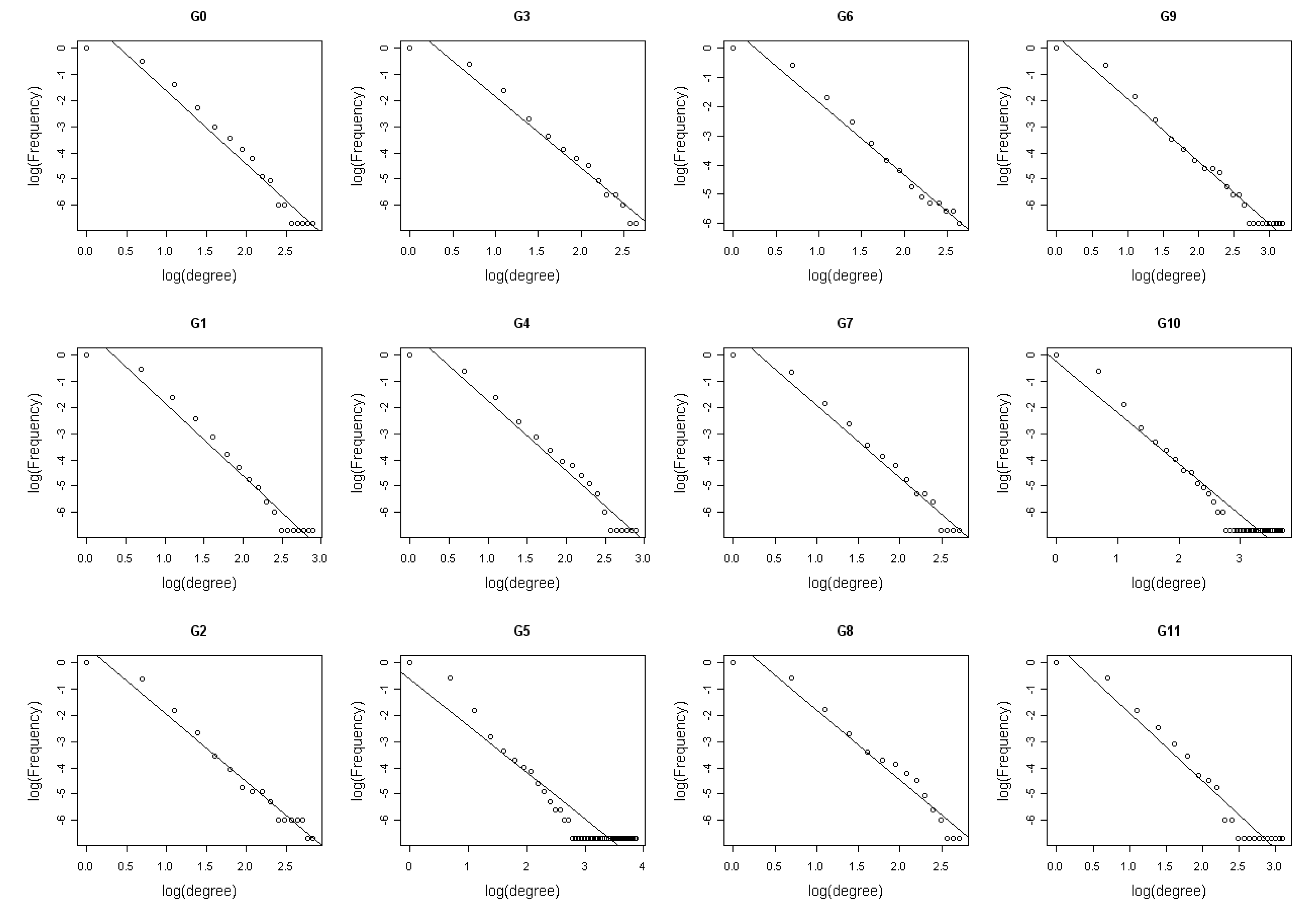
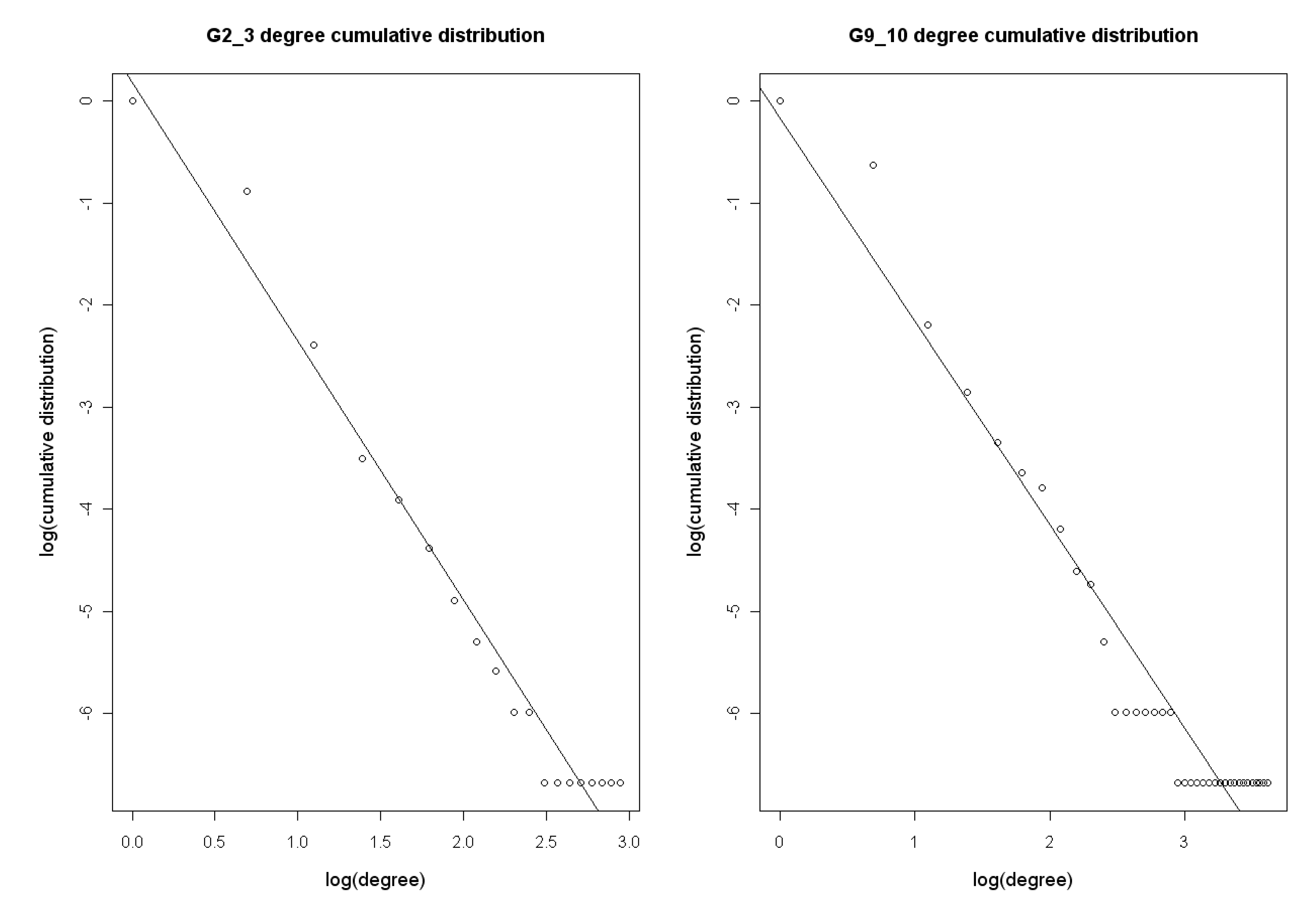
2.2.2. Network structure analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics | Definition | Descriptions |
| Average degree K [29] |  | de(v): the degree of node v |
| N: the number of nodes in network S | ||
| Average path length l[30,32] |  | dij: the shortest path between vi and vj |
| Betweenness Bv[33] |  | givj: the number of shortest paths from i to j that pass through a node v |
| gij : the number of shortest geodesic paths from i to j. | ||
| Clustering coefficient CC [34] |  | Nt: number of closed triplets |
| Ntn: number of connected triples of nodes | ||
| Centralization Ce( S ) [35] |  | C(v): the degree centrality for node v and  |
| Global efficiency of the network E[36] |  | dij : shortest path length |
| Maximum vulnerability of the networks Vu [37] |  | E: the efficiency of the network |
| Ei : the efficiency of the network without the node i and all edges connecting it with other vertices |
2.2.3. Arabidopsis gene regulatory networks reconstruction based on different time point deletion

3. Results and Discussion
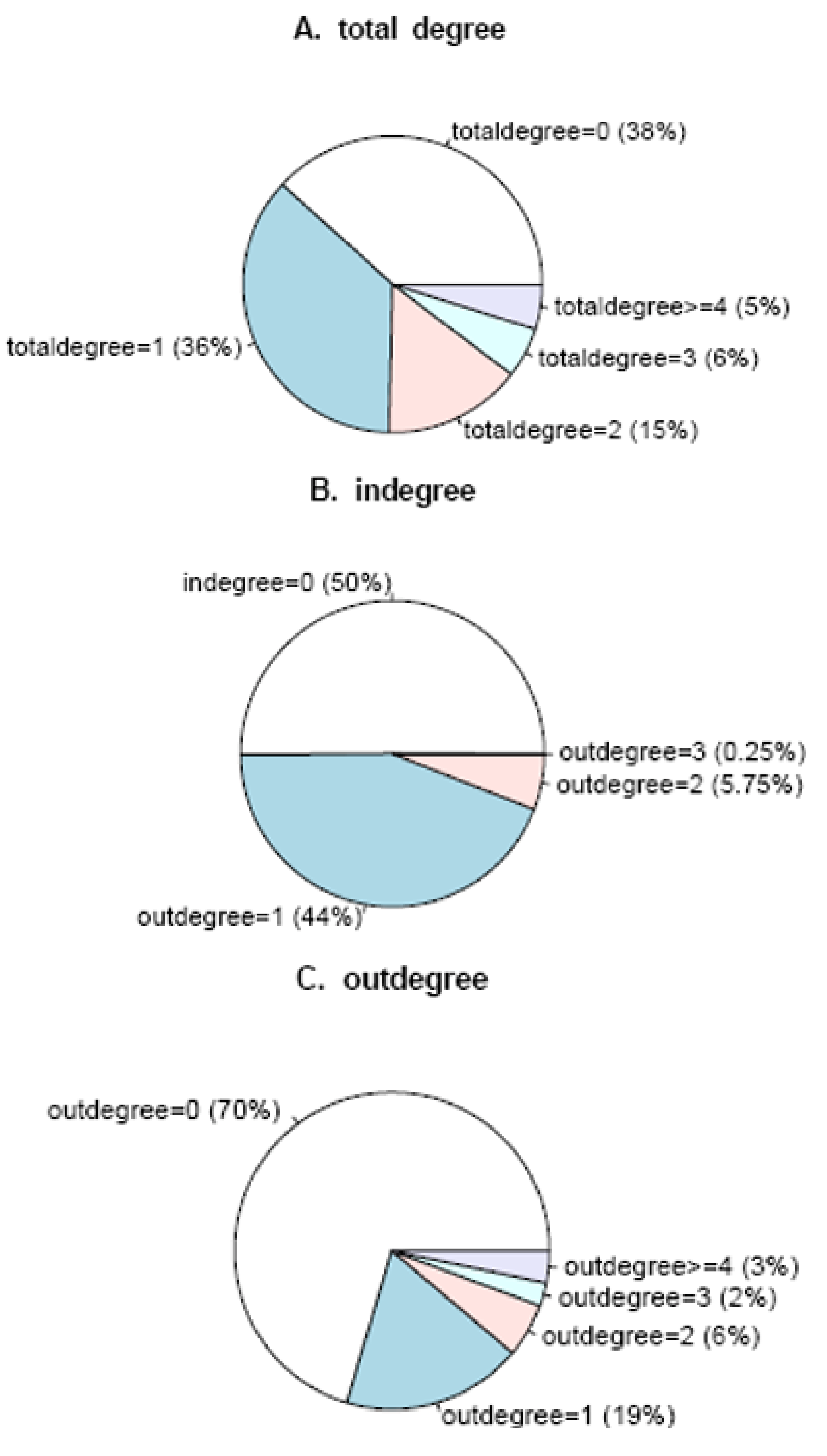
3.1. The analysis of constructed Arabidopsis gene regulatory networks

| K | Dia | l | N0 | Rn | E | Vu | CC | Ce |
|---|---|---|---|---|---|---|---|---|
| 1.1175 | 12 | 3.0467 | 306 | 447 | 0.0013 | 0.0302 | 0.0019 | 0.0093 |

3.2. Identification of network statistics insensitive to time points measurement
| Network | K | Dia | l | Ce | Rn | E | Vu |
| G0 | 1.1175 | 12 | 3.0467 | 0.0093 | 447 | 0.001258 | 0.0302 |
| G1 | 0.9750 | 10 | 2.4462 | 0.0101 | 390 | 0.000944 | 0.0397 |
| G2 | 0.8725 | 6 | 1.6998 | 0.0095 | 349 | 0.000726 | 0.0499 |
| G3 | 0.9175 | 6 | 1.9530 | 0.0076 | 367 | 0.000849 | 0.0366 |
| G4 | 0.9525 | 11 | 2.3965 | 0.0101 | 381 | 0.000859 | 0.0602 |
| G5 | 0.9625 | 5 | 1.8720 | 0.0289 | 385 | 0.000919 | 0.0809 |
| fG6 | 0.9425 | 7 | 2.0107 | 0.0076 | 377 | 0.000811 | 0.0344 |
| G7 | 0.8475 | 10 | 2.5515 | 0.0083 | 339 | 0.000804 | 0.0396 |
| G8 | 0.9250 | 7 | 2.4457 | 0.0082 | 370 | 0.000892 | 0.0472 |
| G9 | 0.8625 | 7 | 2.2134 | 0.0139 | 345 | 0.000784 | 0.0590 |
| G10 | 0.9200 | 7 | 2.0985 | 0.0239 | 368 | 0.000863 | 0.0728 |
| G11 | 0.9500 | 5 | 1.7365 | 0.0126 | 380 | 0.000806 | 0.0466 |
| ave | 0.9371 | 7.7500 | 2.2059 | 0.0125 | 374.8300 | 0.000876 | 0.0497 |
| d_score | 0.5785 | 2.7400 | 1.4780 | 4.6882 | 0.5800 | 1.07808 | 2.5885 |
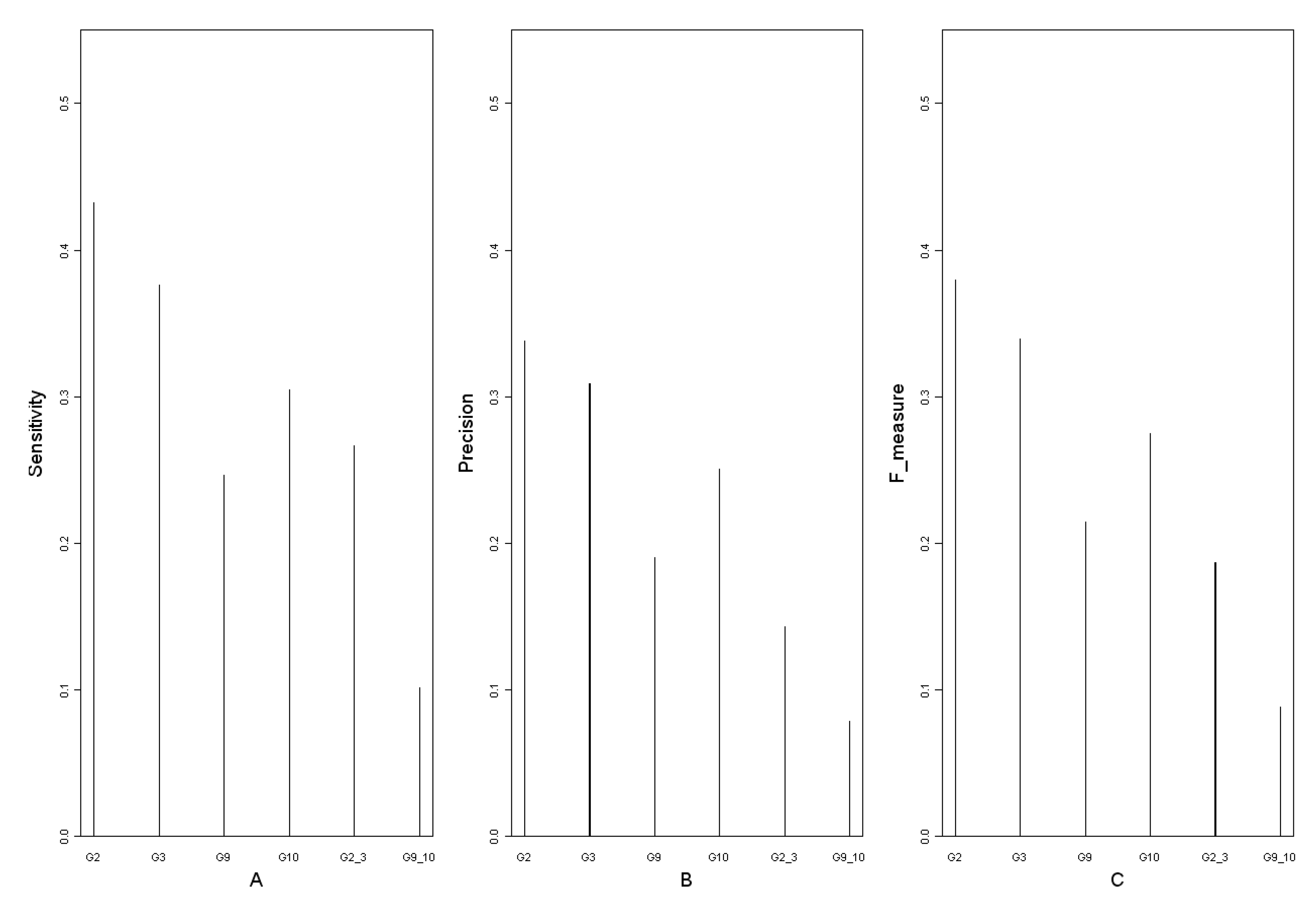
3.3. Comparison of the influence of different time points on the networks reconstruction

| Measurement | Definition | Descriptions | |
|---|---|---|---|
| sensitivity |  | Ntp: number of true positives Nfn: number of false negatives Nfp: number of false positives | |
| precision |  | ||
| F-measure |  | ||




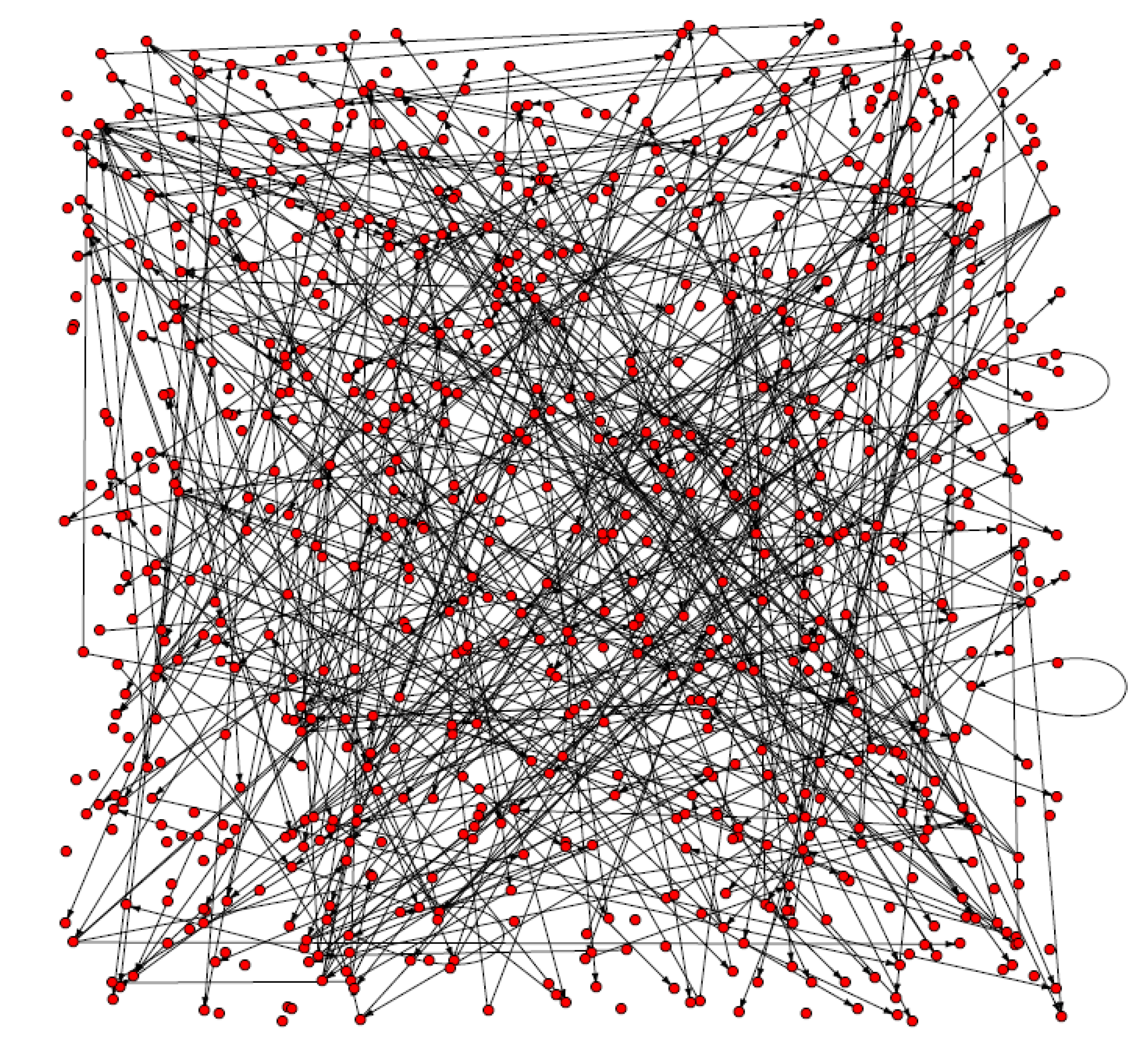
3.4. Detection of key regulatory modules
| Predictor | Target | Networks with the regulation | Network without the regulation |
|---|---|---|---|
| At1g77510 | At1g17430 | G0, G1, G2, G3, G5, G6, G7, G8, G9, G10, G11 | G4 |
| At3g02720 | At2g30010 | G0, G1, G2, G3, G4, G5, G6, G7, G8, G9, G10 | G11 |
| At5g06280 | At1g77510 | G0, G1, G2, G3, G4, G5, G6, G7, G9, G10, G11 | G8 |
| At5g58870 | At5g38510 | G0, G1, G2, G3, G4, G5, G6, G7, G8, G10, G11 | G9 |
| Predictor | Target | Networks with the regulation | Network without the regulation |
|---|---|---|---|
| At1g01250 | At4g16780 | G0, G2, G3, G4, G5, G7, G8, G9, G10, G11 | G1, G6 |
| At1g36390 | At4g09570 | G0, G1, G4, G5, G6, G7, G8, G9, G10, G11 | G2, G3 |
| At1g07180 | At3g01060 | G0, G1, G2, G3, G5, G6, G7, G8, G9, G11 | G4, G10 |
| At1g07180 | At5g35970 | G0, G1, G2, G3, G5, G6, G7, G8, G9, G11 | G4, G10 |
| At3g5490 | At3g10720 | G0, G1, G2, G4, G5, G7, G8, G9, G10, G11 | G3, G6 |
| At5g40890" | At3g11710 | G0, G1, G2, G3, G4, G6, G7, G8, G10, G11 | G5, G9 |
| At5g56900 | At4g02380 | G0, G2, G3, G4, G5, G6, G7, G8, G9, G10 | G1, G11 |
| At5g56900 | At5g66920 | G0, G1, G2, G3, G4, G6, G7, G8, G10, G11 | G5, G9 |
| At1g51110 | At3g12760 | G0, G1, G2, G3, G4, G5, G6, G7, G8, G10 | G9, G11 |
| At2g40890 | At4g35090 | G0, G1, G2, G3, G5, G6, G7, G8, G10, G11 | G4, G9 |
4. Discussion and Conclusions
Acknowledgements
References
- Kato, M.; Tsunoda, T.; Takagi, T. Inferring genetic networks from DNA microarray data by multiple regression analysis. Genome Inform. Ser. Workshop Genome Inform. 2000, 11, 118–128. [Google Scholar]
- Chen, T.; He, H.L.; Church, G.M. Modeling gene expression with differential equations. Pac. Symp. Biocomput. 1999, 29–40. [Google Scholar]
- de Hoon, M.J.; Imoto, S.; Kobayashi, K.; Ogasawara, N.; Miyano, S. Inferring gene regulatory networks from time-ordered gene expression data of Bacillus subtilis using differential equations. Pac. Symp. Biocomput. 2003, 17–28. [Google Scholar]
- Basso, K.; Margolin, A.A.; Stolovitzky, G.; Klein, U.; Dalla-Favera, R.; Califano, A. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 2005, 37, 382–390. [Google Scholar]
- Liang, S.; Fuhrman, S.; Somogyi, R. Reveal, a general reverse engineering algorithm for inference of genetic network architectures. Pac. Symp. Biocomput. 1998, 18–29. [Google Scholar]
- Friedman, N.; Linial, M.; Nachman, I.; Pe'er, D. Using Bayesian networks to analyze expression data. J. Comput. Biol. 2000, 7, 601–620. [Google Scholar] [CrossRef]
- Ong, I.M.; Glasner, J.D.; Page, D. Modelling regulatory pathways in E. coli from time series expression profiles. Bioinformatics 2002, 18 Suppl. 1, S241–S248. [Google Scholar]
- Zou, M.; Conzen, S.D. A new dynamic Bayesian network (DBN) approach for identifying gene regulatory networks from time course microarray data. Bioinformatics 2005, 21, 71–79. [Google Scholar]
- Opgen-Rhein, R.; Strimmer, K. Learning causal networks from systems biology time course data: an effective model selection procedure for the vector autoregressive process. BMC Bioinformatics 2007, 8 Suppl 2, S3. [Google Scholar] [CrossRef]
- Beal, M.J.; Falciani, F.; Ghahramani, Z.; Rangel, C.; Wild, D.L. A Bayesian approach to reconstructing genetic regulatory networks with hidden factors. Bioinformatics 2005, 21, 349–356. [Google Scholar] [CrossRef]
- Perrin, B.E.; Ralaivola, L.; Mazurie, A.; Bottani, S.; Mallet, J.; d'Alche-Buc, F. Gene networks inference using dynamic Bayesian networks. Bioinformatics 2003, 19 Suppl. 2, ii138–ii148. [Google Scholar] [CrossRef]
- Rangel, C.; Angus, J.; Ghahramani, Z.; Lioumi, M.; Sotheran, E.; Gaiba, A.; Wild, D.L.; Falciani, F. Modeling T-cell activation using gene expression profiling and state-space models. Bioinformatics 2004, 20, 1361–1372. [Google Scholar] [CrossRef]
- Wu, F.X.; Zhang, W.J.; Kusalik, A.J. Modeling gene expression from microarray expression data with state-space equations. Pac. Symp. Biocomput. 2004, 581–592. [Google Scholar]
- Smith, S.M.; Fulton, D.C.; Chia, T.; Thorneycroft, D.; Chapple, A.; Dunstan, H.; Hylton, C.; Zeeman, S.C.; Smith, A.M. Diurnal changes in the transcriptome encoding enzymes of starch metabolism provide evidence for both transcriptional and posttranscriptional regulation of starch metabolism in Arabidopsis leaves. Plant Physiol. 2004, 136, 2687–2699. [Google Scholar] [CrossRef]
- Boyes, D.C.; Zayed, A.M.; Ascenzi, R.; McCaskill, A.J.; Hoffman, N.E.; Davis, K.R.; Gorlach, J. Growth stage-based phenotypic analysis of Arabidopsis: A model for high throughput functional genomics in plants. Plant Cell 2001, 13, 1499–1510. [Google Scholar]
- May, S. NASC's International Affymetrix Service. Available online: http://affymetrix.arabidopsis.info/ (accessed on 13 October 2009).
- The Comprehensive R Archive Network. Available online: http://mirrors.geoexpat.com/cran/ (accessed on 10 May 2010).
- Lèbre, S. Inferring Dynamic Genetic Networks with Low Order Independencies. Stat. Appl. Genet. Mol. Biol. 2009, 8, 9. [Google Scholar]
- Murphy, K.; Mian, S. Modelling Gene Expression Data using Dynamic Bayesian Networks; University of California, Computer Science Division: Berkeley, CA, USA, 1999. [Google Scholar]
- Carlson, J.M.; Chakravarty, A.; Khetani, R.S.; Gross, R.H. Bounded search for de novo identification of degenerate cis-regulatory elements. BMC Bioinformatics 2006, 7, 254. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V.; Mannhold, R.; Kubinyi, H.; Timmerman, H. Handbook of Molecular Descriptors; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Gonzalez-Diaz, H.; Gonzalez-Diaz, Y.; Santana, L.; Ubeira, F.M.; Uriarte, E. Proteomics, networks and connectivity indices. Proteomics 2008, 8, 750–778. [Google Scholar] [CrossRef]
- González-Díaz, H.; Munteanu, C.R. Topological Indices for Medicinal Chemistry, Biology, Parasitology, Neurological and Social Networks; Transworld Research Network: Kerala, India, 2010; p. 212. [Google Scholar]
- Stefan, B.; Heinz Georg, S. Handbook of Graphs and Networks: From the Genome to the Internet; John Wiley & Sons, Inc.: New York, NY, USA, 2003; p. 401. [Google Scholar]
- Mrabet, Y.; Semmar, N. Mathematical methods to analysis of topology, functional variability and evolution of metabolic systems based on different decomposition concepts. Curr. Drug Metab. 2010, 11, 315–341. [Google Scholar] [CrossRef]
- Chou, K.C. Graphic rule for drug metabolism systems. Curr. Drug Metab. 2010, 11, 369–378. [Google Scholar]
- Gonzalez-Diaz, H. Network topological indices, drug metabolism, and distribution. Curr. Drug Metab. 2010, 11, 283–284. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Duardo-Sanchez, A.; Ubeira, F.M.; Prado-Prado, F.; Perez-Montoto, L.G.; Concu, R.; Podda, G.; Shen, B. Review of MARCH-INSIDE & complex networks prediction of drugs: ADMET, anti-parasite activity, metabolizing enzymes and cardiotoxicity proteome biomarkers. Curr. Drug Metab. 2010, 11, 379–406. [Google Scholar] [CrossRef]
- Diestel, R. Graph theory. In Graduate Texts in Mathematics; 1997; Volume 173, p. 410. [Google Scholar]
- West, D. Introduction to Graph Theory, 2nd ed; Prentice Hall: Englewood Cliffs, NJ, 1996. [Google Scholar]
- Chia, T.; Thorneycroft, D.; Chapple, A.; Messerli, G.; Chen, J.; Zeeman, S.C.; Smith, S.M.; Smith, A.M. A cytosolic glucosyltransferase is required for conversion of starch to sucrose in Arabidopsis leaves at night. Plant J. 2004, 37, 853–863. [Google Scholar] [CrossRef]
- Albert, R.; Barabasi, A.-L. Statistical mechanics of complex networks. Rev. Mod. Phys. 2002, 74, 47–97. [Google Scholar]
- Freeman, L.C. A set of measures of centrality based on betweenness. Sociometry 1977, 40, 35–41. [Google Scholar] [CrossRef]
- Wasserman, S.; Faust, K. Social Network Analysis: Methods and Applications (Structural Analysis in the Social Sciences), 1st ed; Cambridge University Press: New York, NY, USA, 1994; p. 857. [Google Scholar]
- Freeman, L. Centrality in social networks: Conceptual clarification. Soc. Networks 1979, 1, 215–239. [Google Scholar] [CrossRef]
- Latora, V.; Marchiori, M. Efficient behavior of small-world networks. Phys. Rev. Lett. 2001, 87, 198701. [Google Scholar] [CrossRef]
- Gol'dshtein, V.; Koganov, G.A.; Surdutovich, G.I. Vulnerability and Hierarchy of Complex Networks. arXiv: preprint cond-mat/0409298 2004. [Google Scholar]
- Riechmann, J.L.; Meyerowitz, E.M. The AP2/EREBP family of plant transcription factors. Biol. Chem. 1998, 379, 633–646. [Google Scholar]
- Barabasi, A.L.; Bonabeau, E. Scale-free networks. Sci. Am. 2003, 288, 60–69. [Google Scholar] [CrossRef]
- Altman, D.G.; Bland, J.M. Diagnostic tests. 1: Sensitivity and specificity. BMJ 1994, 308, 1552. [Google Scholar] [CrossRef]
- Dzeroski, S.; Todorovski, L. Equation discovery for systems biology: finding the structure and dynamics of biological networks from time course data. Curr. Opin. Biotechnol. 2008, 19, 360–368. [Google Scholar] [CrossRef]
- Wang, Y.; Joshi, T.; Zhang, X.S.; Xu, D.; Chen, L. Inferring gene regulatory networks from multiple microarray datasets. Bioinformatics 2006, 22, 2413–2420. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yan, W.; Zhu, H.; Yang, Y.; Chen, J.; Zhang, Y.; Shen, B. Effects of Time Point Measurement on the Reconstruction of Gene Regulatory Networks. Molecules 2010, 15, 5354-5368. https://doi.org/10.3390/molecules15085354
Yan W, Zhu H, Yang Y, Chen J, Zhang Y, Shen B. Effects of Time Point Measurement on the Reconstruction of Gene Regulatory Networks. Molecules. 2010; 15(8):5354-5368. https://doi.org/10.3390/molecules15085354
Chicago/Turabian StyleYan, Wenying, Huangqiong Zhu, Yang Yang, Jiajia Chen, Yuanyuan Zhang, and Bairong Shen. 2010. "Effects of Time Point Measurement on the Reconstruction of Gene Regulatory Networks" Molecules 15, no. 8: 5354-5368. https://doi.org/10.3390/molecules15085354