Hammerhead Ribozymes: True Metal or Nucleobase Catalysis? Where Is the Catalytic Power from?

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
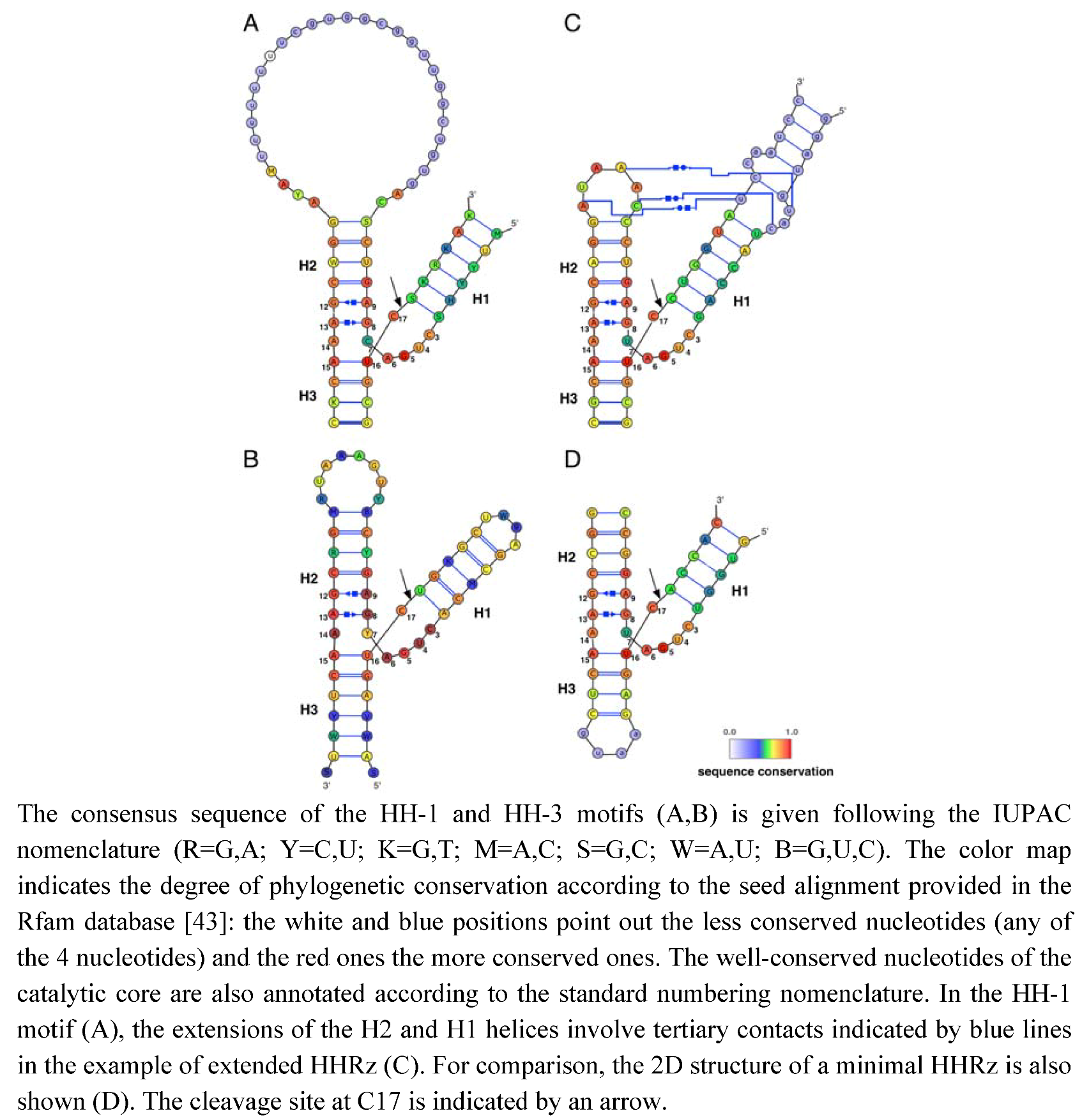
1. Introduction

2. Results and Discussion
2.1. Metal Binding Sites in Self-Cleaving and Self-Splicing Ribozymes

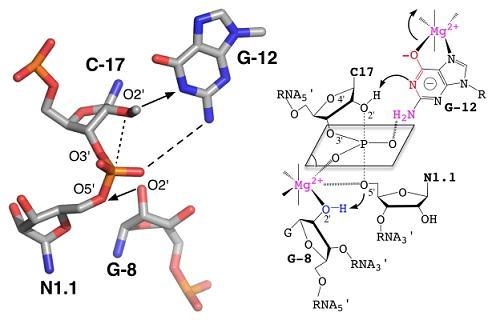
2.2. Metal / Nucleobase in the HHRz Catalysis
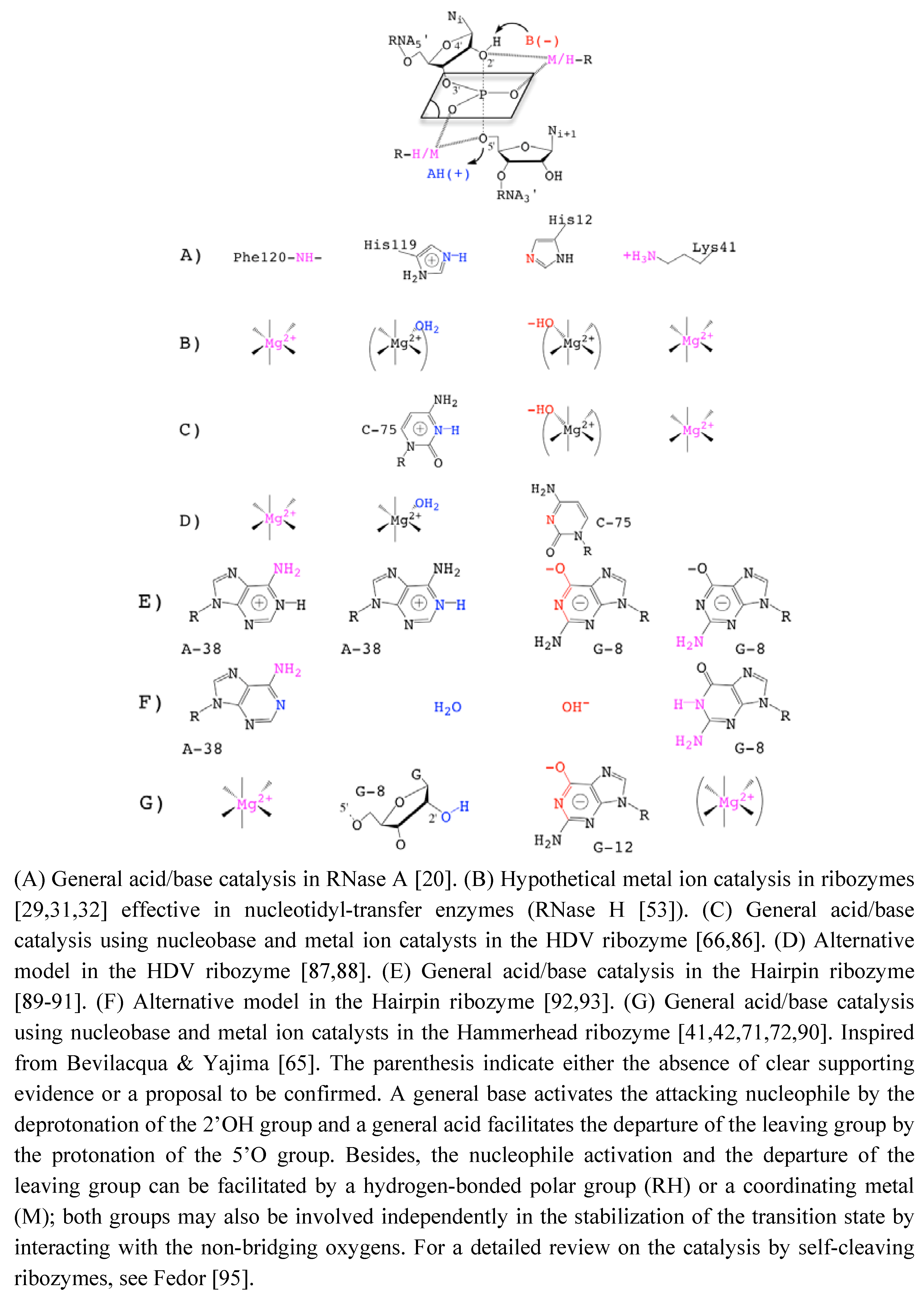
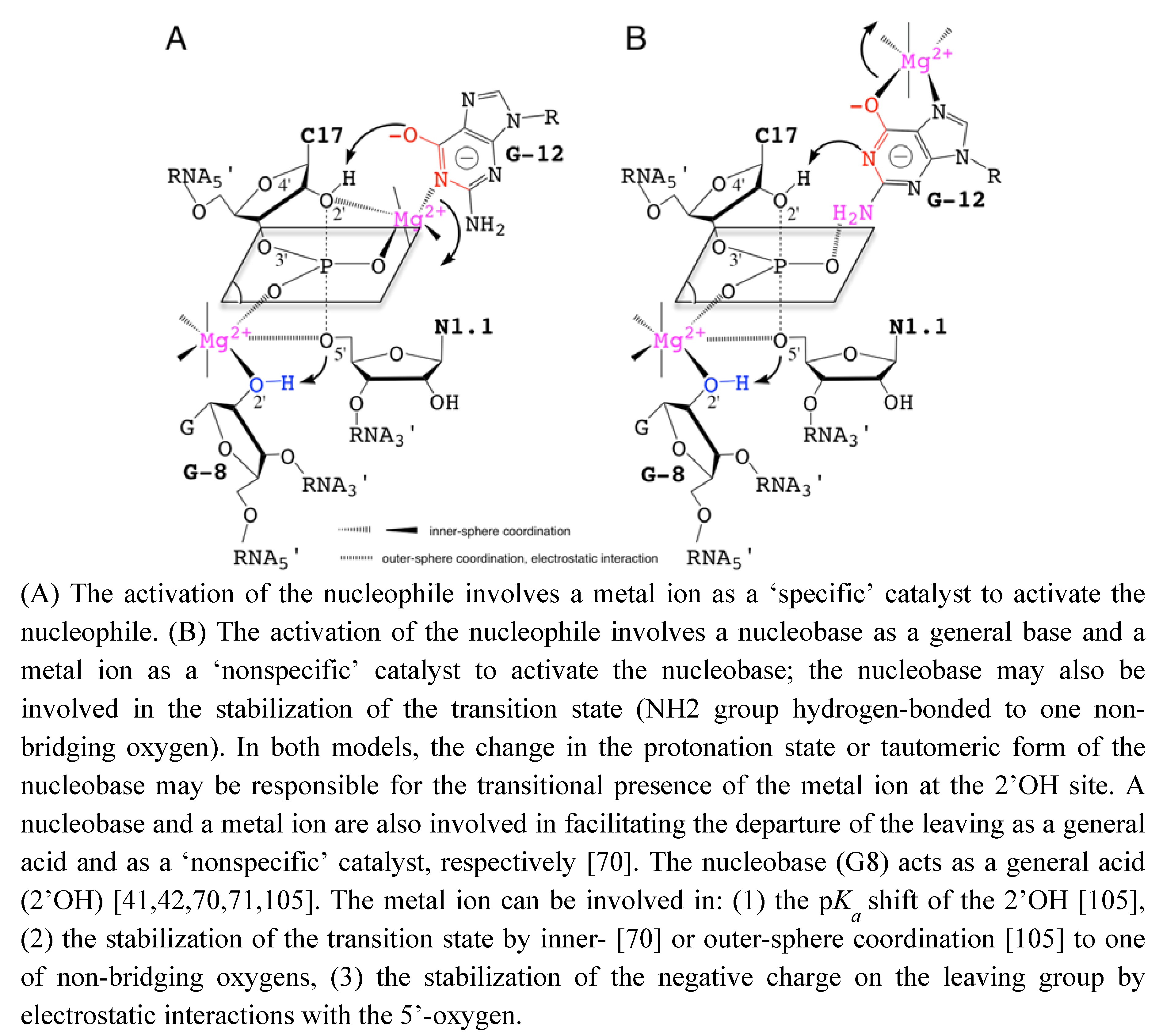
 [24,38,72] without abolishing the catalytic activity was taken as an evidence of the minor or strictly structural role of divalent cations. Those metals would just play a nonspecific role in charge screening (zero-metal mechanism) [73]. In addition, the remaining catalytic activity in the absence of divalent metal ions in various other self-cleaving ribozymes [24,74] was suggesting alternative catalytic strategies that would rely on nucleobases as general acid/base catalysts [65,75] (Figure 3).
[24,38,72] without abolishing the catalytic activity was taken as an evidence of the minor or strictly structural role of divalent cations. Those metals would just play a nonspecific role in charge screening (zero-metal mechanism) [73]. In addition, the remaining catalytic activity in the absence of divalent metal ions in various other self-cleaving ribozymes [24,74] was suggesting alternative catalytic strategies that would rely on nucleobases as general acid/base catalysts [65,75] (Figure 3).
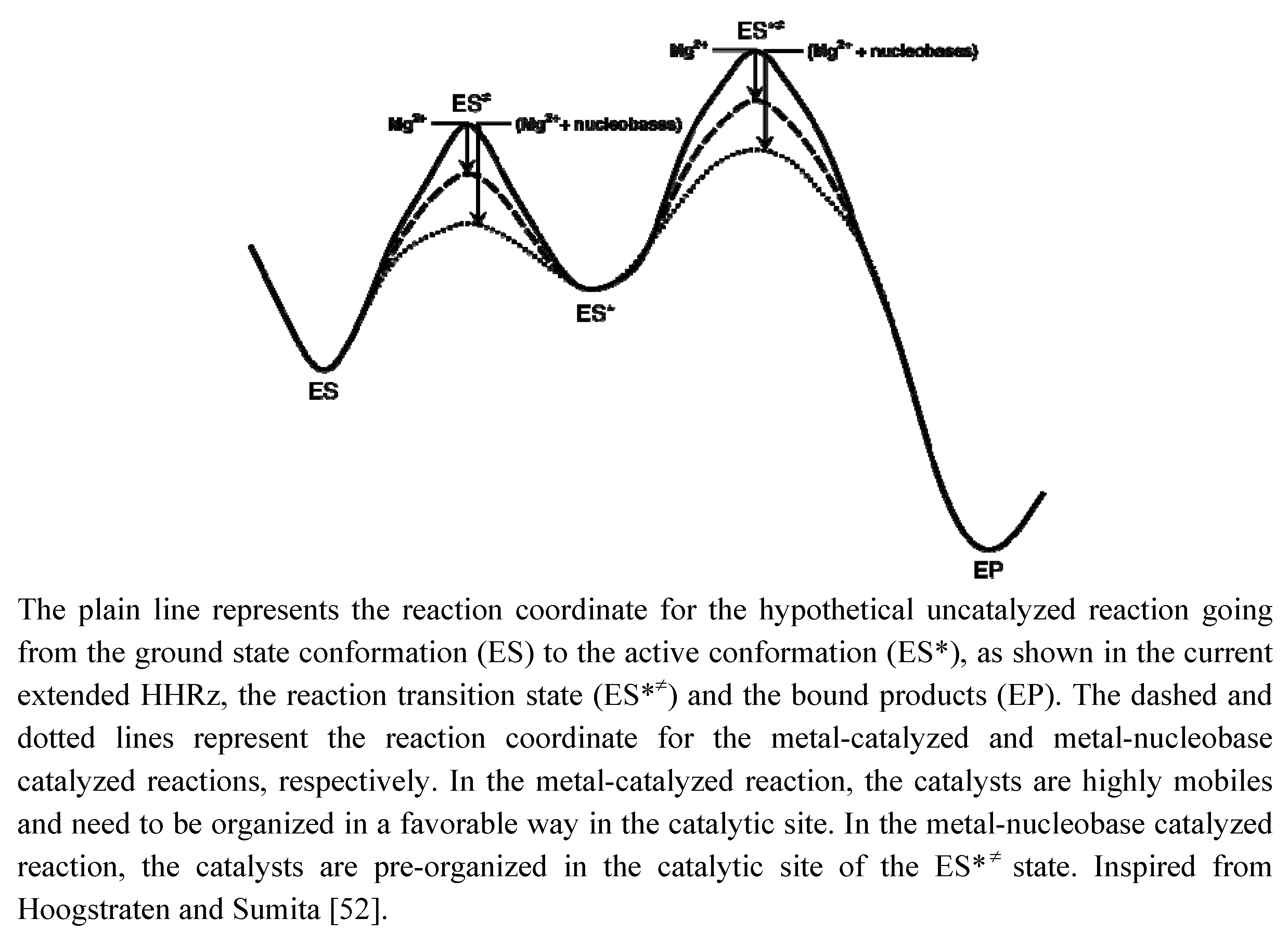
2.3. Metals & Nucleobase in the HHRz Catalysis

2.4. Possible Evolution and Origin of the Catalytic Power in the HHRz

3. Conclusions
Acknowledgements
- Sample Availability: Not available.
References
- Wedekind, J.E.; McKay, D.B. Crystallographic structures of the hammerhead ribozyme: relationship to ribozyme folding and catalysis. Ann. Rev. Biophys. Biomol. Struc. 1998, 27, 475–502. [Google Scholar] [CrossRef]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Lilley, D.M. RNA folding and catalysis. Genetica 1999, 106, 95–102. [Google Scholar] [CrossRef]
- Takagi, Y.; Warashina, M.; Stec, W.J.; Yoshinari, K.; Taira, K. Recent advances in the elucidation of the mechanisms of action of ribozymes. Nucl. Acid. Res. 2001, 29, 1815–1834. [Google Scholar] [CrossRef]
- DeRose, V.J. Two decades of RNA catalysis. Chem. Biol. 2002, 9, 961–969. [Google Scholar] [CrossRef]
- Ferbeyre, G.; Bourdeau, V.; Pageau, M.; Miramontes, P.; Cedergren, R. Distribution of hammerhead and hammerhead-like RNA motifs through the GenBank. Genome Res. 2000, 10, 1011–1019. [Google Scholar] [CrossRef]
- Bourdeau, V.; Ferbeyre, G.; Pageau, M.; Paquin, B.; Cedergren, R. The distribution of RNA motifs in natural sequences. Nucl. Acid. Res. 1999, 27, 4457–4467. [Google Scholar] [CrossRef]
- Hutchins, C.J.; Rathjen, P.D.; Forster, A.C.; Symons, R.H. Self-cleavage of plus and minus RNA transcripts of avocado sunblotch viroid. Nucl. Acid. Res. 1986, 14, 3627–3640. [Google Scholar] [CrossRef]
- Prody, G.A.; Bakos, J.T.; Buzayan, J.M.; Schneider, I.R.; Bruening, G. Autolytic Processing of Dimeric Plant Virus Satellite RNA. Science 1986, 231, 1577–1580. [Google Scholar]
- Forster, A.C.; Symons, R.H. Self-cleavage of virusoid RNA is performed by the proposed 55-nucleotide active site. Cell 1987, 50, 9–16. [Google Scholar] [CrossRef]
- Rojas, A.A.; Vazquez-Tello, A.; Ferbeyre, G.; Venanzetti, F.; Bachmann, L.; Paquin, B.; Sbordoni, V.; Cedergren, R. Hammerhead-mediated processing of satellite pDo500 family transcripts from Dolichopoda cave crickets. Nucl. Acid. Res. 2000, 28, 4037–4043. [Google Scholar] [CrossRef]
- Zhang, Y.; Epstein, L.M. Cloning and characterization of extended hammerheads from a diverse set of caudate amphibians. Gene 1996, 172, 183–190. [Google Scholar] [CrossRef]
- Salehi-Ashtiani, K.; Szostak, J.W. In vitro evolution suggests multiple origins for the hammerhead ribozyme. Nature 2001, 414, 82–84. [Google Scholar] [CrossRef]
- Fedor, M.J.; Williamson, J.R. The catalytic diversity of RNAs. Nat. Rev. Mol. Cell Biol. 2005, 6, 399–412. [Google Scholar] [CrossRef]
- van Tol, H.; Buzayan, J.M.; Feldstein, P.A.; Eckstein, F.; Bruening, G. Two autolytic processing reactions of a satellite RNA proceed with inversion of configuration. Nucl. Acid. Res. 1990, 18, 1971–1975. [Google Scholar] [CrossRef]
- Koizumi, M.; Ohtsuka, E. Effects of phosphorothioate and 2-amino groups in hammerhead ribozymes on cleavage rates and Mg2+ binding. Biochemistry 1991, 30, 5145–5150. [Google Scholar] [CrossRef]
- Slim, G.; Gait, M.J. Configurationally defined phosphorothioate-containing oligoribonucleotides in the study of the mechanism of cleavage of hammerhead ribozymes. Nucl. Acid. Res. 1991, 19, 1183–1188. [Google Scholar] [CrossRef]
- Cassano, A.G.; Anderson, V.E.; Harris, M.E. Analysis of solvent nucleophile isotope effects: evidence for concerted mechanisms and nucleophilic activation by metal coordination in nonenzymatic and ribozyme-catalyzed phosphodiester hydrolysis. Biochemistry 2004, 43, 10547–10559. [Google Scholar]
- Raines, R.T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1066. [Google Scholar]
- Steitz, T.A.; Steitz, J.A. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6498–6502. [Google Scholar] [CrossRef]
- Scott, W.G.; Finch, J.T.; Klug, A. The crystal structure of an all-RNA hammerhead ribozyme: A proposed mechanism for RNA catalytic cleavage. Cell 1995, 81, 991–1002. [Google Scholar] [CrossRef]
- Scott, W.G.; Murray, J.B.; Arnold, J.R.; Stoddard, B.L.; Klug, A. Capturing the structure of a catalytic RNA intermediate: the hammerhead ribozyme. Science 1996, 274, 2065–2069. [Google Scholar] [CrossRef]
- Murray, J.B.; Seyhan, A.A.; Walter, N.G.; Burke, J.M.; Scott, W.G. The hammerhead, hairpin and vs. ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol. 1998, 5, 587–595. [Google Scholar] [CrossRef]
- Murray, J.B.; Szöke, H.; Szöke, A.; Scott, W.G. Capture and visualization of a catalytic RNA enzyme-product complex using crystal lattice trapping and X-ray holographic reconstruction. Mol. Cell 2000, 5, 279–287. [Google Scholar] [CrossRef]
- Kuimelis, R.G.; McLaughlin, L.W. Ribozyme-mediated cleavage of a substrate analogue containing an internucleotide-bridging 5’-phosphorothioate: Evidence for the single-metal model. Biochemistry 1996, 35, 5308–5317. [Google Scholar]
- Hermann, T.; Auffinger, P.; Scott, W.G.; Westhof, E. Evidence for a hydroxide ion bridging two magnesium ions at the active site of the hammerhead ribozyme. Nucl. Acid. Res. 1997, 25, 3421–3427. [Google Scholar]
- Kuimelis, R.G.; McLaughlin, L.W. Application of a 5’-bridging phosphorothioate to probe divalent metal and hammerhead ribozyme mediated RNA cleavage. Bioorg. Med. Chem. 1997, 5, 1051–1061. [Google Scholar] [CrossRef]
- Torres, R.A.; Himo, F.; Bruice, T.C.; Noodleman, L.; Lovell, T. Theoretical examination of Mg(2+)-mediated hydrolysis of a phosphodiester linkage as proposed for the hammerhead ribozyme. J. Am. Chem. Soc. 2003, 125, 9861–9867. [Google Scholar]
- Uebayasi, M.; Uchimaru, T.; Sawata, S.; Shimayama, T. Theoretical and Experimental Considerations on the Hammerhead Ribozyme Reactions: Divalent Magnesium. J. Org. Chem. 1994, 59, 7414–7420. [Google Scholar]
- Boero, M.; Tateno, M.; Terakura, K.; Oshiyama, A. Double-Metal-Ion/Single-Metal-Ion Mechanisms of the Cleavage Reaction of Ribozymes: First-Principles. J. Chem. Theory Comput. 2005, 1, 925–934. [Google Scholar] [CrossRef]
- Leclerc, F.; Karplus, M. Two-metal-ion mechanism for hammerhead-ribozyme catalysis. J. Phys. Chem. B 2006, 110, 3395–3409. [Google Scholar] [CrossRef]
- Khvorova, A.; Lescoute, A.; Westhof, E.; Jayasena, S.D. Sequence elements outside the hammerhead ribozyme catalytic core enable intracellular activity. Nat. Struct. Biol. 2003, 10, 708–712. [Google Scholar] [CrossRef]
- Canny, M.D.; Jucker, F.M.; Kellogg, E.; Khvorova, A.; Jayasena, S.D.; Pardi, A. Fast cleavage kinetics of a natural hammerhead ribozyme. J. Am. Chem. Soc. 2004, 126, 10848–10849. [Google Scholar]
- de la Peña, M.; Gago, S.; Flores, R. Peripheral regions of natural hammerhead ribozymes greatly increase their self-cleavage activity. EMBO J. 2003, 22, 5561–5570. [Google Scholar] [CrossRef]
- Osborne, E.M.; Schaak, J.E.; DeRose, V.J. Characterization of a native hammerhead ribozyme derived from schistosomes. RNA 2005, 11, 187–196. [Google Scholar] [CrossRef]
- Roychowdhury-Saha, M.; Burke, D.H. Extraordinary rates of transition metal ion-mediated ribozyme catalysis. RNA 2006, 12, 1846–1852. [Google Scholar] [CrossRef]
- Curtis, E.A.; Bartel, D.P. The hammerhead cleavage reaction in monovalent cations. RNA 2001, 7, 546–552. [Google Scholar] [CrossRef]
- Han, J.; Burke, J.M. Model for general acid-base catalysis by the hammerhead ribozyme: pH-activity relationships of G8 and G12 variants at the putative active site. Biochemistry 2005, 44, 7864–7870. [Google Scholar] [CrossRef]
- Thomas, J.M.; Perrin, D.M. Probing general base catalysis in the hammerhead ribozyme. J. Am. Chem. Soc. 2008, 130, 15467–15475. [Google Scholar] [CrossRef]
- Thomas, J.M.; Perrin, D.M. Probing general acid catalysis in the hammerhead ribozyme. J. Am. Chem. Soc. 2009, 131, 1135–1143. [Google Scholar] [CrossRef]
- Martick, M.; Scott, W.G. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell 2006, 126, 309–320. [Google Scholar] [CrossRef]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; Bateman, A. Rfam: Updates to the RNA families database. Nucl. Acid. Res. 2009, 37, 136–140. [Google Scholar]
- Pyle, A.M. Metal ions in the structure and function of RNA. J. Biol. Inorg. Chem. 2002, 7, 679–690. [Google Scholar] [CrossRef]
- Sigel, R.K.O.; Pyle, A.M. Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem. Rev. 2007, 107, 97–113. [Google Scholar]
- Jensen, M.R.; Hass, M.A.S.; Hansen, D.F.; Led, J.J. Investigating metal-binding in proteins by nuclear magnetic resonance. Cell Mol. Life Sci. 2007, 64, 1085–1104. [Google Scholar] [CrossRef]
- Houben, K.; Wasielewski, E.; Dominguez, C.; Kellenberger, E.; Atkinson, R.A.; Timmers, H.T.M.; Kieffer, B.; Boelens, R. Dynamics and metal exchange properties of C4C4 RING domains from CNOT4 and the p44 subunit of TFIIH. J. Mol. Biol. 2005, 349, 621–637. [Google Scholar] [CrossRef]
- Stahley, M.R.; Strobel, S.A. Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science 2005, 309, 1587–1590. [Google Scholar] [CrossRef]
- Stahley, M.R.; Adams, P.L.; Wang, J.; Strobel, S.A. Structural metals in the group I intron: A ribozyme with a multiple metal ion core. J. Mol. Biol. 2007, 372, 89–102. [Google Scholar] [CrossRef]
- Toor, N.; Keating, K.S.; Taylor, S.D.; Pyle, A.M. Crystal structure of a self-spliced group II intron. Science 2008, 320, 77–82. [Google Scholar] [CrossRef]
- Pyle, A.M. The tertiary structure of group II introns: Implications for biological function and evolution. Crit.Rev. Biochem. Mol. Biol. 2010, 45, 215–232. [Google Scholar] [CrossRef]
- Hoogstraten, C.; Sumita, M. Structure-function relationships in RNA and RNP enzymes: Recent advances. Biopolymers 2007, 87, 317–328. [Google Scholar]
- Yang, W. An equivalent metal ion in one- and two-metal-ion catalysis. Nat. Struct. Mol. Biol. 2008, 15, 1228–1231. [Google Scholar] [CrossRef]
- Chen, J.H.; Gong, B.; Bevilacqua, P.C.; Carey, P.R.; Golden, B.L. A catalytic metal ion interacts with the cleavage Site G.U wobble in the HDV ribozyme. Biochemistry 2009, 48, 1498–1507. [Google Scholar]
- Hansen, M.R.; Simorre, J.P.; Hanson, P.; Mokler, V.; Bellon, L.; Beigelman, L.; Pardi, A. Identification and characterization of a novel high affinity metal-binding site in the hammerhead ribozyme. RNA 1999, 5, 1099–1104. [Google Scholar]
- Vogt, M.; Lahiri, S.; Hoogstraten, C.G.; Britt, R.D.; DeRose, V.J. Coordination environment of a site-bound metal ion in the hammerhead ribozyme determined by 15N and 2H ESEEM spectroscopy. J. Am. Chem. Soc. 2006, 128, 16764–16770. [Google Scholar]
- Chartrand, P.; Leclerc, F.; Cedergren, R. Relating conformation, Mg2+ binding, and functional group modification in the hammerhead ribozyme. RNA 1997, 3, 692–696. [Google Scholar]
- Hampel, K.J.; Burke, J.M. Solvent protection of the hammerhead ribozyme in the ground state: Evidence for a cation-assisted conformational change leading to catalysis. Biochemistry 2003, 42, 4421–4429. [Google Scholar] [CrossRef]
- Campbell, D.O.; Bouchard, P.; Desjardins, G.; Legault, P. NMR structure of varkud satellite ribozyme stem-loop V in the presence of magnesium ions and localization of metal-binding sites. Biochemistry 2006, 45, 10591–10605. [Google Scholar]
- Izatt, R.M.; Christensen, J.J.; Rytting, J.H. Sites and thermodynamic quantities associated with proton and metal ion interaction with ribonucleic acid, deoxyribonucleic acid, and their constituent bases, nucleosides, and nucleotides. Chem. Rev. 1971, 71, 439–481. [Google Scholar] [CrossRef]
- Schnabl, J.; Sigel, R.K. Controlling ribozyme activity by metal ions. Curr. Opinion Chem. Biol. 2010, 14, 269–275. [Google Scholar] [CrossRef] [Green Version]
- DeRose, V.J. Metal ion binding to catalytic RNA molecules. Curr. Opin. Struct. Biol. 2003, 13, 317–324. [Google Scholar] [CrossRef]
- Forconi, M.; Piccirilli, J.A.; Herschlag, D. Modulation of individual steps in group I intron catalysis by a peripheral metal ion. RNA 2007, 13, 1656–1667. [Google Scholar] [CrossRef]
- Boots, J.L.; Canny, M.D.; Azimi, E.; Pardi, A. Metal ion specificities for folding and cleavage activity in the Schistosoma hammerhead ribozyme. RNA 2008, 14, 2212–2222. [Google Scholar] [CrossRef]
- Bevilacqua, P.C.; Yajima, R. Nucleobase catalysis in ribozyme mechanism. Curr. Opinion Chem. Biol. 2006, 10, 455–464. [Google Scholar] [CrossRef]
- Cerrone-Szakal, A.L.; Siegfried, N.A.; Bevilacqua, P.C. Mechanistic characterization of the HDV genomic ribozyme: Solvent isotope effects and proton inventories in the absence of divalent metal ions support C75 as the general acid. J. Am. Chem. Soc. 2008, 130, 14504–14520. [Google Scholar]
- Ferré-D’Amaré, A.R.; Zhou, K.; Doudna, J.A. Crystal structure of a hepatitis delta virus ribozyme. Nature 1998, 395, 567–574. [Google Scholar]
- Rupert, P.B.; Massey, A.P.; Sigurdsson, S.T.; Ferré-D’Amaré, A.R. Transition state stabilization by a catalytic RNA. Science 2002, 298, 1421–1424. [Google Scholar] [CrossRef]
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal structure of a self-splicing group I intron with both exons. Nature 2004, 430, 45–50. [Google Scholar]
- Martick, M.; Lee, T.S.; York, D.M.; Scott, W.G. Solvent structure and hammerhead ribozyme catalysis. Chem. Biol. 2008, 15, 332–342. [Google Scholar] [CrossRef]
- Chi, Y.I.; Martick, M.; Lares, M.; Kim, R.; Scott, W.G.; Kim, S.H. Capturing hammerhead ribozyme structures in action by modulating general base catalysis. PLoS Biol. 2008, 6, e234. [Google Scholar]
- O’Rear, J.L.; Wang, S.; Feig, A.L.; Beigelman, L.; Uhlenbeck, O.C.; Herschlag, D. Comparison of the hammerhead cleavage reactions stimulated by monovalent and divalent cations. RNA 2001, 7, 537–545. [Google Scholar] [CrossRef]
- Scott, W.G. Biophysical and biochemical investigations of RNA catalysis in the hammerhead ribozyme. Quart. Rev. Biophys. 1999, 32, 241–284. [Google Scholar] [CrossRef]
- Nakano, S.; Proctor, D.J.; Bevilacqua, P.C. Mechanistic characterization of the HDV genomic ribozyme: Assessing the catalytic and structural contributions of divalent metal ions within a multichannel reaction mechanism. Biochemistry 2001, 40, 12022–12038. [Google Scholar] [CrossRef]
- Bevilacqua, P.C. Mechanistic considerations for general acid-base catalysis by RNA: Revisiting the mechanism of the hairpin ribozyme. Biochemistry 2003, 42, 2259–2265. [Google Scholar] [CrossRef]
- Warshel, A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J. Biol. Chem. 1998, 273, 27035–27038. [Google Scholar]
- Warshel, A. Computer simulations of enzyme catalysis: Methods, progress, and insights. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 425–443. [Google Scholar] [CrossRef]
- Garcia-Viloca, M.; Gao, J.; Karplus, M.; Truhlar, D.G. How enzymes work: Analysis by modern rate theory and computer simulations. Science 2004, 303, 186–195. [Google Scholar]
- Kluge, S.; Weston, J. Can a hydroxide ligand trigger a change in the coordination number of magnesium ions in biological systems? Biochemistry 2005, 44, 4877–4885. [Google Scholar] [CrossRef]
- Tommaso, D.D.; de Leeuw, N.H. Structure and dynamics of the hydrated magnesium ion and of the solvated magnesium carbonates: Insights from first principles simulations. Phys. Chem. Chem. Phys. 2010, 12, 894–901. [Google Scholar]
- Walter, N.G. Ribozyme catalysis revisited: Is water involved? Mol. Cell 2007, 28, 923–929. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tagaya, M.; Hori, T.; Sakamoto, T.; Kurihara, Y.; Katahira, M.; Uesugi, S. Cleavage reaction of HDV ribozymes in the presence of Mg2+ is accompanied by a conformational change. Genes Cells 2002, 7, 567–579. [Google Scholar] [CrossRef]
- Tinsley, R.A.; Harris, D.A.; Walter, N.G. Magnesium dependence of the amplified conformational switch in the trans-acting hepatitis delta virus ribozyme. Biochemistry 2004, 43, 8935–8945. [Google Scholar]
- Lambert, D.; Heckman, J.E.; Burke, J.M. Three conserved guanosines approach the reaction site in native and minimal hammerhead ribozymes. Biochemistry 2006, 45, 7140–7147. [Google Scholar] [CrossRef]
- Nelson, J.A.; Uhlenbeck, O.C. Hammerhead redux: Does the new structure fit the old biochemical data? RNA 2008, 14, 605–615. [Google Scholar] [CrossRef]
- Wei, K.; Liu, L.; Cheng, Y.H.; Fu, Y.; Guo, Q.X. Theoretical examination of two opposite mechanisms proposed for hepatitis delta virus ribozyme. J. Phys. Chem. B 2007, 111, 1514–1516. [Google Scholar]
- Ke, A.; Zhou, K.; Ding, F.; Cate, J.H.D.; Doudna, J.A. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature 2004, 429, 201–205. [Google Scholar]
- Banás, P.; Rulìsek, L.; Hánosová, V.; Svozil, D.; Walter, N.G.; Sponer, J.; Otyepka, M. General base catalysis for cleavage by the active-site cytosine of the hepatitis delta virus ribozyme: QM/MM calculations establish chemical feasibility. J. Phys. Chem. B 2008, 112, 11177–11187. [Google Scholar]
- Nam, K.; Gao, J.; York, D.M. Quantum mechanical/molecular mechanical simulation study of the mechanism of hairpin ribozyme catalysis. J. Am. Chem. Soc. 2008, 130, 4680–4691. [Google Scholar] [CrossRef]
- Suydam, I.T.; Levandoski, S.D.; Strobel, S.A. Catalytic importance of a protonated adenosine in the hairpin ribozyme active site. Biochemistry 2010, 49, 3723–3732. [Google Scholar] [CrossRef]
- Mlýnský, V.; Banáš, P.; Hollas, D.; Réblová, K.; Walter, N.G.; Šponer, J.; Otyepka, M. Extensive Molecular Dynamics Simulations Showing That Canonical G8 and Protonated A38H(+) Forms Are Most Consistent with Crystal Structures of Hairpin Ribozyme. J. Phys. Chem. B 2010, 114, 6642–6652. [Google Scholar]
- Rhodes, M.M.; Réblová, K.; Sponer, J.; Walter, N.G. Trapped water molecules are essential to structural dynamics and function of a ribozyme. Proc. Natl. Acad. Sci. USA 2006, 103, 13380–13385. [Google Scholar]
- Nam, K.; Gao, J.; York, D.M. Electrostatic interactions in the hairpin ribozyme account for the majority of the rate acceleration without chemical participation by nucleobases. RNA 2008, 14, 1501–1507. [Google Scholar] [CrossRef]
- Osborne, E.M.; Ward, W.L.; Ruehle, M.Z.; DeRose, V.J. The identity of the nucleophile substitution may influence metal interactions with the cleavage site of the minimal hammerhead ribozyme. Biochemistry 2009, 48, 10654–10664. [Google Scholar] [CrossRef]
- Fedor, M.J. Comparative enzymology and structural biology of RNA self-cleavage. Ann. Rev. Biophys. 2009, 38, 271–299. [Google Scholar]
- Nelson, J.A.; Uhlenbeck, O.C. Minimal and extended hammerheads utilize a similar dynamic reaction mechanism for catalysis. RNA 2008, 14, 43–54. [Google Scholar]
- Roychowdhury-Saha, M.; Burke, D.H. Distinct reaction pathway promoted by non-divalent-metal cations in a tertiary stabilized hammerhead ribozyme. RNA 2007, 13, 841–848. [Google Scholar] [CrossRef]
- Nelson, J.A.; Uhlenbeck, O.C. When to believe what you see. Mol. Cell 2006, 23, 447–50. [Google Scholar] [CrossRef]
- Lippert, B. Ligand-pKa shifts through metals: Potential relevance to ribozyme chemistry. Chem. Biodivers. 2008, 5, 1455–1474. [Google Scholar] [CrossRef]
- Lee, T.S.; López, C.S.; Giambasu, G.M.; Martick, M.; Scott, W.G.; York, D.M. Role of Mg2+ in hammerhead ribozyme catalysis from molecular simulation. J. Am. Chem. Soc. 2008, 130, 3053–3064. [Google Scholar]
- Lee, T.S.; York, D.M. Origin of mutational effects at the C3 and G8 positions on hammerhead ribozyme catalysis from molecular dynamics simulations. J. Am. Chem. Soc. 2008, 130, 7168–7169. [Google Scholar] [CrossRef]
- Smith, M.D.; Mehdizadeh, R.; Olive, J.E.; Collins, R.A. The ionic environment determines ribozyme cleavage rate by modulation of nucleobasepK a. RNA 2008, 14, 1942–1949. [Google Scholar] [CrossRef]
- Tanaka, Y.; Taira, K. Detection of RNA nucleobase metalation by NMR spectroscopy. Chem. Commun. (Camb) 2005, 2069–2079. [Google Scholar] [CrossRef]
- Koutmou, K.S.; Casiano-Negroni, A.; Getz, M.M.; Pazicni, S.; Andrews, A.J.; Penner-Hahn, J.E.; Al-Hashimi, H.M.; Fierke, C.A. NMR and XAS reveal an inner-sphere metal binding site in the P4 helix of the metallo-ribozymeribonuclease P. Proc. Natl. Acad. Sci. 2010, 107, 2479–2484. [Google Scholar]
- Lee, T.S.; Giambaşu, G.M.; Sosa, C.P.; Martick, M.; Scott, W.G.; York, D.M. Threshold occupancy and specific cation binding modes in the hammerhead ribozyme active site are required for active conformation. J. Mol. Biol. 2009, 388, 195–206. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Leclerc, F. Hammerhead Ribozymes: True Metal or Nucleobase Catalysis? Where Is the Catalytic Power from? Molecules 2010, 15, 5389-5407. https://doi.org/10.3390/molecules15085389
Leclerc F. Hammerhead Ribozymes: True Metal or Nucleobase Catalysis? Where Is the Catalytic Power from? Molecules. 2010; 15(8):5389-5407. https://doi.org/10.3390/molecules15085389
Chicago/Turabian StyleLeclerc, Fabrice. 2010. "Hammerhead Ribozymes: True Metal or Nucleobase Catalysis? Where Is the Catalytic Power from?" Molecules 15, no. 8: 5389-5407. https://doi.org/10.3390/molecules15085389