New Catechol Derivatives of Safrole and Their Antiproliferative Activity towards Breast Cancer Cells

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
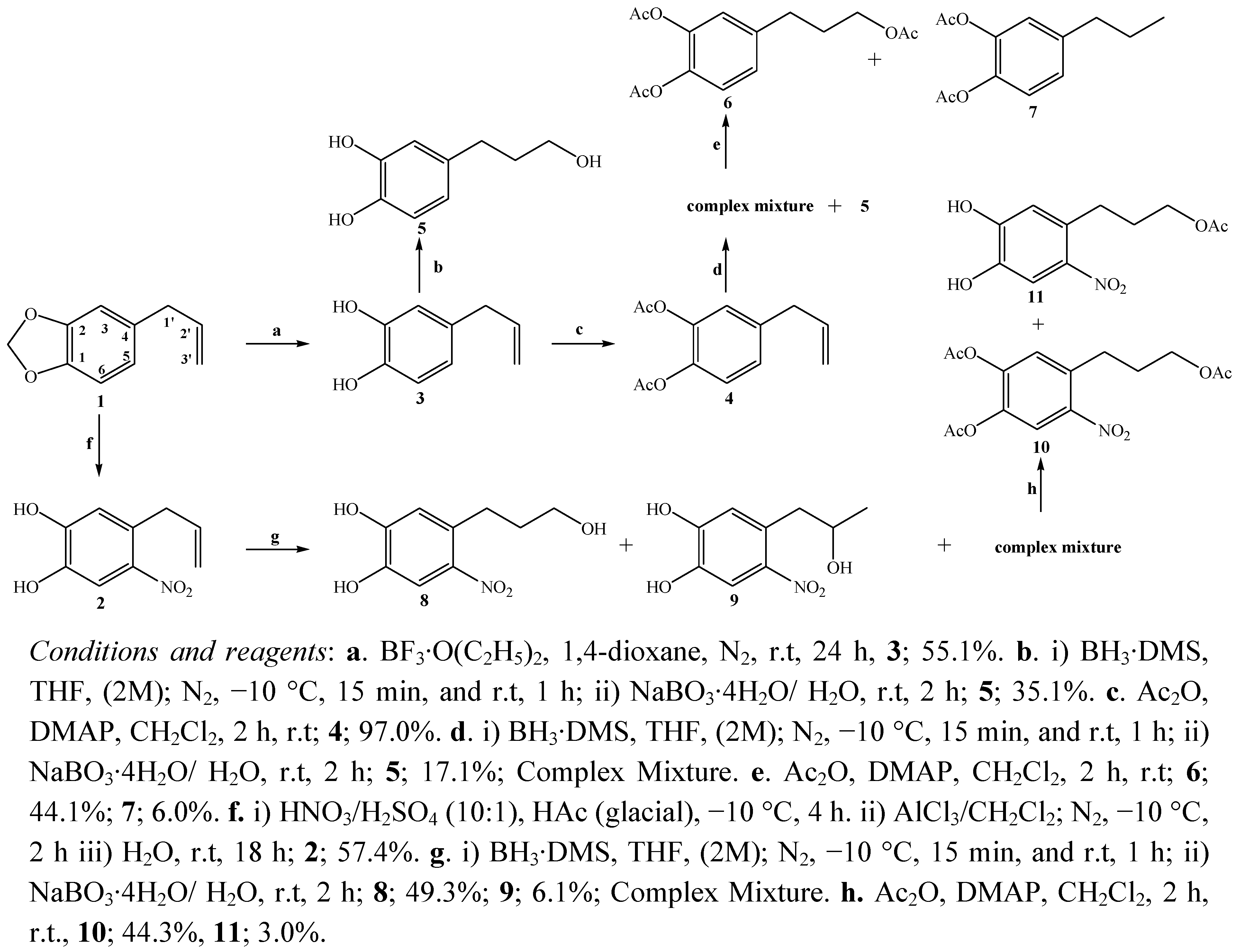
2.1. Chemistry

2.2. Biological Results

{kind=link}
| Compound | IC 50 (μM) | ||
|---|---|---|---|
| MCF-7 | MDA-MB-231 | DHF | |
| 2 | 55.0 ± 7.11 | 37.5 ± 2.65 | >100 |
| 3 | 98.4 ± 11.6 | 40.2 ± 6.9 | >100 |
| 4 | >100 | 54.7 ± 6.6 | >100 |
| 5 | >100 | 54.3 ± 4.1 | >100 |
| 6 | 97.8 ± 12.4 | 5.9 ± 0.8 | >100 |
| 7 | >100 | 41.7 ± 7.4 | >100 |
| 8 | 78.2 ± 8.9 | 57.3 ± 4.1 | >100 |
| 9 | >100 | >100 | >100 |
| 10 | 51.3 ± 2.1 | 33.8 ± 4.9 | >100 |
| 11 | 63.8 ± 5.5 | 27.1 ± 4.4 | >100 |
| safrole | >100 | >100 | >100 |
| Daunorubicin | 0.19 ± 0.01 | 0.38 ± 0.06 | 14.3 ± 1.85 |
3. Experimental
3.1. General
3.2. Cell Lines
3.3. In vitro Growth Inhibition Assay
4. Conclusions
Acknowledgements
References and Notes
- Miller, E.C.; Swanson, A.B.; Phillips, D.H.; Fletcher, T.L.; Liem, A.; Miller, J.A. Structure-activity studies of the carcinogenicities in the mouse and rat of some naturally occurring and synthetic alkenylbenzene derivatives related to safrole and estragole. Cancer Res. 1983, 43, 1124–1134. [Google Scholar]
- Liu, T.Y.; Chen, C.C.; Chen, C.L.; Chi, C.L. Safrole-induced Oxidative Damage in the Liver of Sprague-Dawley Rats. Food Chem. Toxicol. 1999, 37, 697–702. [Google Scholar] [CrossRef]
- Bridges, J.W.; Fennell, T.R. The relationship between the metabolism and toxicity of benzodioxole compounds. Adu. Exp. Med. Biol. 1981, 136, 881–893. [Google Scholar]
- Bolton, J.L.; Acay, N.M.; Vukomanovic, V. Evidence That 4-Allyl-o-quinones Spontaneously Rearrange to Their More Electrophilic Quinone Methides: Potential Bioactivation Mechanism for the Hepatocarcinogen Safrole. Chem. Res. Toricol. 1994, 7, 443–450. [Google Scholar] [CrossRef]
- Miao, J.Y.; Zhao, B.X.; Li, H.H.; Zhang, S.L.; Du, C.Q. Effect of safrole oxide on vascular endothelial cell growth and apoptosis induced by deprivation of fibroblast growth factor. Acta Pharmacol. Sin. 2002, 23, 323–326. [Google Scholar]
- Zhao, J.; Zhao, B.; Miao, J.; Zhang, S.; Yin, D. Safrole Oxide Inhibits Angiogenesis by Inducing Apoptosis. Vasc. Pharmacol. 2005, 43, 69–74. [Google Scholar] [CrossRef]
- Zhao, J.; Miao, J.; Zhao, B.; Zhang, S. Upregulating of Fas, integrin β4 and P53 and depressing of PC-PLC activity and ROS level in VEC apoptosis by safrole oxide. FEBS Lett. 2005, 25, 5809–5813. [Google Scholar]
- Espinoza, L.; Madrid, A.; Taborga, L.; Villena, J.; Cuellar, M.; Carrasco, H. Synthesis of Nine Safrole Derivatives and their Antiproliferative Activity Towards Human Cancer Cells. J. Chil. Chem. Soc. 2010, 55, 219–222. [Google Scholar]
- Madrid, A. Synthesis of Compounds Similar Structures of ent-Labdane from Diterpenhydroquinones and Safrole, with possible Biological Activity. PhD Thesis, Joint Doctoral Program Universidad Técnica Federico Santa María- Universidad de Valparaíso, Valparaíso, Chile, 2009. [Google Scholar]
- Rosa, F.; Rebelo, R.; Nascimento, M. Synthesis of new indolecarboxylic acids related to the plant hormone indoleacetic acid IAA. J. Braz. Chem. Soc. 2003, 14, 11–15. [Google Scholar]
- Khadem, S.; Joseph, R.; Rategar, M.; Leek, D.M.; Outdatchin, K.A.; Arya, P. A Solution- and Solid-Phase approach to Tetrahydroquinoline-derived Polycyclics having a 10-Membered Ring. J. Comb. Chem. 2004, 6, 724–734. [Google Scholar] [CrossRef] [Green Version]
- Leite, A.; Peixoto da Silva, K.; de Souza, I.A.; Magali de Araujo, J.; Brondani, D.J. Synthesis, antitumour and antimicrobial activities of new peptidyl derivatives containing the 1,3-benzodioxole system. Eur. J. Med. Chem. 2004, 39, 1059–1065. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Shoup, T.M.; Goudgaon, N.M. Sodium perborate: A mild and convenient reagent for efficiently oxidizing organoboranes. J. Org. Chem. 1989, 54, 5930–5933. [Google Scholar] [CrossRef]
- Avery, M.A.; Verlander, M.S.; Goodman, M. Synthesis of 6-aminoisoproterenol. J. Org. Chem. 1980, 45, 2750–2753. [Google Scholar] [CrossRef]
- Amorim, M.; Da Silva, A.J.; Costa, P.R. The reaction of safrole derivatives with aluminum chloride: Improved procedures for the preparation of catechols or their mono-O-Methylated Derivatives and a mechanistic interpretation. J. Braz. Chem. Soc. 2001, 12, 346–353. [Google Scholar] [CrossRef]
- Costa, P.R.R. Safrol E Eugenol: Estudo Da Reatividade Química E Uso Em Síntese De Produtos Naturais Biologicamente Ativos E Seus Derivados. Quím. Nova 2000, 23, 357–369. [Google Scholar] [CrossRef]
- Cox, M.; Klass, G. Synthesis by-products from the Wacker oxidation of safrole in methanol using p-benzoquinone and palladium chloride. Forensic Sci. Int. 2006, 164, 138–147. [Google Scholar] [CrossRef]
- Araújo, J.X., Jr.; Barreiro, E.J.; Parente, J.P.; Fraga, C. Synthesis of Piperamides from Natural Safrole. Synth. Commun. 1999, 29, 2263–2273. [Google Scholar]
- Barreiro, J.; Fraga, C. A utilização do safrol, principal componente químico do óleo de sassafráz, na síntese de substâncias bioativas na cascata do ácido araquidônico: Antiinflamatórios, analgésicos e anti-trombóticos. Quím. Nova 1999, 22, 744–759. [Google Scholar] [CrossRef]
- Hung, S.L.; Chen, Y.L. Effects of Safrole on the Defensive Functions of Human Neutrophils. J. Period. Res. 2003, 38, 130–134. [Google Scholar] [CrossRef]
- Uhl, M.; Helma, C.; Knasmuller, S. Evaluation of the single cell gel electrophoresis assay with human hepatoma (Hep G2) cells. Mutat. Res. 2000, 468, 213–225. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screenin. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Chen, W.; Cheng, H.; Lu, Y.H.; Chen, I.; Liu, S.; Hsu, S.; Chang, H.; Huang, H.; Chen, J.; Jan, C. The Carcinogen Safrole Increases Intracellular Free Ca2+ Levels and Causes Death in MDCK Cells. Chin. J. Physiol. 2007, 50, 34–40. [Google Scholar]
- Sample Availability: Samples of the compounds 1, 2, 3 and 4 are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Madrid Villegas, A.; Espinoza Catalán, L.; Montenegro Venegas, I.; Villena García, J.; Carrasco Altamirano, H. New Catechol Derivatives of Safrole and Their Antiproliferative Activity towards Breast Cancer Cells. Molecules 2011, 16, 4632-4641. https://doi.org/10.3390/molecules16064632
Madrid Villegas A, Espinoza Catalán L, Montenegro Venegas I, Villena García J, Carrasco Altamirano H. New Catechol Derivatives of Safrole and Their Antiproliferative Activity towards Breast Cancer Cells. Molecules. 2011; 16(6):4632-4641. https://doi.org/10.3390/molecules16064632
Chicago/Turabian StyleMadrid Villegas, Alejandro, Luis Espinoza Catalán, Iván Montenegro Venegas, Joan Villena García, and Héctor Carrasco Altamirano. 2011. "New Catechol Derivatives of Safrole and Their Antiproliferative Activity towards Breast Cancer Cells" Molecules 16, no. 6: 4632-4641. https://doi.org/10.3390/molecules16064632