Discovery and Development of Anti-HBV Agents and Their Resistance

Abstract
:
Abbreviations
| cccDNA | covalently closed circular DNA |
| FDA | U.S. Food and Drug Administration |
| GASP | Genetic Algorithm Similarity Program |
| HBsAg | hepatitis B surface antigen |
| HBeAg | hepatitis B e antigen |
| HBV | hepatitis B virus |
| HBx | HBV X protein |
| HCC | hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HIV | human immunodeficiency virus |
| HNF | hepatocyte nuclear factor |
| RT | reverse transcriptase |
| WHO | World Health Organization |
| WHV | woodchuck hepatitis virus |
1. Introduction
2. Anti-HBV Drugs
2.1. Current anti-HBV drugs
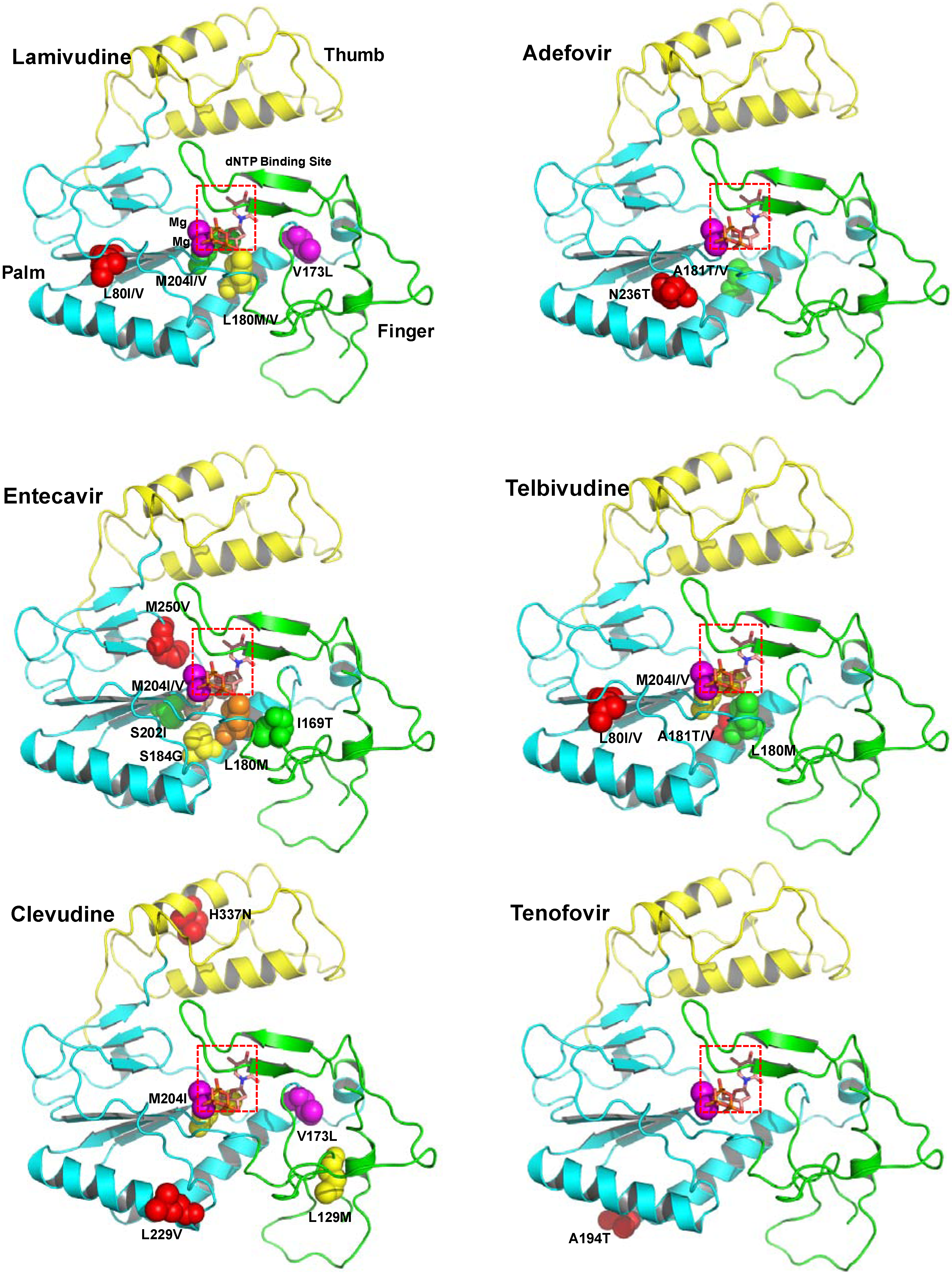
2.1.1. Lamivudine

{kind=link}
{kind=link}
| Molecule | Structure | Brand name | Mechanism | Company | Year of FDA approval |
|---|---|---|---|---|---|
| Lamivudine | 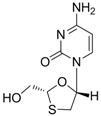 | Zeffix, Heptovir, Epivir, and Epivir-HBV | Nucleoside analogue/RT Inhibitor | GlaxoSmithKline | 1998 (for adults) and 2000 (for children) in US |
| Adefovir |  | Preveon and Hepsera | Nucleotide analogue/RT Inhibitor | Gilead | 2002 (US) |
| Entecavir | 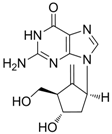 | Baraclude | Nucleoside analogue/RT Inhibitor | Bristol Meyers Squibb | 2005 (US) |
| Telbivudine | 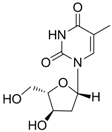 | Sebivo (Europe) Tyzeka (US) | Nucleoside analogue/RT Inhibitor | Idenix, Novartis | 2006 (US) |
| Clevudine | 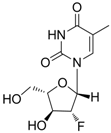 | Levovir and Revovir | Nucleoside analogue/RT Inhibitor. | Bukwang Pharm | 2006 (KOREA) |
| Tenofovir | 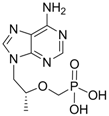 | Viread | Nucleotide analogue/RT Inhibitor | Gilead | 2008 (US) |
2.1.2. Adefovir Dipivoxil
2.1.3. Entecavir
2.1.4. Telbivudine
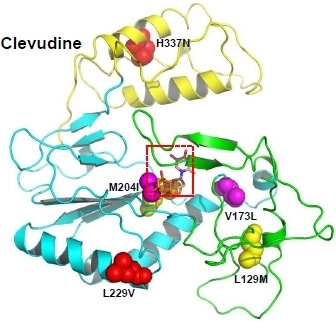
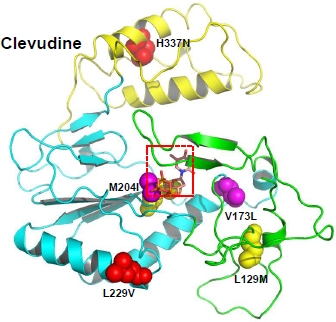
2.1.5. Clevudine
2.1.6. Tenofovir
2.2. Adverse effects of current HBV drugs
3. Viral Resistance to HBV Drugs
3.1. Molecular mechanism of resistance
3.1.1. Lamivudine resistance
3.1.2. Adefovir Dipivoxil resistance
3.1.3. Entecavir resistance
3.1.4. Telbivudine resistance
3.1.5. Clevudine resistance
3.1.6. Tenofovir resistance
3.2. Molecular modeling study of drug-resistant HBV

4. Development of Anti-HBV Agents
4.1. Drugs in clinical trials for HBV
| Molecule | Structure | Brand name | Mechanism | Company | Year |
|---|---|---|---|---|---|
| Lagociclovir valactate | 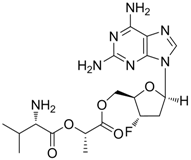 | - | Nucleoside analogue/RT inhibitor/Prodrug | Medvir AB | Phase II 2005 (Sweden) |
| Elvucitabine | 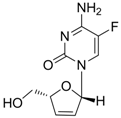 | - | Nucleoside analogue/RT Inhibitor | Achillion Pharmaceuticals | 2001 (US) |
| B-80380 | 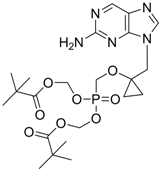 | - | Nucleotide analogue/RT Inhibitor | LG Chem Ltd | Phase II 2003 (KOREA) |
| radefovir | 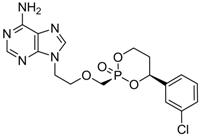 | - | Nucleotide analogue/RT inhibitor/Prodrug | Metabasis Therapeutics | Phase II 2007 (US) |
| altorcitabine |  | Nucleoside analogue/RT inhibitor/Prodrug | Idenix Pharmaceuticals/Novartis | Phase II 2003 (US) |
4.2. Novel non-nucleoside HBV inhibitors
| Compound (Target) | Structure | Activity | Mechanism of Action | Reference |
|---|---|---|---|---|
| 1 (HBsAg, HBeAg) | 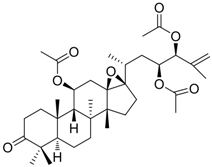 | CC50 : >2.6mM HBsAg(IC50): 0.024 mM HBeAg(IC50): 0.028 mM | Inhibition of HBsAg and HBeAg secretion | [85] |
| 2 (HBeAg) | 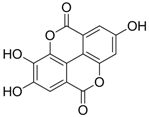 | IC50: 0.07 μg/mL | Inhibition of HBeAg secretion in HepG2 2.2.15 cell line | [88] |
| 3 (HBsAg) |  | EC50: 1.5 μM | Inhibition of HBsAg secretion | [89] |
| 4 (HBsAg) | 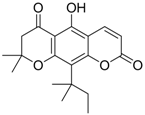 | EC50: 1.14 μM | Inhibition of HBsAg secretion | [90] |
| 5 (Virion secretion) | 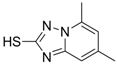 | IC50: 0.12 μM | Inhibition of the interaction between core and surface protein | [91] |
| 6 (Virion secretion) | 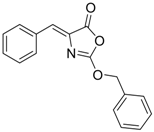 | IC50: 5.4 μM | Inhibition of the interaction between core and surface protein | [91] |
| 7 (Capsid formation) | 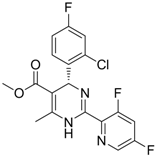 | IC50: 0.05 μM | Inhibition of replication by nucleocapsid depletion | [92] |
| Compound | Structure | Activity | Mechanism of Action | Reference |
|---|---|---|---|---|
| 8 |  | IC50: 2.4 μM in HepG2 cells | Inhibition of replication by blocking RNA packaging | [93,94,95] |
| 9 | 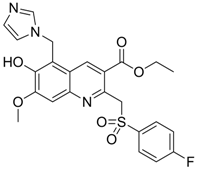 | IC50: 4.7 μM (Replication)
IC50: 26.2 μM (HBsAg) IC50: 98.1 μM (HBeAg) in HepG2 2.2.15 cells | Inhibition of replication, HBsAg and HBeAg secretion | [96] |
| 10 | 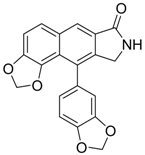 | IC50: 0.08 μM in HepG2 2.2.15 cells | Inhibition of replication by down-regulation of HNF-3 and 4 | [97,98] |
| 11 | 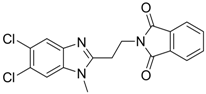 | IC50: 0.9 μM in HepG2 cells
CC50: >1000 μM | Inhibition of replication | [99] |
| 12 |  | IC50: 0.7 μM
CC50: 192 μM in HepG2 2.2.15 cells | Inhibition of replication | [100] |
| 13 | 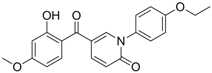 | IC50: 0.206 μM
CC50: >109.6 μM in HepG2 cells | Inhibition of replication | [101] |
| 14 | 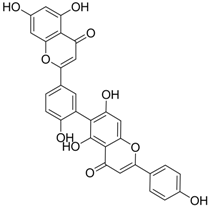 | IC50: 0.25 μM in HepG2 2.2.15 cells | Inhibition of replication | [102] |
| 15 | 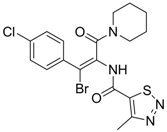 | IC50: 3.59 μg/mL in HepG2 2.2.15 cells | Inhibition of replication | [103] |
| 16 | 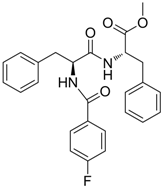 | IC50: 1.40 μM in HepG2 2.2.15 cells | Inhibition of replication, HBsAg and HBeAg secretion | [104] |
| 17 | 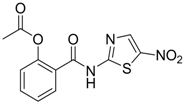 | EC50: 0.12 μM in HepG2 2.2.15 cells | Inhibition of replication | [105] |
| 18 | 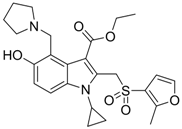 | IC50: 1.52 μg/mL in HepG2 2.2.15 | Inhibition of replication | [106] |
| 19 |  | IC50: 0.57 μg/mL in DHBV replication.
IC50: 4.0 μg/mL (HBsAg, HBeAg) | Inhibition of replication, HBsAg and HBeAg secretion | [107] |
| 20 |  | IC50: 2.3 μM (HBsAg)
IC50: 0.5 μM (replication) | Inhibition of replication and HBsAg secretion | [108] |
| 21 |  | EC50: 1.7 μM,
CC50: 286 μM in HepG2 2.2.15 cells | Inhibition of virion secretion and replication | [109] |
| 22 |  | IC50: ~0.01 μg/mL | Inhibition of RT | [110] |
| 23 | 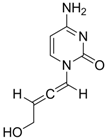 | IC50:0.08 μM in HepG2 2.2.15 | Inhibition of replication | [111] |
4.3. Virtual screening, modeling and rational drug design
4.4. Targets for future drugs
5. Conclusions
Acknowledgements
References
- Tiollais, P.; Charnay, P.; Vyas, G.N. Biology of hepatitis B virus. Science 1981, 213, 406–411. [Google Scholar]
- Robinson, W.S. Molecular events in the pathogenesis of hepadnavirus-associated hepatocellular carcinoma. Annu. Rev. Med. 1994, 45, 297–323. [Google Scholar] [CrossRef]
- Kremsdorf, D.; Soussan, P.; Paterlini-Brechot, P.; Brechot, C. Hepatitis B virus-related hepatocellular carcinoma: Paradigms for viral-related human carcinogenesis. Oncogene 2006, 25, 3823–3833. [Google Scholar] [CrossRef]
- Brechot, C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: Old and new paradigms. Gastroenterology 2004, 127, S56–S61. [Google Scholar] [CrossRef]
- Seeger, C.; Zoulim, F.; Mason, W.S. Hepadnaviruses. In Fields Virology, 5th; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2977–3029. [Google Scholar]
- Ganem, D.; Schneider, R.J. The molecular biology of the hepatitis B viruses. In Fields Virology, 4th; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2, pp. 2923–2970. [Google Scholar]
- Wang, G.H.; Seeger, C. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 1993, 67, 6507–6512. [Google Scholar]
- Wang, G.H.; Seeger, C. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 1992, 71, 663–670. [Google Scholar] [CrossRef]
- Kwon, S.Y.; Park, Y.K.; Ahn, S.H.; Cho, E.S.; Choe, W.H.; Lee, C.H.; Kim, B.K.; Ko, S.Y.; Choi, H.S.; Park, E.S.; Shin, G.C.; Kim, K.H. Identification and characterization of clevudine-resistant mutants of hepatitis B virus isolated from chronic hepatitis B patients. J. Virol. 2010, 84, 4494–4503. [Google Scholar]
- De Clercq, E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 2002, 1, 13–25. [Google Scholar] [CrossRef]
- Mazzucco, C.E.; Hamatake, R.K.; Colonno, R.J.; Tenney, D.J. Entecavir for treatment of hepatitis B virus displays no in vitro mitochondrial toxicity or DNA polymerase gamma inhibition. Antimicrob. Agents Chemother. 2008, 52, 598–605. [Google Scholar] [CrossRef]
- Innaimo, S.F.; Seifer, M.; Bisacchi, G.S.; Standring, D.N.; Zahler, R.; Colonno, R.J. Identification of BMS-200475 as a potent and selective inhibitor of hepatitis B virus. Antimicrob. Agents Chemother. 1997, 41, 1444–1448. [Google Scholar]
- Levine, S.; Hernandez, D.; Yamanaka, G.; Zhang, S.; Rose, R.; Weinheimer, S.; Colonno, R.J. Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob. Agents Chemother. 2002, 46, 2525–2532. [Google Scholar] [CrossRef]
- Chang, T.T. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 2006, 354, 1001–1010. [Google Scholar] [CrossRef]
- Lai, C.L.; Shouval, D.; Lok, A.S.; Chang, T.T.; Cheinquer, H.; Goodman, Z.; DeHertogh, D.; Wilber, R.; Zink, R.C.; Cross, A.; Colonno, R.; Fernandes, L. BEHoLD AI463027 Study Group. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 2006, 354, 1011–1020. [Google Scholar] [CrossRef]
- Sherman, M.; Yurdaydin, C.; Sollano, J.; Silva, M.; Liaw, Y.F.; Cianciara, J.; Boron-Kaczmarska, A.; Martin, P.; Goodman, Z.; Colonno, R.; Cross, A.; Denisky, G.; Kreter, B.; Hindes, R. AI463026 BEHoLD Study Group. Entecavir for the treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B. Gastroenterology 2006, 130, 2039–2049. [Google Scholar] [CrossRef]
- Sherman, M.; Yurdaydin, C.; Simsek, H.; Silva, M.; Liaw, Y.F.; Rustgi, V.K.; Sette, H.; Tsai, N.; Tenney, D.J.; Vaughan, J.; Kreter, B.; Hindes, R. AI463026 Benefits of Entecavir for Hepatitis B Liver Disease (BEHoLD) Study Group. Entecavir therapy for lamivudine-refractory chronic hepatitis B: Improved virologic, biochemical, and serology outcomes through 96 weeks. Hepatology 2008, 48, 99–108. [Google Scholar] [CrossRef]
- Bryant, M.L.; Bridges, E.G.; Placidi, L.; Faraj, A.; Loi, A.G.; Pierra, C.; Dukhan, D.; Gosselin, G.; Imbach, J.L.; Hernandez, B.; Juodawlkis, A.; Tennant, B.; Korba, B.; Cote, P.; Marion, P.; Cretton-Scott, E.; Schinazi, R.F.; Sommadossi, J.P. Antiviral L-nucleosides specific for hepatitis B virus infection. Antimicrob. Agents Chemother. 2001, 45, 229–235. [Google Scholar]
- Liaw, Y.F.; Gane, E.; Leung, N.; Zeuzem, S.; Wang, Y.; Lai, C.L.; Heathcote, E.J.; Manns, M.; Bzowej, N.; Niu, J.; Han, S.H.; Hwang, S.G.; Cakaloglu, Y.; Tong, M.J.; Papatheodoridis, G.; Chen, Y.; Brown, N.A.; Albanis, E.; Galil, K.; Naoumov, N.V. GLOBE Study Group. 2-Year GLOBE trial results: Telbivudine is superior to lamivudine in patients with chronic hepatitis B. Gastroenterology 2009, 136, 486–495. [Google Scholar]
- Lai, C.L.; Gane, E.; Liaw, Y.F.; Hsu, C.W.; Thongsawat, S.; Wang, Y.; Chen, Y.; Heathcote, E.J.; Rasenack, J.; Bzowej, N.; Naoumov, N.V.; Di Bisceglie, A.M.; Zeuzem, S.; Moon, Y.M.; Goodman, Z.; Chao, G.; Constance, B.F.; Brown, N.A. Globe Study Group. Telbivudine versu lamivudine in patients with chronic hepatitis B. N. Engl. J. Med. 2007, 357, 2576–2588. [Google Scholar]
- Lai, C.L.; Leung, N.; Teo, E.K.; Tong, M.; Wong, F.; Hann, H.W.; Han, S.; Poynard, T.; Myers, M.; Chao, G.; Lloyd, D.; Brown, N.A. Telbivudine Phase II Investigator Group. A 1-year trial of telbivudine, lamivudine, and the combination in patients with hepatitis B e antigen-positive chronic hepatitis B. Gastroenterology 2005, 129, 528–536. [Google Scholar]
- Chu, CK.; Ma, T.; Shanmuganathan, K.; Wang, C.; Xiang, Y.; Pai, S.B.; Yao, G.Q.; Sommadossi, J.P.; Cheng, Y.C. Use of 2'-fluoro-5-methyl-beta-L-arabinofuranosyluracil as a novel antiviral agent for hepatitis B virus and Epstein-Barr virus. Antimicrob. Agents Chemother. 1995, 39, 979–981. [Google Scholar] [CrossRef]
- Balakrishna Pai, S.; Liu, S.H.; Zhu, Y.L.; Chu, C.K.; Cheng, Y.C. Inhibition of hepatitis B virus by a novel L-nucleoside, 2’-fluor-5-methyl-beta L-arabinofuranosyl uracil. Antimicrob. Agents Chemother. 1996, 40, 380–386. [Google Scholar]
- Liu, S.H.; Grove, K.L.; Cheng, Y.C. Unique metabolism of a novel antiviral L-nucleoside analog, 2'-fluoro-5-methyl-beta-L-arabinofuranosyluracil: A substrate for both thymidine kinase and deoxycytidine kinase. Antimicrob. Agents Chemother. 1998, 42, 833–839. [Google Scholar]
- Yoo, B.C.; Kim, J.H; Kim, T.H.; Koh, K.C.; Um, S.H.; Kim, Y.S.; Lee, K.S.; Han, B.H.; Chon, C.Y.; Han, J.Y.; Ryu, S.H.; Kim, H.C.; Byun, K.S.; Hwang, S.G.; Kim, B.I.; Cho, M.; Yoo, K.; Lee, H.J.; Hwang, J.S.; Kim, Y.S.; Lee, Y.S.; Choi, S.K.; Lee, Y.J.; Yang, J.M.; Park, J.W.; Lee, M.S.; Kim, D.G.; Chung, Y.H.; Cho, S.H.; Choi, J.Y.; Kweon, Y.O.; Lee, H.Y.; Jeong, S.H.; Yoo, H.W.; Lee, H.S. Clevudine is highly efficacious in hepatitis B e antigen-negative chronic hepatitis B with durable off-therapy viral suppression. Hepatology 2007, 46, 1041–1048. [Google Scholar]
- Yoo, B.C.; Kim, J.H.; Chung, Y.H.; Lee, K.S.; Paik, S.W.; Ryu, S.H.; Han, B.H.; Han, J.Y.; Byun, K.S.; Cho, M.; Lee, H.J.; Kim, T.H.; Cho, S.H.; Park, J.W.; Um, S.H.; Hwang, S.G.; Kim, Y.S.; Lee, Y.J.; Chon, C.Y.; Kim, B.I.; Lee, Y.S.; Yang, J.M.; Kim, H.C.; Hwang, J.S.; Choi, S.K.; Kweon, Y.O.; Jeong, S.H.; Lee, M.S.; Choi, J.Y.; Kim, D.G.; Kim, Y.S.; Lee, H.Y.; Yoo, K.; Yoo, H.W.; Lee, H.S. Twenty-four-week clevudine therapy showed potent and sustained antiviral activity in HBeAg-positive chronic hepatitis B. Hepatology 2007, 45, 1172–1178. [Google Scholar]
- van Bommel, F.; Wunsche, T.; Mauss, S.; Reinke, P.; Bergk, A.; Schurmann, D.; Wiedenmann, B.; Berg, T. Comparison of adefovir and tenofovir in the treatment of lamivudine-resistant hepatitis B virus infection. Hepatology 2004, 40, 1421–1425. [Google Scholar] [CrossRef]
- van Bommel, F.; Zollner, B.; Sarrazin, C.; Spengler, U.; Huppe, D.; Moller, B.; Feucht, H.H.; Wiedenmann, B.; Berg, T. Tenofovir for patients with lamivudine-resistant hepatitis B virus (HBV) infection and high HBV DNA level during adefovir therapy. Hepatology 2006, 44, 318–325. [Google Scholar] [CrossRef]
- Delaney, W.E.; Ray, A.S.; Yang, H.; Qi, X.; Xiong, S.; Zhu, Y.; Miller, M.D. Intracellular metabolism and in vitro activity of tenofovir against hepatitis B virus. Antimicrob. Agents Chemother. 2006, 50, 2471–2477. [Google Scholar] [CrossRef]
- Marcellin, P.; Heathcote, E.J.; Buti, M.; Gane, E.; de Man, R.A.; Krastev, Z.; Germanidis, G.; Lee, S.S.; Flisiak, R.; Kaita, K.; Manns, M.; Kotzev, I.; Tchernev, K.; Buggisch, P.; Weilert, F.; Kurdas, O.O.; Shiffman, M.L.; Trinh, H.; Washington, M.K.; Sorbel, J.; Anderson, J.; Snow-Lampart, A.; Mondou, E.; Quinn, J.; Rousseau, F. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N. Engl. J. Med. 2008, 359, 2442–2455. [Google Scholar]
- Fung, S.K.; Lok, A.S. Drug insight: Nucleoside and nucleotide analog inhibitors for hepatitis B. Nat. Clin. Pract. Gastroenterol. Hepatol. 2004, 1, 90–97. [Google Scholar] [CrossRef]
- Fleischer, R.D.; Lok, A.S. Myopathy and neuropathy associated with nucleos(t)ide analog therapy for hepatitis B. J. Hepatol. 2009, 51, 787–791. [Google Scholar] [CrossRef]
- Rho, M.; Perazella, M.A. Nephrotoxicity associated with antiretroviral therapy in HIV-infected patients. Curr. Drug Saf. 2007, 2, 147–154. [Google Scholar] [CrossRef]
- Tanji, N.; Tanji, K.; Kambham, N.; Markowitz, G.S.; Bell, A.; D'agati, V.D. Adefovir nephrotoxicity: Possible role of mitochondrial DNA depletion. Hum. Pathol. 2001, 32, 734–740. [Google Scholar] [CrossRef]
- Kahn, J.; Lagakos, S.; Wulfsohn, M.; Cherng, D.; Miller, M.; Cherrington, J.; Hardy, D.; Beall, G.; Cooper, R.; Murphy, R.; Basgoz, N.; Ng, E.; Deeks, S.; Winslow, D.; Toole, J.J.; Coakley, D. Efficacy and safety of adefovir dipivoxil with antiretroviral therapy: A randomized controlled trial. JAMA 1999, 282, 2305–2312. [Google Scholar]
- Martin, J.L.; Brown, C.E.; Matthews-Davis, N.; Reardon, J.E. Effects of antiviral nucleoside analogs on human DNA polymerases and mitochondrial DNA synthesis. Antimicrob. Agents Chemother. 1994, 38, 2743–2749. [Google Scholar] [CrossRef]
- Kakuda, T.N. Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clin. Ther. 2000, 22, 685–708. [Google Scholar] [CrossRef]
- Lewis, W.; Day, B.J.; Copeland, W.C. Mitochondrial toxicity of NRTI antiviral drugs: An integrated cellular perspective. Nat. Rev. Drug. Discov. 2003, 2, 812–822. [Google Scholar] [CrossRef]
- Seok, J.I.; Lee, D.K.; Lee, C.H.; Park, M.S.; Kim, S.Y.; Kim, H.S.; Jo, H.Y.; Lee, C.H.; Kim, D.S. Long-term therapy with clevudine for chronic hepatitis B can be associated with myopathy characterized by depletion of mitochondrial DNA. Hepatology 2009, 49, 2080–2086. [Google Scholar] [CrossRef]
- Girones, R.; Miller, R.H. Mutation rate of the hepadnavirus genome. Virology 1989, 170, 595–597. [Google Scholar] [CrossRef]
- Nowak, M.A.; Bonhoeffer, S.; Hill, A.M.; Boehme, R.; Thomas, H.C.; McDade, H. Viral dynamics in hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 1996, 93, 4398–4402. [Google Scholar] [CrossRef]
- Ngui, S.L.; Hallet, R.; Teo, C.G. Natural and iatrogenic variation in hepatitis B virus. Rev. Med. Virol. 1999, 9, 183–209. [Google Scholar] [CrossRef]
- Ghany, M.; Liang, T.J. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology 2007, 132, 1574–1585. [Google Scholar] [CrossRef]
- Zoulim, F.; Locarnini, S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009, 137, 1593–1608. [Google Scholar] [CrossRef]
- Lok, A.S.; Lai, C.L.; Leung, N.; Yao, G.B.; Cui, Z.Y.; Schiff, E.R.; Dienstag, J.L.; Heathcote, E.J.; Little, N.R.; Griffiths, D.A.; Gardner, S.D.; Castiglia, M. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology 2003, 125, 1714–1722. [Google Scholar] [CrossRef]
- Allen, M.I.; Deslauriers, M.; Andrews, C.W.; Tipples, G.A.; Walters, K.A.; Tyrrell, D.L.; Brown, N.; Condreay, L.D. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998, 27, 1670–1677. [Google Scholar] [CrossRef]
- Stuyver, L.J.; Locarnini, S.A.; Lok, A.; Richman, D.D.; Carman, W.F.; Dienstag, J.L.; Schinazi, R.F. Nomenclature for antiviral-resistant human hepatitis B virus mutations in the polymerase region. Hepatology 2001, 33, 751–757. [Google Scholar] [CrossRef]
- Warner, N.; Locarnini, S.; Kuiper, M.; Bartholomeusz, A.; Ayres, A.; Yuen, L.; Shaw, T. The L80I substitution in the reverse transcriptase domain of the hepatitis B virus polymerase is associated with lamivudine resistance and enhanced viral replication in vitro. Antimicrob. Agents Chemother. 2007, 51, 2285–2292. [Google Scholar] [CrossRef]
- Delaney, W.E., 4th; Yang, H.; Westland, C.E.; Das, K.; Arnold, E.; Gibbs, C.S.; Miller, M.D.; Xiong, S. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J. Virol. 2003, 77, 11833–11841. [Google Scholar]
- Huang, H.; Chopra, R.; Verdine, G.L.; Harrison, S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase transcriptase: Implications for drug resistance. Science 1998, 282, 1669–1675. [Google Scholar] [CrossRef]
- Das, K.; Xiong, X.; Yang, H.; Westland, C.E.; Gibbs, C.S.; Sarafianos, S.G.; Arnold, E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol. 2001, 75, 4771–4779. [Google Scholar]
- Yang, H.; Westland, C.E.; Delaney, W.E.; Heathcote, E.J.; Ho, V.; Fry, J.; Brosgart, C.; Gibbs, C.S.; Miller, M.D.; Xiong, S. Resistance surveillance in chronic hepatitis B patients treated with adefovir dipivoxil for up to 60 weeks. Hepatology 2002, 36, 464–473. [Google Scholar] [CrossRef]
- Hadziyannis, S.J.; Tassopoulos, N.C.; Heathcote, E.J.; Chang, T.T.; Kitis, G.; Rizzetto, M.; Marcellin, P.; Lim, S.G.; Goodman, Z.; Ma, J.; Brosgart, C.L.; Borroto-Esoda, K.; Arterburn, S.; Chuck, S.L. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology 2006, 131, 1743–1751. [Google Scholar] [CrossRef]
- Angus, P.; Vaughan, R.; Xiong, S.; Yang, H.; Delaney, W.; Gibbs, C.; Brosgart, C.; Colledge, D.; Edwards, R.; Ayres, A.; Bartholomeusz, A. Locarnini, S. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology 2003, 125, 292–297. [Google Scholar] [CrossRef]
- Brunelle, M.N.; Jacquard, A.C.; Pichoud, C.; Durantel, D.; Carrouee-Durantel, S.; Villeneuve, J.P.; Trepo, C.; Zoulim, F. Susceptibility to antivirals of a human HBV strain with mutations conferring resistance to both lamivudine and adefovir. Hepatology 2005, 41, 1391–1398. [Google Scholar] [CrossRef]
- Villet, S.; Pichoud, C.; Billioud, G.; Barraud, L.; Durantel, S.; Trépo, C.; Zoulim, F. Impact of hepatitis B virus rtA181V/T mutants on hepatitis B treatment failure. J Hepatol. 2008, 48, 747–755. [Google Scholar] [CrossRef]
- Colonno, R.J.; Rose, R.; Baldick, C.J.; Levine, S.; Pokornowski, K.; Yu, C.F.; Walsh, A.; Fang, J.; Hsu, M.; Mazzucco, C.; Eggers, B.; Zhang, S.; Plym, M.; Klesczewski, K.; Tenney, D.J. Entecavir resistance is rare in nucleoside naive patients with hepatitis B. Hepatology 2006, 44, 1656–1665. [Google Scholar] [CrossRef]
- Tenney, D.J.; Rose, R.E; Baldick, C.J.; Pokornowski, K.A.; Eggers, B.J.; Fang, J.; Wichroski, M.J.; Xu, D.; Yang, J.; Wilber, R.B.; Colonno, R.J. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleosidenaive patients is rare through 5 years of therapy. Hepatology 2009, 49, 1503–1514. [Google Scholar] [CrossRef]
- Tenney, D.J.; Levine, S.M.; Rose, R.E.; Walsh, A.W.; Weinheimer, S.P.; Discotto, L.; Plym, M.; Pokornowski, K.; Yu, C.F.; Angus, P.; Ayres, A.; Bartholomeusz, A.; Sievert, W.; Thompson, G.; Warner, N.; Locarnini, S.; Colonno, R.J. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to lamivudine. Antimicrob. Agents Chemother. 2004, 48, 3498–3507. [Google Scholar]
- Nash, K. Telbivudine in the treatment of chronic hepatitis B. Adv. Ther. 2009, 26, 155–169. [Google Scholar] [CrossRef]
- Locarnini, S.; Mason, W.S. Cellular and virological mechanisms of HBV drug resistance. J. Hepatol. 2006, 44, 422–431. [Google Scholar] [CrossRef]
- Seifer, M.; Patty, A.; Serra, I.; Li, B.; Standring, D.N. Telbivudine, a nucleoside analog inhibitor of HBV polymerase, has a different in vitro cross-resistance profile than the nucleotide analog inhibitors adefovir and tenofovir. Antiviral Res. 2009, 81, 147–155. [Google Scholar] [CrossRef]
- Yamamoto, T.; Litwin, S.; Zhou, T.; Zhu, Y.; Condreay, L.; Furman, P.; Mason, W.S. Mutations of the woodchuck hepatitis virus polymerase gene that confer resistance to lamivudine and 2'-fluoro-5-methyl-beta-L-arabinofuranosyluracil. J. Virol. 2002, 76, 1213–1223. [Google Scholar] [CrossRef]
- Ono, S.K.; Kato, N.; Shiratori, Y.; Kato, J.; Goto, T.; Schinazi, R.F.; Carrilho, F.J.; Omata, M. The polymerase L528M mutation cooperates with nucleotide binding-site mutations, increasing hepatitis B virus replication and drug resistance. J. Clin. Invest. 2001, 107, 449–455. [Google Scholar] [CrossRef]
- Chin, R.; Shaw, T.; Torresi, J.; Sozzi, V.; Trautwein, C.; Bock, T.; Manns, M.; Isom, H.; Furman, P.; Locarnini, S. In vitro susceptibilities of wild-type or drug-resistant hepatitis B virus to (-)-beta-D-2,6-diaminopurine dioxolane and 2'-fluoro-5-methyl-beta-L-arabinofuranosyluracil. Antimicrob. Agents Chemother. 2001, 45, 2495–2501. [Google Scholar] [CrossRef]
- Sheldon, J.; Camino, N.; Rodés, B.; Bartholomeusz, A.; Kuiper, M.; Tacke, F.; Núñez, M.; Mauss, S.; Lutz, T.; Klausen, G.; Locarnini, S.; Soriano, V. Selection of hepatitis B virus polymerase mutations in HIV-coinfected patients treated with tenofovir. Antivir. Ther. 2005, 10, 727–734. [Google Scholar]
- Amini-Bavil-Olyaee, S.; Herbers, U.; Sheldon, J.; Luedde, T.; Trautwein, C.; Tacke, F. The rtA194T polymerase mutation impacts viral replication and susceptibility to tenofovir in hepatitis B e antigen-positive and hepatitis B e antigen-negative hepatitis B virus strains. Hepatology 2009, 49, 1158–1165. [Google Scholar] [CrossRef]
- Langley, D.R.; Walsh, A.W.; Baldick, C.J.; Eggers, B.J.; Rose, R.E.; Levine, S.M.; Kapur, A.J.; Colonno, R.J.; Tenney, D.J. Inhibition of hepatitis B virus polymerase by entecavir. J. Virol. 2007, 81, 3992–4001. [Google Scholar] [CrossRef]
- Lin, X.; Yuan, Z.H.; Wu, L.; Ding, J.P.; Wen, Y.M. A single amino acid in the reverse transcriptase domain of hepatitis B virus affects virus replication efficiency. J. Virol. 2001, 75, 11827–11833. [Google Scholar] [CrossRef]
- Sharon, A.; Chu, C.K. Understanding the molecular basis of HBV drug resistance by molecular modeling. Antiviral Res. 2008, 80, 339–353. [Google Scholar] [CrossRef]
- Bartholomeusz, A.; Tehan, B.G.; Chalmers, D.K. Comparisons of the HBV and HIV polymerase, and antiviral resistance mutations. Antivir. Ther. 2004, 9, 149–160. [Google Scholar]
- Daga, P.R.; Duan, J.; Doerksen, R.J. Computational model of hepatitis B virus DNA polymerase: Molecular dynamics and docking to understand resistant mutations. Protein Sci. 2010, 19, 796–807. [Google Scholar] [CrossRef]
- Xiong, X.; Flores, C.; Yang, H.; Toole, J.J.; Gibbs, C.S. Mutations in hepatitis B DNA polymerase associated with resistance to lamivudine do not confer resistance to adefovir in vitro. Hepatology 1998, 28, 1669–1673. [Google Scholar] [CrossRef]
- Aloman, C.; Wands, J.R. Resistance of HBV to adefovir dipivoxil: A case for combination antiviral therapy? Hepatology 2003, 38, 1584–1587. [Google Scholar] [CrossRef]
- Dando, T.; Plosker, G. Adefovir dipivoxil: A review of its use in chronic hepatitis B. Drugs 2003, 63, 2215–2234. [Google Scholar] [CrossRef]
- Yadav, V.; Chu, C.K. Molecular mechanisms of adefovir sensitivity and resistance in HBV polymerase mutants: A molecular dynamics study. Bioorg. Med. Chem. Lett. 2004, 14, 4313–4317. [Google Scholar] [CrossRef]
- Michalak, T.I.; Zhang, H.; Churchill, N.D.; Larsson, T.; Johansson, N.G.; Oberg, B. Profound antiviral effect of oral administration of MIV-210 on chronic hepadnaviral infection in a woodchuck model of hepatitis B. Antimicrob. Agents Chemother. 2009, 53, 3803–3814. [Google Scholar] [CrossRef]
- Zhu, Y.L.; Dutschman, D.E.; Liu, S.H.; Bridges, E.G.; Cheng, Y.C. Anti-hepatitis B virus activity and metabolism of 2',3'-dideoxy-2',3'-didehydro-beta-L(-)-5-fluorocytidine. Antimicrob. Agents Chemother. 1998, 42, 1805–1810. [Google Scholar]
- Choi, J.R.; Cho, D.G.; Roh, K.Y.; Hwang, J.T.; Ahn, S.; Jang, H.S.; Cho, W.Y.; Kim, K.W.; Cho, Y.G.; Kim, J.; Kim, Y.Z. A novel class of phosphonate nucleosides. 9-[(1-phosphonomethoxycyclopropyl)-methyl]guanine as a potent and selective anti-HBV agent. J. Med. Chem. 2004, 47, 2864–2869. [Google Scholar] [CrossRef]
- Fung, J.; Lai, C.L.; Yuen, M.F. LB80380: A promising new drug for the treatment of chronic hepatitis B. Expert Opin. Investig. Drugs. 2008, 17, 1581–1588. [Google Scholar] [CrossRef]
- Yuen, M.F.; Lee, S.H.; Kang, H.M.; Kim, C.R.; Kim, J.; Ngai, V.; Lai, C.L. Pharmacokinetics of LB80331 and LB80317 following oral administration of LB80380, a new antiviral agent for chronic hepatitis B (CHB), in healthy adult subjects, CHB patients, and mice. Antimicrob. Agents Chemother. 2009, 53, 1779–1785. [Google Scholar] [CrossRef]
- Yuen, M.F.; Han, K.H.; Um, S.H.; Yoon, S.K.; Kim, H.R.; Kim, J.; Kim, C.R.; Lai, C.L. Antiviral activity and safety of LB80380 in hepatitis B e antigen-positive chronic hepatitis B patients with lamivudine-resistant disease. Hepatology 2010, 51, 767–776. [Google Scholar] [CrossRef]
- Reddy, K.R.; Matelich, M.C.; Ugarkar, B.G.; Gómez-Galeno, J.E.; DaRe, J.; Ollis, K.; Sun, Z.; Craigo, W.; Colby, T.J.; Fujitaki, J.M.; Boyer, S.H.; van Poelje, P.D.; Erion, M.D. Pradefovir: A prodrug that targets adefovir to the liver for the treatment of hepatitis B. J. Med. Chem. 2008, 14, 666–676. [Google Scholar]
- Lim, S.G.; Lai, C.L.; Myers, M. Final results of a phase I/II dose escalation trial of valtorcitabine in patients with chronic hepatitis B (abstract). J. Hepatol. 2005, 42 Suppl 2, 16. [Google Scholar]
- Zhang, Q.; Jiang, Z.Y.; Luo, J.; Cheng, P.; Ma, Y.B.; Zhang, X.M.; Zhang, F.X.; Zhou, J.; Chen, J.J. Anti-HBV agents. Part 1: Synthesis of alisol A derivatives: A new class of hepatitis B virus inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4647–4650. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, Z.Y.; Luo, J.; Liu, J.F.; Ma, Y.B.; Guo, R.H.; Zhang, X.M.; Zhou, J.; Chen, J.J. Anti-HBV agents. Part 2: Synthesis and in vitro anti-hepatitis B virus activities of alisol A derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 2148–2153. [Google Scholar]
- Zhang, Q.; Jiang, Z.Y.; Luo, J.; Ma, Y.B.; Liu, J.F.; Guo, R.H.; Zhang, X.M.; Zhou, J.; Niu, W.; Du, F.F.; Li, L.; Li, C.; Chen, J.J. Anti-HBV agents. Part 3: Preliminary structure-activity relationships of tetra-acylalisol A derivatives as potent hepatitis B virus inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6659–6665. [Google Scholar]
- Shin, M.S.; Kang, E.H.; Lee, Y.I. A flavonoid from medicinal plants blocks hepatitis B virus-e antigen secretion in HBV-infected hepatocytes. Antiviral Res. 2005, 67, 163–168. [Google Scholar] [CrossRef]
- Dougherty, A.M.; Guo, H.; Westby, G.; Liu, Y.; Simsek, E.; Guo, J.T.; Mehta, A.; Norton, P.; Gu, B.; Block, T.; Cuconati, A. A substituted tetrahydro-tetrazolo-pyrimidine is a specific and novel inhibitor of hepatitis B virus surface antigen secretion. Antimicrob. Agents Chemother. 2007, 51, 4427–4437. [Google Scholar]
- Su, C.R.; Yeh, S.F.; Liu, C.M.; Damu, A.G.; Kuo, T.H.; Chiang, P.C.; Bastow, K.F.; Lee, K.H.; Wu, T.S. Anti-HBV and cytotoxic activities of pyranocoumarin derivatives. Bioorg. Med. Chem. 2009, 17, 6137–6143. [Google Scholar]
- Asif-Ullah, M.; Choi, K.J.; Choi, K.I.; Jeong, Y.J.; Yu, Y.G. Identification of compounds that inhibit the interaction between core and surface protein of hepatitis B virus. Antiviral Res. 2006, 70, 85–90. [Google Scholar] [CrossRef]
- Deres, K.; Schröder, C.H.; Paessens, A.; Goldmann, S.; Hacker, H.J.; Weber, O.; Krämer, T.; Niewöhner, U.; Pleiss, U.; Stoltefuss, J.; Graef, E.; Koletzki, D.; Masantschek, R.N.; Reimann, A.; Jaeger, R.; Gross, R.; Beckermann, B.; Schlemmer, K.H.; Haebich, D.; Rübsamen-Waigmann, H. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 2003, 299, 893–896. [Google Scholar]
- Delaney, W.E., 4th; Edwards, R.; Colledge, D.; Shaw, T.; Furman, P.; Painter, G.; Locarnini, S. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob. Agents Chemother. 2002, 46, 3057–3560. [Google Scholar] [CrossRef]
- King, R.W.; Ladner, S.K.; Miller, T.J.; Zaifert, K.; Perni, R.B.; Conway, S.C.; Otto, M.J. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (-)beta-L-2',3'-dideoxy-3'-thiacytidine. Antimicrob. Agents Chemother. 1998, 42, 3179–31486. [Google Scholar]
- Feld, J.J.; Colledge, D.; Sozzi, V.; Edwards, R.; Littlejohn, M.; Locarnini, S.A. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res. 2007, 76, 168–177. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Y.; Zhai, X.; Feng, X.; Wang, J.; Gong, P. Synthesis and anti-hepatitis B virus evaluation of novel ethyl 6-hydroxyquinoline-3-carboxylates in vitro. Bioorg. Med. Chem. 2008, 16, 6522–6527. [Google Scholar] [CrossRef]
- Li, Y.; Fu, L.; Yeo, H.; Zhu, J.L.; Chou, C.K.; Kou, Y.H.; Yeh, S.F.; Gullen, E.; Austin, D.; Cheng, Y.C. Inhibition of hepatitis B virus gene expression and replication by helioxanthin and its derivative. Antivir. Chem. Chemother. 2005, 16, 193–201. [Google Scholar]
- Ying, C.; Li, Y.; Leung, C.H.; Robek, M.D.; Cheng, Y.C. Unique antiviral mechanism discovered in anti-hepatitis B virus research with a natural product analogue. Proc. Natl. Acad. Sci. USA 2007, 104, 8526–8531. [Google Scholar]
- Li, Y.F.; Wang, G.F.; He, P.L.; Huang, W.G.; Zhu, F.H.; Gao, H.Y.; Tang, W.; Luo, Y.; Feng, C.L.; Shi, L.P.; Ren, Y.D.; Lu, W.; Zuo, J.P. Synthesis and anti-hepatitis B virus activity of novel benzimidazole derivatives. J. Med. Chem. 2006, 49, 4790–4794. [Google Scholar]
- Li, Y.F.; Wang, G.F.; Luo, Y.; Huang, W.G.; Tang, W.; Feng, C.L.; Shi, L.P.; Ren, Y.D.; Zuo, J.P.; Lu, W. Identification of 1-isopropylsulfonyl-2-amine benzimidazoles as a new class of inhibitors of hepatitis B virus. Eur. J. Med. Chem. 2007, 42, 1358–1364. [Google Scholar] [CrossRef]
- Lv, Z.; Sheng, C.; Wang, T.; Zhang, Y.; Liu, J.; Feng, J.; Sun, H.; Zhong, H.; Niu, C.; Li, K. Design, synthesis, and antihepatitis B virus activities of novel 2-pyridone derivatives. J. Med. Chem. 2010, 53, 660–668. [Google Scholar] [CrossRef]
- Zembower, D.E.; Lin, Y.M.; Flavin, M.T.; Chen, F.C.; Korba, B.E. Robustaflavone, a potential non-nucleoside anti-hepatitis B agent. Antiviral Res. 1998, 39, 81–88. [Google Scholar] [CrossRef]
- Dong, W.L.; Liu, Z.X.; Liu, X.H.; Li, Z.M.; Zhao, W.G. Synthesis and antiviral activity of new acrylamide derivatives containing 1,2,3-thiadiazole as inhibitors of hepatitis B virus replication. Eur. J. Med. Chem. 2010, 45, 1919–1926. [Google Scholar] [CrossRef]
- Xu, B.; Huang, Z.; Liu, C.; Cai, Z.; Pan, W.; Cao, P.; Hao, X.; Liang, G. Synthesis and anti-hepatitis B virus activities of Matijing-Su derivatives. Bioorg. Med. Chem. 2009, 17, 3118–3125. [Google Scholar] [CrossRef]
- Korba, B.E.; Montero, A.B.; Farrar, K.; Gaye, K.; Mukerjee, S.; Ayers, M.S. Rossignol, J.F. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 2008, 77, 56–63. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, Y.; Chai, H.; Gong, P. Synthesis and in vitro anti-hepatitis B virus activities of some ethyl 5-hydroxy-1H-indole-3-carboxylates. Bioorg. Med. Chem. 2006, 14, 2552–2558. [Google Scholar] [CrossRef]
- Guo, Q.; Zhao, L.; You, Q.; Yang, Y.; Gu, H.; Song, G.; Lu, N.; Xin, J. Anti-hepatitis B virus activity of wogonin in vitro and in vivo. Antiviral Res. 2007, 74, 16–24. [Google Scholar] [CrossRef]
- Romero, M.R.; Efferth, T.; Serrano, M.A.; Castaño, B.; Macias, R.I.; Briz, O.; Marin, J.J. Effect of artemisinin/artesunate as inhibitors of hepatitis B virus production in an "in vitro" replicative system. Antiviral Res. 2005, 68, 75–83. [Google Scholar]
- Zhang, P.; Zhang, N.; Korba, B.E.; Hosmane, R.S. Synthesis and in vitro anti-hepatitis B and C virus activities of ring-expanded ('fat') nucleobase analogues containing the imidazo[4,5-e][1,3]diazepine-4,8-dione ring system. Bioorg. Med. Chem. Lett. 2005, 15, 5397–5401. [Google Scholar] [CrossRef]
- Lee, J.; Shim, H.; Park, Y.; Park, S.; Shin, J.; Yang, W.; Lee, H.; Park, W.; Chung, Y.; Lee, S. 2,5-Pyridinedicarboxylic acid derivatives as non-nucleosidic reverse transcriptase inhibitors of hepatitis B virus. Bioorg. Med. Chem. Lett. 2002, 12, 2715–2717. [Google Scholar] [CrossRef]
- Zhu, Y.L.; Pai, S.B.; Liu, S.H.; Grove, K.L.; Jones, B.C.; Simons, C.; Zemlicka, J.; Cheng, Y.C. Inhibition of replication of hepatitis B virus by cytallene in vitro. Antimicrob. Agents Chemother. 1997, 41, 1755–1760. [Google Scholar]
- Kim, K.H.; Kim, N.D.; Seong, B.L. Pharmacophore-based virtual screening: A review of recent applications. Expert Opin. Drug Discov. 2010, 5, 205–222. [Google Scholar] [CrossRef]
- Musmuca, I.; Caroli, A.; Mai, A.; Kaushik-Basu, N.; Arora, P.; Ragno, R. Combining 3-D quantitative structure-activity relationship with ligand based and structure based alignment procedures for in silico screening of new hepatitis C virus NS5B polymerase inhibitors. J. Chem. Inf. Model. 2010, 50, 662–676. [Google Scholar] [CrossRef]
- Ryu, K.; Kim, N.D.; Choi, S.I.; Han, C.K.; Yoon, J.H.; No, K.T.; Kim, K.H.; Seong, B.L. Identification of novel inhibitors of HCV RNA-dependent RNA polymerase by pharmacophore-based virtual screening and in vitro evaluation. Bioorg. Med. Chem. 2009, 17, 2975–2982. [Google Scholar]
- Kim, J.; Kim, K.S.; Kim, D.E.; Chong, Y. Identification of novel HCV RNA-dependent RNA polymerase inhibitors using pharmacophore-guided virtual screening. Chem. Biol. Drug. Des. 2008, 72, 585–591. [Google Scholar] [CrossRef]
- Bustanji, Y.; Al-Masri, I.M.; Qasem, A.; Al-Bakri, A.G.; Taha, M.O. In silico screening for non-nucleoside HIV-1 reverse transcriptase inhibitors using physicochemical filters and high-throughput docking followed by in vitro evaluation. Chem. Biol. Drug. Des. 2009, 74, 258–265. [Google Scholar] [CrossRef]
- De Luca, L.; Barreca, M.L.; Ferro, S.; Christ, F.; Iraci, N.; Gitto, R.; Monforte, A.M.; Debyser, Z.; Chimirri, A. Pharmacophore-based discovery of small-molecule inhibitors of protein-protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. ChemMedChem 2009, 4, 1311–1316. [Google Scholar] [CrossRef]
- Carrieri, A.; Pérez-Nueno, V.I.; Fano, A.; Pistone, C.; Ritchie, D.W.; Teixidó, J. Biological profiling of anti-HIV agents and insight into CCR5 antagonist binding using in silico techniques. ChemMedChem 2009, 4, 1153–1163. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C. A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J. Comput. Aided. Mol. Des. 1995, 9, 532–549. [Google Scholar] [CrossRef]
- Walsh, A.W.; Langley, D.R.; Colonno, R.J.; Tenney, D.J. Mechanistic characterization and molecular modeling of hepatitis B virus polymerase resistance to entecavir. PLoS One. 2010, 5, e9195. [Google Scholar]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Y.; Mu, S.; Zhang, J.; Yan, Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J. Med. Virol. 2009, 81, 1177–1183. [Google Scholar] [CrossRef]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar]
- Petersen, J.; Dandri, M.; Mier, W.; Lütgehetmann, M.; Volz, T.; von Weizsäcker, F.; Haberkorn, U.; Fischer, L.; Pollok, J.M.; Erbes, B.; Seitz, S.; Urban, S. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 2008, 26, 335–341. [Google Scholar]
- McCaffrey, A.P.; Nakai, H.; Pandey, K.; Huang, Z.; Salazar, F.H.; Xu, H.; Wieland, S.F.; Marion, P.L.; Kay, M.A. Inhibition of hepatitis B virus in mice by RNA interference. Nat. Biotechnol. 2003, 21, 639–644. [Google Scholar] [CrossRef]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; Vaish, N.; Zinnen, S.; Vargeese, C.; Bowman, K.; Shaffer, C.S.; Jeffs, L.B.; Judge, A.; MacLachlan, I.; Polisky, B. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar]
- Wu, H.L.; Huang, L.R.; Huang, C.C.; Lai, H.L.; Liu, C.J.; Huang, Y.T.; Hsu, Y.W.; Lu, C.Y.; Chen, D.S.; Chen, P.J. RNA interference-mediated control of hepatitis B virus and emergence of resistant mutant. Gastroenterology 2005, 128, 708–716. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Wang, L.H.; Schneider, R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kennedy, W.D.; Yin, Y.W. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 2009, 139, 312–324, Erratum in: Cell 2009, 139, 828. [Google Scholar] [CrossRef]
- Falkenberg, M.; Larsson, N.G. Structure casts light on mtDNA replication. Cell 2009, 139, 231–233. [Google Scholar] [CrossRef]
- Sample Availability: Not Available.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, K.-H.; Kim, N.D.; Seong, B.-L. Discovery and Development of Anti-HBV Agents and Their Resistance. Molecules 2010, 15, 5878-5908. https://doi.org/10.3390/molecules15095878
Kim K-H, Kim ND, Seong B-L. Discovery and Development of Anti-HBV Agents and Their Resistance. Molecules. 2010; 15(9):5878-5908. https://doi.org/10.3390/molecules15095878
Chicago/Turabian StyleKim, Kyun-Hwan, Nam Doo Kim, and Baik-Lin Seong. 2010. "Discovery and Development of Anti-HBV Agents and Their Resistance" Molecules 15, no. 9: 5878-5908. https://doi.org/10.3390/molecules15095878
APA StyleKim, K.-H., Kim, N. D., & Seong, B.-L. (2010). Discovery and Development of Anti-HBV Agents and Their Resistance. Molecules, 15(9), 5878-5908. https://doi.org/10.3390/molecules15095878