Using Topological Indices to Predict Anti-Alzheimer and Anti-Parasitic GSK-3 Inhibitors by Multi-Target QSAR in Silico Screening

Abstract
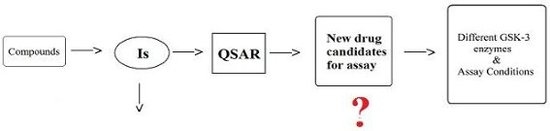
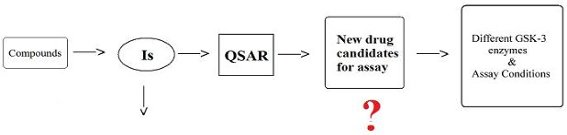
:
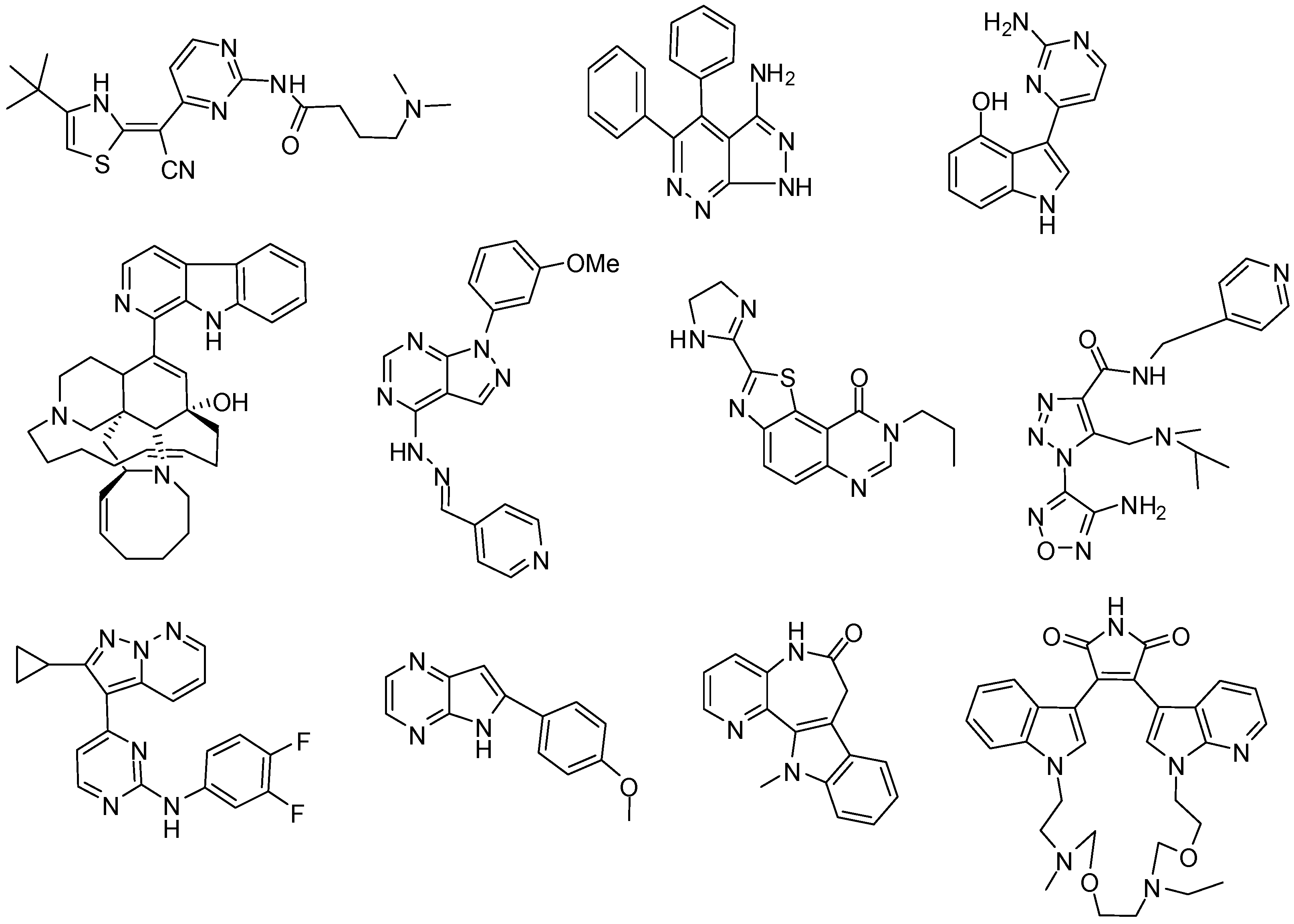
1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Index | Description |
|---|---|
| χ(P), χ(C), χ(PC), χ(Ch) | Randic branching index |
| χv(P), χv (C), χv (PC), χv (Ch) | Valence connectivity |
| e(P), e(C), e(pC), e(Ch) | Epsilon index |
| 1κ, 2κ, 3κ | Kappa index |
| 1κ(alpha), 2κ(alpha), 3κ(alpha) | Kappa (alpha) index |
| Ø | Flexibility index |
| M1 | Zagreb M1 index |
| M2 | Zagreb M2 index |
| H | Harary number |
| J | Balaban index |
2. Results and Discussion
2.1. General QSAR for GSK-3 Inhibitors


| Group | Parameter | % | GSKI-3 | Non-active |
|---|---|---|---|---|
| Training | ||||
| GSKI-3 | Sensitivity | 95.3 | 854 | 42 |
| Non-active | Specificity | 82.8 | 77 | 371 |
| Total | Accuracy | 91.1 | ||
| Validation | ||||
| GSKI-3 | Sensitivity | 95.3 | 282 | 14 |
| Non-active | Specificity | 84.6 | 179 | 985 |
| Total | Accuracy | 86.8 |

3. Materials and Methods
3.1. Computational Methods

3.2. Multi-target Linear Discriminant Analysis (LDA)
- (1).
- We created a raw data representing each compound input as a vector made up of 1 output variable, 189 structural variables (inputs) divided in values (see the first term of the Equation (1)), averages (see the second term of the Equation (1)) and differences between values and averages (see the third term of the Equation (1)); and the CACq variable. CACq is an auxiliary not used to construct the model.
- (2).
- The first element (output) is a dummy variable (Boolean) called Observed Group (OG); OG = 0 if the compound belongs to the class to which we refer in CACq and 1 otherwise (OG = 1). We could repeat each compound more than once in the raw data. In fact, we could repeat each compound 43 times corresponding to 43 CACq Assay Conditions (seeTable 3). The first time we used the CACq = CAC number. It means that we used the real CAC class of the compound in CACq. In this case, the LDA model had to give the highest probability to the group OG = 0 because it had to predict the real class of the compound. The remnant 43 times we use an CAC class number different to the real in CACq and then the LDA model had to predict the highest probability for the group OG = 1. This indicated that the compound did not belong to this group.
| Group | Parameter | Enzyme | Isoform | Enzyme/ Organism | Species | Activity (1,0) | Condition | Observ. |
|---|---|---|---|---|---|---|---|---|
| 1 | % | GSK-3 | beta | enzyme | no | 0 | 0 | = |
| 2 | cKi | GSK-3 | alfa | enzyme | no | 0 | 100 | > |
| 3 | EC50 (μM) | no | no | no | Cell Efficacy | 0 | 2 | > |
| 4 | EC50 (μM) | no | no | no | glycogen synthesis stimulation | 0 | inactive | = |
| 5 | EC50 (μM) | no | no | no | β-catenin synthesis | 0 | 2 | > |
| 6 | EC50 (μM) | no | no | parasite | T. brucei | 0 | 2 | > |
| 7 | EC50 (μM) | no | no | virus | HIV-1 | 0 | NA | = |
| 8 | ED50 (μM) | no | no | parasite | L. donovani | 0 | 5 | < |
| 9 | IC50 (ng/mL) | no | no | parasite | P. falciparum (chloroquine resistant W2 clone) | 0 | NA | = |
| 10 | IC50 (ng/mL) | no | no | parasite | P. falciparum (chloroquine sensitive D6 clone) | 0 | NA | = |
| 11 | IC50 (nM) | GSK-3 | alfa | enzyme | no | 0 | 2000 | > |
| 12 | IC50 (nM) | GSK-3 | beta | enzyme | no | 0 | 2000 | > |
| 13 | IC50 (nM) | GSK-3 | nd | enzyme | no | 0 | 2000 | > |
| 14 | IC50 (μg/mL) | no | no | bacterium | M. intracellulare | 0 | NA | = |
| 15 | IC50 (μg/mL) | no | no | bacterium | MRS | 0 | NA | = |
| 16 | IC50 (μg/mL) | no | no | bacterium | S. aureus | 0 | NA | = |
| 17 | IC50 (μg/mL) | no | no | cell line | Human Vero cells | 0 | NC | <> |
| 18 | IC50 (μg/mL) | no | no | fungus | C. neoformans | 0 | NA | = |
| 19 | IC50 (μg/mL) | no | no | parasite | L. donovani | 0 | NA | = |
| 20 | IC50 (μM) | GSK-3 | beta | enzyme | no | 0 | 2 | > |
| 21 | IC50 (μM) | GSK-3 | nd | enzyme | no | 0 | 2 | > |
| 22 | IC50 (μM) | GSK-3 | no | parasite | P. falciparum | 0 | 20 | > |
| 23 | IC50 (μM) | GSK-3 | α/β | enzyme | no | 0 | 2 | > |
| 24 | IC50 (μM) | no | no | bacterium | M. intracellulare | 0 | — | = |
| 25 | IC50 (μM) | no | no | bacterium | MRSA | 0 | — | = |
| 26 | IC50 (μM) | no | no | cell line | Hep2 | 0 | NA | = |
| 27 | IC50 (μM) | no | no | cell line | HT29 | 0 | NA | = |
| 28 | IC50 (μM) | no | no | cell line | Human Vero cells | 0 | NA | = |
| 29 | IC50 (μM) | no | no | cell line | Human Vero cells | 0 | NC | <> |
| 30 | IC50 (μM) | no | no | cell line | LMM3 | 0 | NA | = |
| 31 | IC50 (μM) | no | no | cell line | PTP | 0 | 2 | > |
| 32 | IC50 (μM) | no | no | fungus | C. albicans | 0 | — | = |
| 33 | IC50 (μM) | no | no | fungus | C. neoformans | 0 | — | = |
| 34 | IC50 (μM) | no | no | parasite | L. mexicana | 0 | 2 | > |
| 35 | IC50 (μM) | no | no | parasite | P. falciparum | 0 | 2 | > |
| 36 | IC50 (μM) | no | no | parasite | P. falciparum D6 | 0 | NA | = |
| 37 | IC50 (μM) | no | no | parasite | P. falciparum W2 | 0 | NA | = |
| 38 | IC50E-9 (M) | GSK-3 | nd | enzyme | no | 0 | 2 | > |
| 39 | IC90 (μg/mL) | no | no | parasite | L. donovani | 0 | NA | = |
| 40 | MIC (μg/mL) | no | no | bacterium | M. tuberculosis (H37Rv) | 0 | NA | = |
| 41 | pIC50 | GSK-3 | beta | enzyme | no | 0 | 0 | = |
| 42 | pIC50 | GSK-3 | nd | enzyme | no | 0 | 0 | = |
| 43 | IC50 (μM) | no | no | no | Cell Efficacy | 0 | 2 | > |

3.3. Data Set
4. Conclusions
Acknowledgements
References
- Olson, R.E. Secretase inhibitors as therapeutics for Alzheimer’s disease. Annu. Rep. Med. Chem. 2000, 35, 31–40. [Google Scholar] [CrossRef]
- Brion, J.P.; Anderton, B.H.; Authelet, M.; Dayanandan, R.; Leroy, K.; Lovestone, S.; Ocatve, J.N.; Pradier, L.; Touchet, N.; Tremp, G. Neurofibrillary Tangles and Tau Phosphorilation. Biochem. Soc. Symp. 2001, 67, 81–88. [Google Scholar]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso, A.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; TAnimukai, H.; Grundke-Iqbal, I. Tau pathology in Alzheimer disease and other tauopathies. BBA-Mol. Basis Dis. 2005, 1739, 198–210. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Woodgett, J.R.; Cohen, P. Multisite phosphorilation of glycogen synthase. Molecular basis for the sustrate specificity of glycogen synthase kinase-3 and casein kinase-II (glycogen synthase kinase-5). Biochim. Biophys. Acta 1984, 788, 339–347. [Google Scholar] [CrossRef]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudailar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 2000, 49, 263–271. [Google Scholar] [CrossRef]
- Lovestone, S.; Reynolds, C.H.; Latimer, D.; Davis, D.R.; Anderton, B.H.; Gallo, J.M.; Hanger, D.; Mulot, S.; Marquardt, B. Alzheimer's disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 1994, 4, 1077–1086. [Google Scholar] [CrossRef]
- Imahori, K.; Uchida, T. Physiology and pathology of tau protein kinases in relation to Alzheimer's disease. J. Biochem. 1997, 121, 179–188. [Google Scholar]
- Takashima, A.; Murayama, M.; Yasutake, K.; Takahashi, H.; Yokoyama, M.; Ishiguro, K. Involvement of cyclin dependent kinase5 activator p25 on tau phosphorylation in mouse brain. Neurosci. Lett. 2001, 306, 37–40. [Google Scholar] [CrossRef]
- Ryves, W.J.; Harwood, A.J. Lithium Inhibits Glycogen Synthase Kinase-3 by Competition for Magnesium. Biochem. Biophys. Res. Commun. 2001, 280, 720–725. [Google Scholar] [CrossRef]
- Ishiguro, K.; Ihara, Y.; Uchida, T.; Imahori, K.A. Novel Tubulin-Dependent Protein Kinase Forming a Paired Helical Filament Epitope on Tau. J. Bio. Chem. 1988, 104, 319–321. [Google Scholar]
- Arana, B.; Rizzo, N.; Diaz, A. Chemotherapy of cutaneous leishmaniasis: A review. Med. Microbiol. Immunol. 2001, 190, 93–95. [Google Scholar]
- Bryceson, A. Current issues in the treatment of visceral leishmaniasis. Med. Microbiol. Immunol. 2001, 190, 85–87. [Google Scholar]
- Sundar, S. Treatment of visceral leishmaniasis. Med. Microbiol. Immunol. 2001, 190, 89–92. [Google Scholar]
- Fairlamb, A.H. Chemotherapy on human African trypanosomiasis: Current and future prospects. Trends Parasitol. 2003, 19, 488–494. [Google Scholar] [CrossRef]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006, 5, 730–732. [Google Scholar] [CrossRef]
- Liao, J.J. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007, 50, 409–424. [Google Scholar] [CrossRef]
- Pink, R.; Hudson, A.; Mouries, M.A.; Bending, M. Opportunities and chanllenges in antiparasitic drug discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740. [Google Scholar] [CrossRef]
- Plyte, S.E.; Hughes, K.; Nilkolakaki, E.; Pulverer, B.J.; Woodgett, J.R. Glycogen Synthase Kinase-3: Functions in oncogenesis and development. Biochim. Biophys. Acta 1114, 147–162. [Google Scholar]
- Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal structure of glycogen synthase kinase-3 beta: Structural basic for phosphate-primed subtrate specificity and autoinhibition. Cell (Cambridge, Mass.) 2001, 105, 721–732. [Google Scholar] [CrossRef]
- Ojo, K.K.; Gillespie, R.G.; Riechers, A.; Napuli, A.J.; Verlinde, C.L.; Buckner, F.S.; Gelb, M.H.; Domostoj, M.M.; Wells, S.J.; Scheer, A.; Wells, T.N.C.; Voorhis, C.V. Glycogen Synthase Kinase 3 is a potential drug target for african trypanosomiasis therapy. Antimicrob. Agents Chemother. 2008, 52, 3710–3717. [Google Scholar]
- González-Díaz, H.; Munteanu, C.R. Topological Indices for Medicinal Chemistry, Biology, Parasitology, Neurological and Social Networks; Transworld Research Network: Kerala, India, 2010. [Google Scholar]
- Gonzalez-Díaz, H.; Prado-Prado, F.; Ubeira, F.M. Predicting antimicrobial drugs and targets with the MARCH-INSIDE approach. Curr. Top Med. Chem. 2008, 8, 1676–1690. [Google Scholar] [CrossRef]
- González-Díaz, H.; González-Díaz, Y.; Santana, L.; Ubeira, F.M.; Uriarte, E. Proteomics, networks and connectivity indices. Proteomics 2008, 8, 750–778. [Google Scholar] [CrossRef]
- González-Díaz, H.; Vilar, S.; Santana, L.; Uriarte, E. Medicinal chemistry and bioinformatics – current trends in drugs discovery with networks topological indices. Curr. Top Med. Chem. 2007, 7, 1015–1029. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Duardo-Sanchez, A.; Ubeira, F.M.; Prado-Prado, F.; Perez-Montoto, L.G.; Concu, R.; Podda, G.; Shen, B. Review of MARCH-INSIDE & complex networks prediction of drugs: ADMET, anti-parasite activity, metabolizing enzymes and cardiotoxicity proteome biomarkers. Curr. Drug Metab. 2010, 11, 379–406. [Google Scholar] [CrossRef]
- Helguera, A.M.; Combes, R.D.; Gonzalez, M.P.; Cordeiro, M.N. Applications of 2D descriptors in drug design: A DRAGON tale. Curr. Top Med. Chem. 2008, 8, 1628–1655. [Google Scholar] [CrossRef]
- Caballero, J.; Fernandez, M. Artificial neural networks from MATLAB in medicinal chemistry. Bayesian-regularized genetic neural networks (BRGNN): Application to the prediction of the antagonistic activity against human platelet thrombin receptor (PAR-1). Curr. Top Med. Chem. 2008, 8, 1580–1605. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Khan, M. T. Predictions of the ADMET properties of candidate drug molecules utilizing different QSAR/QSPR modelling approaches. Curr. Drug Metab. 2010, 11, 285–295. [Google Scholar]
- Garcia, I.; Diop, Y.F.; Gomez, G. QSAR & complex network study of the HMGR inhibitors structural diversity. Curr. Drug Metab. 2010, 11, 307–314. [Google Scholar] [CrossRef]
- Martinez-Romero, M.; Vazquez-Naya, J.M.; Rabunal, J.R.; Pita-Fernandez, S.; Macenlle, R.; Castro-Alvarino, J.; Lopez-Roses, L.; Ulla, J.L.; Martinez-Calvo, A.V.; Vazquez, S.; et al. Artificial intelligence techniques for colorectal cancer drug metabolism: Ontology and complex network. Curr. Drug Metab. 2010, 11, 347–368. [Google Scholar] [CrossRef]
- Mao, B.Y.; Chou, K.C.; Maggiora, G.M. Topological analysis of hydrogen bonding in protein structure. Eur. J. Biochem. 1990, 188, 361–365. [Google Scholar] [CrossRef]
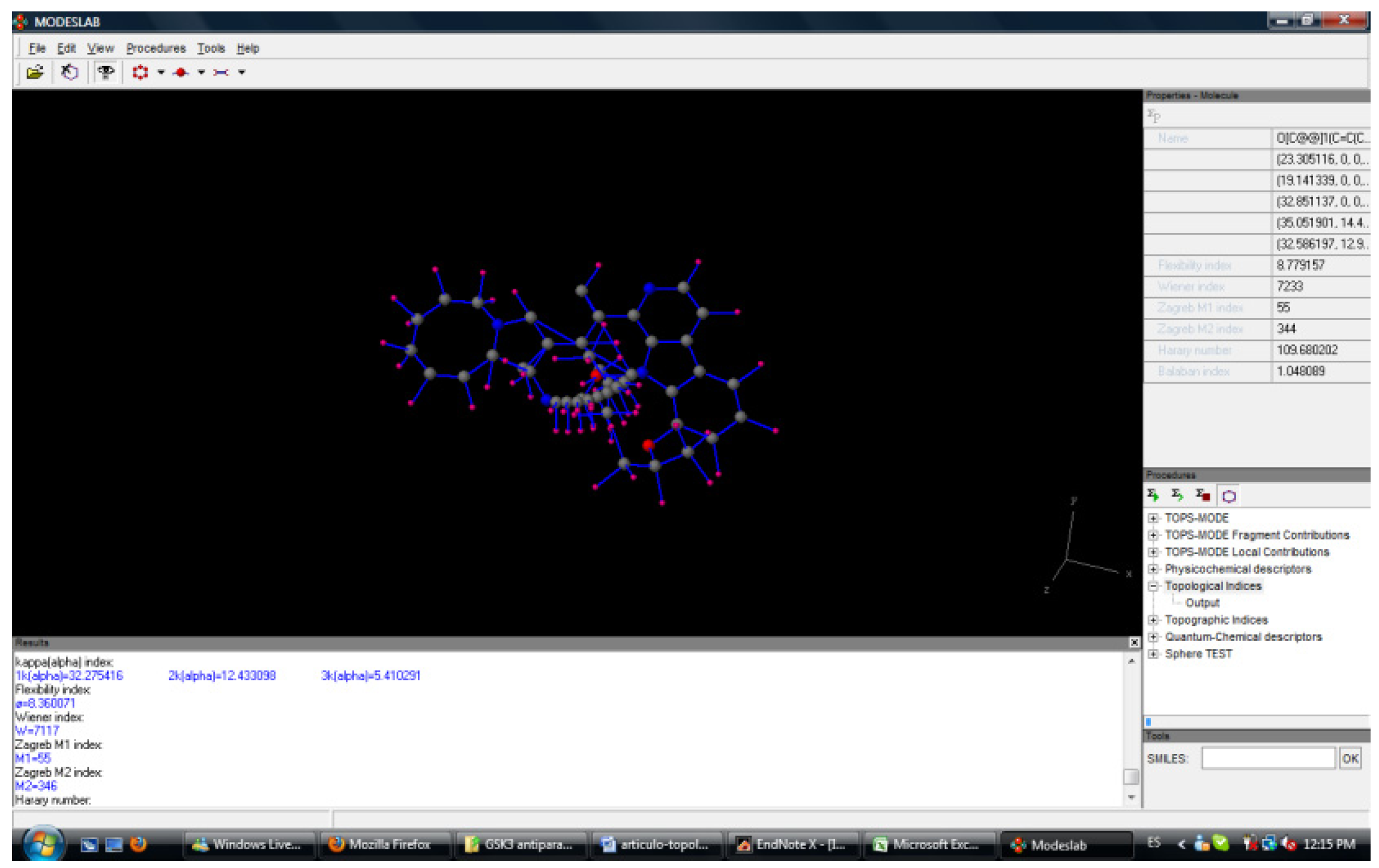
- Estrada, E.; Gutiérrez, Y. ModesLab, versión 1.5, 2002-2004.
- Nunez, M.B.; Maguna, F.P.; Okulik, N.B.; Castro, E.A. QSAR modeling of the MAO inhibitory activity of xanthones derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 5611–5617. [Google Scholar]
- Todeschini, R.; Consonni, V. Handbook of molecular descriptors; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Freund, J.A.; Poschel, T. Stochastic processes in physics, chemistry, and biology. In Lect. Notes Phys. Springer-Verlag: Berlin, Germany, 2000. [Google Scholar]
- Estrada, E.; Uriarte, E. Recent advances on the role of topological indices in drug discovery research. Curr. Med. Chem. 2001, 8, 1573–1588. [Google Scholar] [CrossRef]
- Estrada, E.; Uriarte, E.; Montero, A.; Teijeira, M.; Santana, L.; De Clercq, E. A Novel Approach for the Virtual Screening and Rational Design of Anticancer Compounds. J. Med. Chem. 2001, 43, 1975–1985. [Google Scholar]
- Van Waterbeemd, H. Discriminant Analysis for Activity Prediction. In Chemometric Methods in Molecular Design; Van Waterbeemd, H., Ed.; Wiley-VCH: New York, NY, USA, 1995; Volume 2, pp. 265–282. [Google Scholar]
- Concu, R.; Dea-Ayuela, M.A.; Perez-Montoto, L.G.; Prado-Prado, F.J.; Uriarte, E.; Bolas-Fernandez, F.; Podda, G.; Pazos, A.; Munteanu, C.R.; Ubeira, F.M.; Gonzalez-Diaz, H. 3D entropy and moments prediction of enzyme classes and experimental-theoretic study of peptide fingerprints in Leishmania parasites. Biochim. Biophys. Acta. 2009, 1794, 1784–1794. [Google Scholar]
- Concu, R.; Dea-Ayuela, M.A.; Perez-Montoto, L.G.; Bolas-Fernandez, F.; Prado-Prado, F.J.; Podda, G.; Uriarte, E.; Ubeira, F.M.; Gonzalez-Diaz, H. Prediction of Enzyme Classes from 3D Structure: A General Model and Examples of Experimental-Theoretic Scoring of Peptide Mass Fingerprints of Leishmania Proteins. J. Proteome Res. 2009, 8, 4372–4382. [Google Scholar] [CrossRef]
- Konda, V.R.; Desai, A.; Darland, G.; Bland, J.S.; Tripp, M.L. Rho iso-alpha acids from hops inhibit the GSK-3/NF-kappaB pathway and reduce inflammatory markers associated with bone and cartilage degradation. J. Inflamm. 2009, 6, 26–34. [Google Scholar] [CrossRef]
- Rochais, C.; Duc, N.V.; Lescot, E.; Sopkova-de Oliveira Santos, J.; Bureau, R.; Meijer, L.; Dallemagne, P.; Rault, S. Synthesis of new dipyrrolo- and furopyrrolopyrazinones related to tripentones and their biological evaluation as potential kinases (CDKs1-5, GSK-3) inhibitors. Eur. J. Med. Chem. 2009, 44, 708–716. [Google Scholar] [CrossRef]
- Simon, D.; Benitez, M.J.; Gimenez-Cassina, A.; Garrido, J.J.; Bhat, R.V.; Diaz-Nido, J.; Wandosell, F. Pharmacological inhibition of GSK-3 is not strictly correlated with a decrease in tyrosine phosphorylation of residues 216/279. J. Neurosci. Res. 2008, 86, 668–674. [Google Scholar] [CrossRef]
- Patel, D.S.; Bharatam, P.V. Selectivity criterion for pyrazolo[3,4-b]pyrid[az]ine derivatives as GSK-3 inhibitors: CoMFA and molecular docking studies. Eur. J. Med. Chem. 2008, 43, 949–957. [Google Scholar] [CrossRef]
- Jacquemard, U.; Dias, N.; Lansiaux, A.; Bailly, C.; Loge, C.; Robert, J.M.; Lozach, O.; Meijer, L.; Merour, J.Y.; Routier, S. Synthesis of 3,5-bis(2-indolyl)pyridine and 3-[(2-indolyl)-5-phenyl]pyridine derivatives as CDK inhibitors and cytotoxic agents. Bioorg. Med. Chem. 2008, 16, 4932–4953. [Google Scholar] [CrossRef]
- Xiao, J.; Guo, Z.; Guo, Y.; Chu, F.; Sun, P. Inhibitory mode of N-phenyl-4-pyrazolo[1,5-b] pyridazin-3-ylpyrimidin-2-amine series derivatives against GSK-3: Molecular docking and 3D-QSAR analyses. Protein Eng. Des. Sel. 2006, 19, 47–54. [Google Scholar]
- Tavares, F.X.; Boucheron, J.A.; Dickerson, S.H.; Griffin, R.J.; Preugschat, F.; Thomson, S.A.; Wang, T.Y.; Zhou, H.Q. N-Phenyl-4-pyrazolo[1,5-b]pyridazin-3-ylpyrimidin-2-amines as potent and selective inhibitors of glycogen synthase kinase 3 with good cellular efficacy. J. Med. Chem. 2004, 47, 4716–4730. [Google Scholar] [CrossRef]
- Olesen, P.H.; Sorensen, A.R.; Urso, B.; Kurtzhals, P.; Bowler, A.N.; Ehrbar, U.; Hansen, B.F. Synthesis and in vitro characterization of 1-(4-aminofurazan-3-yl)-5-dialkylaminomethyl-1H-[1,2,3]triazole-4-carboxyl ic acid derivatives. A new class of selective GSK-3 inhibitors. J. Med. Chem. 2003, 46, 3333–3341. [Google Scholar] [CrossRef]
- Calabuig, C.; Anton-Fos, G.M.; Galvez, J.; Garcia-Domenech, R. New hypoglycaemic agents selected by molecular topology. Int. J. Pharm. 2004, 278, 111–118. [Google Scholar] [CrossRef]
- Garcia-Garcia, A.; Galvez, J.; de Julian-Ortiz, J.V.; Garcia-Domenech, R.; Munoz, C.; Guna, R.; Borras, R. New agents active against Mycobacterium avium complex selected by molecular topology: A virtual screening method. J. Antimicrob. Chemother. 2004, 53, 65–73. [Google Scholar]
- Prado-Prado, F.J.; Ubeira, F.M.; Borges, F.; Gonzalez-Diaz, H. Unified QSAR & network-based computational chemistry approach to antimicrobials. II. Multiple distance and triadic census analysis of antiparasitic drugs complex networks. J. Comput. Chem. 2009, 31, 164–173. [Google Scholar]
- Prado-Prado, F.J.; Martinez de la Vega, O.; Uriarte, E.; Ubeira, F.M.; Chou, K.C.; Gonzalez-Diaz, H. Unified QSAR approach to antimicrobials. 4. Multi-target QSAR modeling and comparative multi-distance study of the giant components of antiviral drug-drug complex networks. Bioorg. Med. Chem. 2009, 17, 569–575. [Google Scholar]
- Prado-Prado, F.J.; de la Vega, O.M.; Uriarte, E.; Ubeira, F.M.; Chou, K.C.; Gonzalez-Diaz, H. Unified QSAR approach to antimicrobials. 4. Multi-target QSAR modeling and comparative multi-distance study of the giant components of antiviral drug-drug complex networks. Bioorg. Med. Chem. 2009, 17, 569–575. [Google Scholar] [CrossRef]
- Prado-Prado, F.J.; Borges, F.; Perez-Montoto, L.G.; Gonzalez-Diaz, H. Multi-target spectral moment: QSAR for antifungal drugs vs. different fungi species. Eur. J. Med. Chem. 2009, 44, 4051–4056. [Google Scholar] [CrossRef]
- Prado-Prado, F.J.; Gonzalez-Diaz, H.; de la Vega, O.M.; Ubeira, F.M.; Chou, K.C. Unified QSAR approach to antimicrobials. Part 3: First multi-tasking QSAR model for input-coded prediction, structural back-projection, and complex networks clustering of antiprotozoal compounds. Bioorg. Med. Chem. 2008, 16, 5871–5880. [Google Scholar]
- Prado-Prado, F.J.; Gonzalez-Diaz, H.; Santana, L.; Uriarte, E. Unified QSAR approach to antimicrobials. Part 2: Predicting activity against more than 90 different species in order to halt antibacterial resistance. Bioorg. Med. Chem. 2007, 15, 897–902. [Google Scholar]
- Marrero-Ponce, Y.; Khan, M.T.; Casanola Martin, G.M.; Ather, A.; Sultankhodzhaev, M.N.; Torrens, F.; Rotondo, R. Prediction of Tyrosinase Inhibition Activity Using Atom-Based Bilinear Indices. Chem. Med. Chem. 2007, 2, 449–478. [Google Scholar]
- Marrero-Ponce, Y.; Meneses-Marcel, A.; Castillo-Garit, J.A.; Machado-Tugores, Y.; Escario, J.A.; Barrio, A.G.; Pereira, D.M.; Nogal-Ruiz, J.J.; Aran, V.J.; Martinez-Fernandez, A.R.; Torrens, F.; Rotondo, R.; Ibarra-Velarde, F.; Alvarado, Y.J. Predicting antitrichomonal activity: A computational screening using atom-based bilinear indices and experimental proofs. Bioorg. Med. Chem. 2006, 14, 6502–6524. [Google Scholar]
- Meneses-Marcel, A.; Marrero-Ponce, Y.; Machado-Tugores, Y.; Montero-Torres, A.; Pereira, D.M.; Escario, J.A.; Nogal-Ruiz, J.J.; Ochoa, C.; Aran, V.J.; Martinez-Fernandez, A.R.; et al. A linear discrimination analysis based virtual screening of trichomonacidal lead-like compounds: Outcomes of in silico studies supported by experimental results. Bioorg. Med. Chem. Lett. 2005, 15, 3838–3843. [Google Scholar]
- Marrero-Ponce, Y.; Diaz, H.G.; Zaldivar, V.R.; Torrens, F.; Castro, E.A. 3D-chiral quadratic indices of the 'molecular pseudograph's atom adjacency matrix' and their application to central chirality codification: Classification of ACE inhibitors and prediction of sigma-receptor antagonist activities. Bioorg. Med. Chem. 2004, 12, 5331–5342. [Google Scholar] [CrossRef]
- Murcia-Soler, M.; Perez-Gimenez, F.; Garcia-March, F.J.; Salabert-Salvador, M.T.; Diaz-Villanueva, W.; Medina-Casamayor, P. Discrimination and selection of new potential antibacterial compounds using simple topological descriptors. J. Mol. Graph. Model. 2003, 21, 375–390. [Google Scholar]
- Cercos-del-Pozo, R.A.; Perez-Gimenez, F.; Salabert-Salvador, M.T.; Garcia-March, F.J. Discrimination and molecular design of new theoretical hypolipaemic agents using the molecular connectivity functions. J. Chem. Inf. Comput. Sci. 2000, 40, 178–184. [Google Scholar] [CrossRef]
- Estrada, E.; Vilar, S.; Uriarte, E.; Gutierrez, Y. In silico studies toward the discovery of new anti-HIV nucleoside compounds with the use of TOPS-MODE and 2D/3D connectivity indices. 1. Pyrimidyl derivatives. J. Chem. Inf. Comput. Sci. 2002, 42, 1194–1203. [Google Scholar]
- Cronin, M.T.; Aptula, A.O.; Dearden, J.C.; Duffy, J.C.; Netzeva, T.I.; Patel, H.; Rowe, P.H.; Schultz, T.W.; Worth, A.P.; Voutzoulidis, K.; Schuurmann, G. Structure-based classification of antibacterial activity. J. Chem. Inf. Comput. Sci. 2002, 42, 869–878. [Google Scholar] [CrossRef]
- Santana, L.; Uriarte, E.; González-Díaz, H.; Zagotto, G.; Soto-Otero, R.; Mendez-Alvarez, E.A. QSAR model for in silico screening of MAO-A inhibitors. Prediction, synthesis, and biological assay of novel coumarins. J. Med. Chem. 2006, 49, 1149–1156. [Google Scholar] [CrossRef]
- Kutner, M.H.; Nachtsheim, C.J.; Neter, J.; Li, W. Standardized Multiple Regression Model. In Applied Linear Statistical Models, 5th ed; McGraw Hill: New York, NY, USA, 2005; pp. 271–277. [Google Scholar]
- Budavari, S. The Merck Index, 12th ed; Merck & Co, Inc: Whitehouse Station, NJ, USA, 1996. [Google Scholar]
- Sample Availability: Not available.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
García, I.; Fall, Y.; Gómez, G. Using Topological Indices to Predict Anti-Alzheimer and Anti-Parasitic GSK-3 Inhibitors by Multi-Target QSAR in Silico Screening. Molecules 2010, 15, 5408-5422. https://doi.org/10.3390/molecules15085408
García I, Fall Y, Gómez G. Using Topological Indices to Predict Anti-Alzheimer and Anti-Parasitic GSK-3 Inhibitors by Multi-Target QSAR in Silico Screening. Molecules. 2010; 15(8):5408-5422. https://doi.org/10.3390/molecules15085408
Chicago/Turabian StyleGarcía, Isela, Yagamare Fall, and Generosa Gómez. 2010. "Using Topological Indices to Predict Anti-Alzheimer and Anti-Parasitic GSK-3 Inhibitors by Multi-Target QSAR in Silico Screening" Molecules 15, no. 8: 5408-5422. https://doi.org/10.3390/molecules15085408