1. Introduction
Diabetes mellitus (DM), considered a lifestyle related diseases, is a metabolic disease with hyper-glycemia as a symptom and causes many complications [
1]. Recently, DM is becoming a serious problem around the World, and according to World Health Organization, it affects approximately 171 million people worldwide and the number is expected to reach to 366 million over the next 20 years [
2]. Many researchers have enthusiastically studied the development of antidiabetic agents, however, many potential therapeutics have a number of serious adverse effects [
3,
4], therefore there is a growing trend toward using natural products as treatment [
5]. China has a long history of using herbs for the treatment of human diseases and several medicinal plants are used for the treatment of diabetes.
S. tenuifolia is one such plant.
S. tenuifolia (Rosaceae) is a perennial herb, which is widely distributed in China’s Heilongjiang, Liaoning, and Jilin provinces and Inner Mongolia. The residents in Northeast China regard
S. tenuifolia as a substitute for
S. officinalis, and apply its roots for the treatment of diarrhea, chronic intestinal infections, duodenal ulcers, diabetes mellitus and bleeding [
6,
7]. Our studies indicated that ethyl acetate fraction of
a S. tenuifolia root ethanol extract contains plenty of triterpenes, which can inhibit plasma glucose levels in alloxan-induced diabetic rats.
α-Glucosidase inhibitors are oral anti-diabetic drugs used for diabetes mellitus type 2. They can significantly delay the absorption of carbohydrates from the small intestine and thus have a lowering effect on postprandial blood glucose and insulin levels [
8]. Based on a bioassay-guided isolation, a phytochemical study of
S. tenuifolia was performed and two new triterpenoids were isolated from its ethyl acetate fraction, along with thirteen other known triterpenoids. The new compounds were identified as 2-oxo-3
β,19
α-dihydroxy-olean-12-en-28-oic acid
β-
D-glucopyranosyl ester (
1) and 2
α,19
α-dihydroxy-3-oxo-12-ursen-28-oic acid
β-
D-glucopyranosyl ester (
4), respectively. In the present report, we describe the structural elucidation of
1 and
4, together with the
α-glucosidase inhibitory activity data of all the compounds
1-
15 (
Figure 1).
Figure 1.
Structures of 1-15.
Figure 1.
Structures of 1-15.
2. Results and Discussion
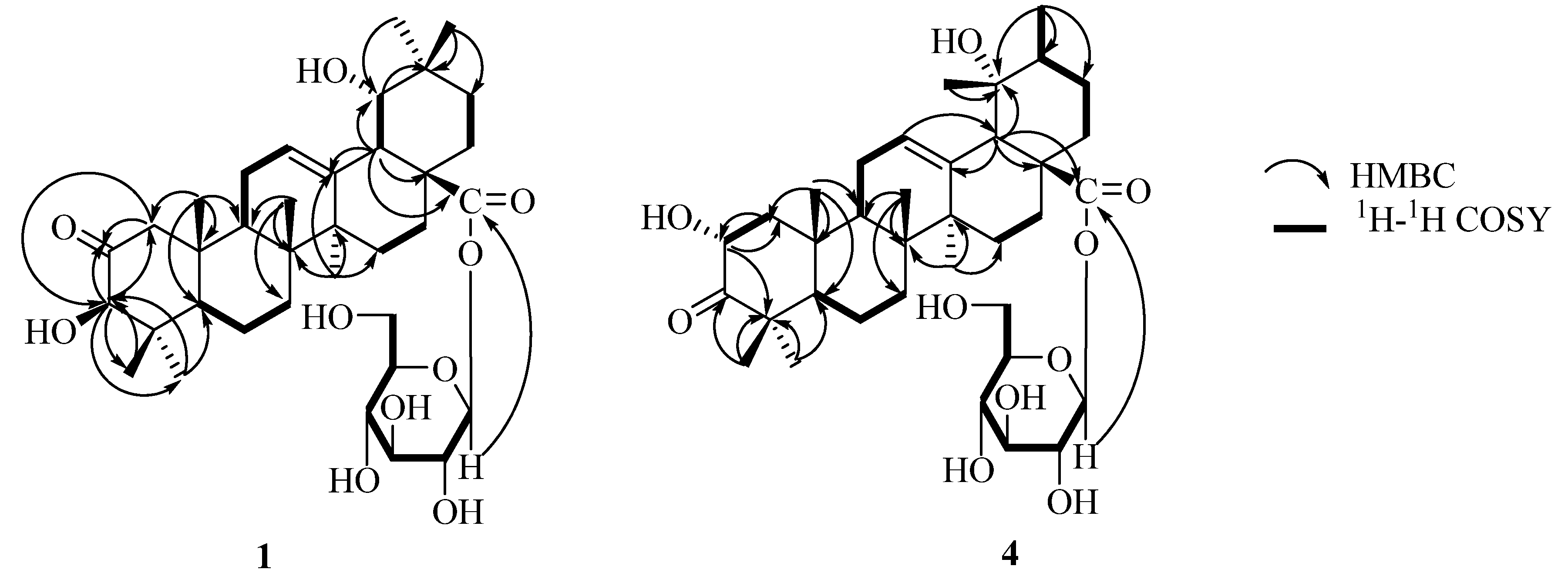
Compound 1 was obtained as a white amorphous powder. The HR-ESI-MS data indicated a molecular formula of C36H56O10, based on the [M + H]+ ion signal at m/z 649.3953 (calc. C36H57O10, 649.3952), [M + NH4]+ ion signal at m/z 666.4221 (calc. C36H60O10N, 666.4217) and [M + Na]+ ion signal at m/z 671.3779 (calc. C36H56O10Na, 671.3771). The IR spectrum showed the presence of hydroxyl groups (3,419.6 cm−1), ester carbonyl (1,735.3 cm−1), carbonyl (1,711.3 cm−1) and double bond (1,640.8 cm−1).
The
13C-NMR spectrum and DEPT of
1 showed seven methyl, nine methylene, eleven methine, and nine quaternary carbon signals, including one ester carbonyl at
δC 177.3, a quaternary olefinic carbonyl at
δC 144.5, one anomeric carbon signal at
δC 95.9, a ketone carbonyl at
δC 213.4. The
1H-NMR spectrum exhibited seven singlet methyl signals at
δH 1.21, 0.92, 1.01, 1.15, 1.52, 1.12 and 0.95, an anomeric proton signal at
δH 6.37 (d,
J = 8.0 Hz), two methine proton signals at
δH 3.89 (s) and 3.54 (d,
J = 2.8 Hz), and an olefinic proton signal at
δH 5.45 (br s), which were characteristic of the oleanolic acid skeleton. Comparison of the data
1 with those of oleanolic acid [
9,
10], suggested that the aglycone of
1 was an oleanolic acid derivative with one hydroxyl group at the ring E portion, as well as one ketone carbonyl group. The proton signal at
δH 3.50 showed long-rang correlations with C-13, C-17, and C-28 in the HMBC spectrum, and was assigned to the H-18 (
Figure 2). This proton had a proton spin-coupling correlation with the signal at
δH 3.54, which was associated with the carbon signal at
δC 81.0 (CH) in the HSQC spectrum. Thus, the presence of a hydroxyl group at C-19 was evident. The
3JH,H value of 2.8 Hz between H-18 and H-19, and NOE correlations from H-19 to Me-29 and Me-30 gave evidence for the C-19
α hydroxy orientation [
11]. There were long-range correlations between protons and carbons: H-3 (
δH 3.89)/C-23 (
δC 27.6), C-24 (
δC 21.7), and ketone carbonyl (
δC 213.4); H-23 (
δH 1.21), H-24 (
δH 0.92)/C-3 (
δC 83.2) in the HMBC spectrum, which indicated that the ketone carbonyl group must be either at position C-1 or C-2. Furthermore, the long-range correlations were observed between protons and carbons: H-3 (
δH 3.89), H-25 (
δH 1.01)/C-1 (
δC 51.5), H-1 (
δH 3.00, 2.27)/ketone carbonyl (
δC 213.4) in the HMBC spectrum (
Figure 2). The ketone must be at the C-2 position based on comparison of the NMR spectral data for C-1, C-2 and C-3 of
1 with that of the similar compound 2-oxopomolic acid (
2) [C-1 (
δC 53.6
δH 2.46, 2.15), C-2 (
δC 211.2) and C-3 (
δC 83.5;
δH 4.17 s)] and 2
α,19
α-dihydroxy-3-oxo-olean-12-en-28-oic acid
β-
D-glucopyranosyl ester (
6) [C-2 (
δC 216.5) and C-3 (
δC 69.7;
δH 4.80 s)] [
12,
13]. Based on these findings, the structure of the aglycone part of
1 was established to be 2-oxo-3
β,19
α-dihydroxyolean-12-en-28-oic acid, a new triterpene. The configuration of the sugar unit was assigned after hydrolysis of
1 with 1 M HCl. The acid hydrolysis gave D-glucose. The data of anomeric carbon signal at
δC 95.9 and anomeric proton signal at
δH 6.37 (d,
J = 8.0 Hz) indicated the glucose was in the
β form and was bound to the aglycone by a glycosidic linkage at C-28 in the HMBC spectrum (
Figure 2). Therefore, the structure of compound
1 was elucidated as 2-oxo-3
β,19
α-dihydroxy-olean-12-en-28- oic acid
β-
D-glucopyranosyl ester.
Compound 4 was obtained as a white amorphous powder. The HR-ESI-MS data indicated a molecular formula of C36H56O10based on the [M + H]+ ion signal at m/z 649.3945 (calc. 649.3952) in the. The IR spectrum showed the presence of hydroxyl groups (3431.1 cm−1), ester carbonyl (1,737.1 cm−1), carbonyl (1,714.3cm−1), and double bond (1,644.6 cm−1).
Figure 2.
Key HMBC and 1H-1H COSY correlations of 1 and 4.
Figure 2.
Key HMBC and 1H-1H COSY correlations of 1 and 4.
The
13C-NMR spectrum shows seven methyl, nine methylene, eleven methine, and nine quaternary carbon signals, including one ester carbonyl at
δC 177.0, a quaternary olefinic carbonyl at
δC 139.5, one anomeric carbon signal at
δC 95.9, a ketone carbonyl at
δC 216.6. Its
1H-NMR spectrum shows the presence of a hydroxymethine proton at
δH 4.82 (1H, dd,
J = 12.5, 6.3 Hz), one trisubstituted olefinic proton at (
δH 5.50, br s), six singlets at
δH 1.19, 0.99, 1.18, 1.15, 1.59, 1.37 for six tertiary methyl groups, one secondary methyl group (
δH 1.05, d,
J = 6.6Hz), one methine proton characteristic of H-18 of pomolic acid (
δH 2.91, s), and one anomeric proton (
δH 6.30 d,
J = 8.0 Hz). The secondary methyl signal on ring E provides a most useful indicator for the presence of an urs-12-ene skeleton [
10]. Additionally, the signals in its
13C-NMR at
δC 128.0 and 139.5 are characteristic for a C-12/C-13 double bond in the ursene-type structure [
14]. Acid hydrolysis of
4 with 1 M HCl (5 mL) gave a D-glucose molecule and a triterpene (C
30H
46O
5,
5). The latter was identified as 2
α,19
α-dihydroxy-3-oxo-12-ursen-28-oic acid by comparing its spectral and physical data with literature values [
15]. When the
1H- and
13C-NMR spectra of
4 were compared with those of
5, an upfield shift due to the glycoside was detected at the C-28 signal at
δC 177.0. The linked site of glycosyl group in
4 was further established from correlations between the anomeric proton H-1' at
δH 6.30 and C-28 at
δC 177.0 in the HMBC spectrum (
Figure 2). Therefore, the structure of
4 was determined as 2
α,19
α-dihydroxy-3-oxo-12-ursen-28-oic acid
β-
D-glucopyranosyl ester.
Table 1.
NMR data of 1 and 4 in pyridine-d5 (δ in ppm, J in Hz, recorded at 400 MHz and 100 MHz, respectively).
Table 1.
NMR data of 1 and 4 in pyridine-d5 (δ in ppm, J in Hz, recorded at 400 MHz and 100 MHz, respectively).
| No. | 1 | | 4 | |
|---|
| | δC (DEPT) | δH (J, Hz) | δC (DEPT) | δH (J, Hz) |
|---|
| 1 | 51.5 (CH2) | 3.00 d (12.4 ), 2.27 d (12.4) | 50.3 (CH2) | 2.48 dd (12.5, 6.3), 1.37 m |
| 2 | 213.4 (C) | | 69.8 (CH) | 4.82 dd (12.5, 6.3) |
| 3 | 83.2 (CH) | 3.89 s | 216.6 (C) | |
| 4 | 42.2 (C) | | 48.2 (C) | |
| 5 | 50.2 (CH) | 2.02 m | 57.7 (CH) | 1.23 m |
| 6 | 19.3 (CH2) | 1.46 m, 1.31 m | 19.6 (CH2) | 1.34 m, 1.29 m |
| 7 | 33.2 (CH2) | 1.43 m, 1.70 m | 33.2 (CH2) | 1.43 m, 1.33 m |
| 8 | 40.3 (C) | | 40.6 (C) | |
| 9 | 48.1 (CH) | 1.83 m | 47.4 (CH) | 1.83 m |
| 10 | 42.8 (C) | | 37.8 (C) | |
| 11 | 24.3 (CH2) | 2.02 m | 24.2 (CH2) | 2.08 m |
| 12 | 123.0 (CH) | 5.45 br s | 128.0 (CH) | 5.50 br s |
| 13 | 144.5 (C) | | 139.5 (C) | |
| 14 | 42.3 (C) | | 42.2 (C) | |
| 15 | 29.0 (CH2) | 2.46 m, 1.20 m | 29.2 (CH2) | 2.49 m, 1.22 m |
| 16 | 27.9 (CH2) | 2.81 m, 2.12 m | 26.1 (CH2) | 3.09 m, 2.05 m |
| 17 | 46.5 (C) | | 48.6 (C) | |
| 18 | 44.6 (CH) | 3.50 d (2.8) | 54.4 (CH) | 2.91 s |
| 19 | 81.0 (CH) | 3.54 d (2.8) | 72.7 (CH) | |
| 20 | 35.6 (CH) | | 42.2 (CH) | 1.39 m |
| 21 | 29.0 (CH2) | 1.24 m, 2.35 m | 26.7 (CH2) | 1.24 m, 2.02 m |
| 22 | 33.0 (CH2) | 2.04 m, 1.93 m | 37.9 (CH2) | 2.03 m, 1.83 m |
| 23 | 27.6 (CH3) | 1.21 s | 25.4 (CH3) | 1.19 s |
| 24 | 21.7 (CH3) | 0.92 s | 21.8 (CH3) | 0.99 s |
| 25 | 16.9 (CH3) | 1.01 s | 17.6 (CH3) | 1.18 s |
| 26 | 17.1 (CH3) | 1.15 s | 16.1 (CH3) | 1.15 s |
| 27 | 24.9 (CH3) | 1.52 s | 24.6 (CH3) | 1.59 s |
| 28 | 177.3 (C) | | 177.0 (C) | |
| 29 | 28.7 (CH3) | 1.12 s | 27.0 (CH3) | 1.37 s |
| 30 | 24.4 (CH3) | 0.95 s | 16.8 (CH3) | 1.05 d (6.6) |
| 1' | 95.9 (CH) | 6.37 d (8.0) | 95.9 (CH) | 6.30 d (8.0) |
| 2' | 74.1 (CH) | 4.22 t (8.4) | 74.1 (CH) | 4.24 t (8.4) |
| 3' | 79.3 (CH) | 4.29 t (8.8) | 79.4 (CH) | 4.34 t (8.7) |
| 4' | 71.2 (CH) | 4.38 t (9.0) | 71.2 (CH) | 4.39 t (9.3) |
| 5' | 79.0 (CH) | 4.04 m | 79.0 (CH) | 4.05 (m) |
| 6' | 62.2 (CH2) | 4.42 br d (12.1), 4.46 dd (12.1, 3.8) | 62.4 (CH2) | 4.42 br d (11.7), 4.49 dd (11.7, 4.4) |
The other compounds were characterized as 2-oxopomolic acid (
2) [
12], 2-oxopomolic acid
β-
D-glucopyranoside (
3) [
12], 2
α,19
α-dihydroxy-3-oxo-12-ursen-28-oic acid (
5) [
15], 2
α,19
α-dihydroxy-3-oxo-olean-12-en-28-oic acid
β-
D-glucopyranosyl ester (
6) [
13], rosamutin (
7) [
16], euscaphic acid (
8) [
17], kaji-ichigoside Fl (
9) [
16], 24-deoxysericoside (
10) [
17], ursolic acid (
11) [
10],
p-coumaroylursolic acid (
12) [
18], ziyu-glycoside I (
13) [
17], 3
β-[(
α-L-arabinopyranosyl) oxy] urs-12,19(29)-dien-28-oic acid 28-
β-
D-glucopyranosyl ester (
14) [
11] and pomolic acid (
15) [
17] by comparing their NMR spectroscopic data with the literature values. All these known compounds are reported for the first time in
S. tenuifolia.
We next evaluated the isolated compounds for their inhibitory activity against
α-glucosidase since some compounds are known
α-glucosidase inhibitors [
19]. The results are shown in
Table 2, with acarbose used as a positive control. Compounds
1-
15 exhibited dose-dependent
α-glucosidase inhibitory activities with IC
50 values of 0.62-3.62 mM. Compounds
8 and
12 showed the most potent activity (IC
50 0.67 and 0.62 mM, respectively), comparable with the positive control. Triterpenoids of the EtOAc-soluble fraction may be the potential anti-hypoglycemic agents in this plant, as they have been shown to induce an anti-diabetic effect.
Table 2.
In vito α-glucosidase inhibitory assay.
Table 2.
In vito α-glucosidase inhibitory assay.
| Compound | IC50 (mM ± SEM, mM) |
|---|
| 1 | 1.88 ± 0.28 |
| 2 | 1.35 ± 0.04 |
| 3 | 2.22 ± 0.06 |
| 4 | 1.56 ± 0.04 |
| 5 | 1.23 ± 0.09 |
| 6 | 2.01 ± 0.06 |
| 7 | 3.28 ± 0.08 |
| 8 | 0.67 ± 0.09 |
| 9 | 3.10 ± 0.24 |
| 10 | 3.52 ± 0.16 |
| 11 | 1.69 ± 0.04 |
| 12 | 0.62 ± 0.06 |
| 13 | 3.62 ± 0.21 |
| 14 | 2.87 ± 0.06 |
| 15 | 1.84 ± 0.12 |
| Acarbose | 0.79 ± 0.13 |
3. Experimental Section
3.1. General
Open column chromatography (CC) was carried out using silica gel (200-300 mesh, Qingdao Marine Chemical Co., Qingdao, China) or octadecyl silica gel (ODS, 25-40 μm, Fuji) as stationary phases. TLC employed precoated silica gel plates (5-7 μm, Qingdao Marine). Preparative HPLC was carried out on a Waters 600 instument equipped with a Waters UV-2487 detector. A Waters Sunfire prep C18 OBD (19 × 250 mm i.d.) column was used for this purpose. The IR spectra were recorded as KBr pellets on a Jasco 302-A spectrometer. Optical rotation was recorded on a Jasco P-2000 polarimeter. HRESIMS were measured on a FTMS-7 instrument (Bruker Daltonics). Melting points were determined on a Gallenkemp apparatus and are uncorrected. The 1H-, 13C- and 2D (1H-1H COSY, HSQC, HMBC, NOESY) NMR spectra were recorded on a Bruker AMX-400 spectrometer using standard pulse sequences. Chemical shifts are reported in ppm (δ), and scalar coupling are reported in Hz. GC analyses were carried out using a Fuli 9790 instrument equipped with a DM-5 column (0.25 μm, 30 m × 0.25 mm, Dikma, China). α-Glucosidase (EC.3.2.1.20) from Saccharomyces sp. was purchased from Wako Pure Chemical Indutries Ltd. (Wako 076-02841). Other reagents were purchased from various commercial sources.
3.2. Plant Material
The roots of S. tenuifolia were collected in October 2008 from Fangzheng of Heilongjiang Province, China, and identified by Zhenyue Wang, of Heilongjiang University of Chinese Medicine. A voucher specimen (20081023) was deposited at the herbarium of Heilongjiang University of Chinese Medicine, Harbin, China.
3.3. Extraction and Isolation
The dried roots of S. tenuifolia (5.0 kg) were extracted with 70% EtOH (3 × 10 L) to afford the EtOH extract (1.3 kg) which was then suspended in water (10 L) and then extracted with petroleum ether and ethyl acetate (EtOAc) (3 × 10 L each), yielding petroleum ether (10.2 g) and ethyl acetate (222.5 g) extracts. The EtOAc fraction (222.5 g) was subjected to silica gel column with a stepwise CH2Cl2-MeOH gradient (30:1; 20:1; 10:1; 5:1, v/v), and finally with MeOH alone, to give five fractions I-V. Fraction I (40.8 g) was separated using silica gel CC eluting with CH2Cl2-MeOH (50:1, 30:1, 10:1, v/v) to obtain three sub-fractions, I1-I3. Sub-fraction I2 (10.6 g) was further separated by ODS silica gel CC with MeOH-H2O (9:1, v/v) and to 11 (33.2 mg), 12 (37.5 mg) and 15 (25.5 mg); Fraction II (38.3 g) was subjected to silica gel CC eluting with CH2Cl2-MeOH (30:1, 20:1, 10:1, v/v) to afford four sub-fractions, II1-II4. Sub-fraction II1 (13.3 g) afforded compounds 2 (21.0 mg), 5 (44.5 mg) and 8 (24.6 mg, tR = 50.5 min), after subjecting it to ODS silica gel CC eluting with MeOH-H2O (3:1, 3:2, v/v), followed by preparative HPLC with MeOH-H2O (4:1, v/v).Fraction III (31.3 g) was subjected to silica gel CC eluting with CH2Cl2-MeOH (8:1, 5:1, 1:1, v/v) to afford four sub-fractions, III1-III4. Sub-fraction III2 (7.3 g) afforded 4 (43.5 mg) and a mixture of 1, 3 and 6 by ODS silica gel CC using MeOH-H2O (2:1, v/v) as eluent. The mixture was separated into 1 (25.2 mg, tR = 45.3 min), 3 (23.4 mg tR = 43.2 min) and 6 (12.5 mg, tR = 48.5 min) by preparative HPLC using MeOH-H2O (3:2, v/v). Fraction IV (43.1 g) was applied to a silica gel column which was eluted with CH2Cl2-MeOH-H2O (8:1:0.1, 6:1:0.1, 3:1:0.1, v/v) to afford four sub-fractions, IV1-IV4. Sub-fraction IV2 (16.3 g) afforded a mixture of 7, 9 and 10, along with a few impurities, after ODS silica gel CC with MeOH-H2O (2:1, v/v). The mixture was separated by preparative HPLC using MeOH-H2O (3:2, v/v) into 7 (27.5 mg, tR = 49.2 min), 9 (25.2 mg, tR = 43.2 min) and 10 (30.4 mg, tR = 45.2 min). Fraction V (40.1 g) was applied to a silica gel column eluted with CH2Cl2-MeOH-H2O (6:1:0.1, 3:1:0.1, v/v) to afford three sub-fractions, V1-V2. Sub-fraction V1 (13.3 g) afforded 13 (60.8 mg) and 14 (19.4 mg) by ODS silica gel CC eluting with MeOH-H2O (2:1, v/v).
2-Oxo-3β,19α-dihydroxyolean-12-en-28-oic acid β-D-glucopyranosyl ester (
1). White amorphous powder.
![Molecules 16 04642 i001]()
+ 16.5° (
c 1.05, MeOH). IR (KBr): 3419.6, 1735.7, 1711.3, 1640.8, 1070.4, 1029.9, 993.3 cm
-1. HR-ESI-MS
m/z 671.3779 [
M + Na]
+ (calc. C
36H
56O
10Na, 671.3771), 649.3953 [
M + H]
+ (calc. C
36H
57O
10, 649.3952), 666.4221 [
M + NH
4]
+ (calc. C
36H
60O
10N, 666.4217);
1H- and
13C-NMR (pyridine-
d5) data, see
Table 1.
2
α,19
α-Dihydroxy-3-oxo-12-ursen-28-oic acid
β-
D-glucopyranosyl ester (
4). White amorphous powder.
![Molecules 16 04642 i001]()
+ 21.5° (
c 1.25, MeOH). IR (KBr): 3431.1, 1737.1, 1714.3, 1644.6, 1070.4, 1029.9, 991.3 cm
-1. HR-ESI-MS
m/z 649.3945 [
M + H]
+ (calc. C
36H
57O
10, 649.3952);
1H- and
13C-NMR (pyridine-
d5) data, see
Table 1.
3.3.1. Acid Hydrolysis of 1 and 4 and Determination of the Absolute Configuration of the Mono-saccharides
1 (5 mg) in 1 M HCl (dioxane-H2O, 1:1, 5 mL) was heated at 90 °C for 3 h under an Ar atmosphere. After the dioxane was removed, the solution was extracted with EtOAc (3 mL × 3) to remove the aglycone. The aqueous layer was neutralized by passing through an ion-exchange resin column (Amberlite MB-3, Organo, Tokyo, Japan) and concentrated to dryness under reduced pressure to give the sugar fraction. The residue was dissolved in pyridine (0.1 mL) to which 0.1 M L-cysteine methyl ester hydrochloride in pyridine (0.1 mL) was added. The mixture was heated at 60 °C for 1 h. After the reaction mixture was dried in vacuo, the residue was trimethylsilylated with l-trimethylsilylimidazole (0.2 mL) for 2 h. The mixture was partitioned between hexane and H2O (0.6 mL, each), and the hexane extracted was analyzed by GC under the following conditions: capillary column, DM-5 (0.25 mm × 30 m × 0.25 μm); detector, FID; injector temperature, 280 °C, detector temperature, 280 °C; initial temperature was maintained at 160 °C for 2 min and then raised to 195 °C at a rate of 10 °C/min; carrier gas, N2. In the acid hydrolysate of 1, D-glucose was confirmed by comparison of the retention time of their derivatives with those of D-glucose and L-glucose derivatives prepared in a similar way, which showed retention times of 28.56 and 27.72 min, respectively. The sugar from 4 (30 mg) was also identified by the same method.
3.3.2. α-Glucosidase Inhibition Assay
α-Glucosidase (EC.3.2.1.20) enzyme inhibition assay has been performed according to the literature [
19].
α-Glucosidase (25 μL, 0.2 U/mL), various concentrations of samples (25 μL), and 67 mM phosphate buffer (pH 6.8, 175 μL) were mixed at room temperature for 10 min. Reactions were initiated by the addition of 23.2 mM
p-nitrophenyl-
α-
D-glucopyranoside (25 μL). The reaction mixtures were incubated at 37 °C for 15 min in a final volume of 250 μL, and then 1 M Na
2CO
3 (50 μL) was added to the incubation solution to stop the reaction. The activities of glucosidase were detected in a 96-well plate, and the absorbance was read at 405 nm by a microplate spectrophotometer (Spectra Max, Molecular Devices, USA). The negative control was prepared by adding phosphate buffer instead of the sample in the same way as the test. Acarbose was utilized as the positive control. The blank was prepared by adding phosphate buffer instead of
α-glucosidase using the same method. The inhibition rates (%) were calculated from the following formula:




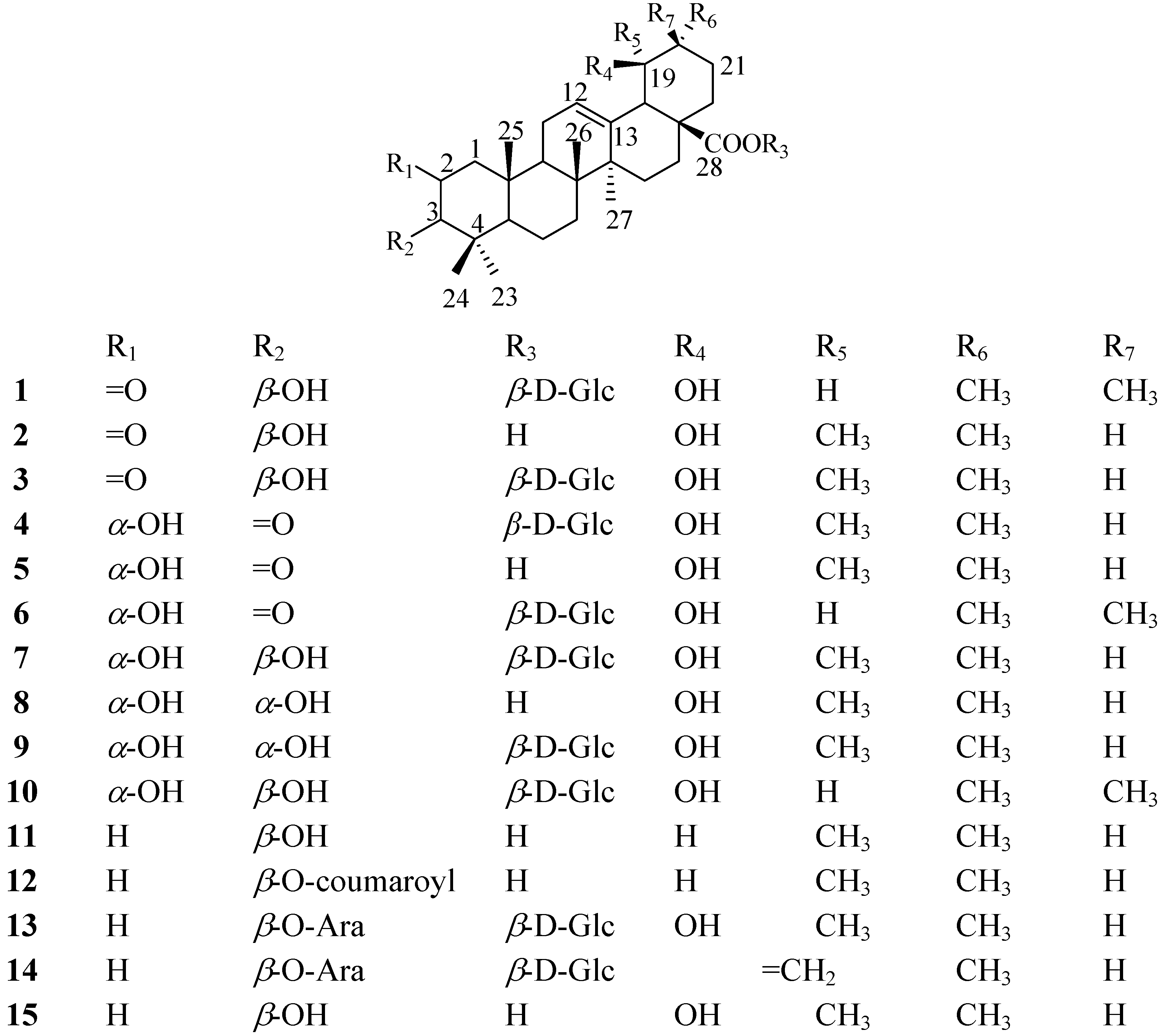{kind=link}
{kind=link}
 + 16.5° (c 1.05, MeOH). IR (KBr): 3419.6, 1735.7, 1711.3, 1640.8, 1070.4, 1029.9, 993.3 cm-1. HR-ESI-MS m/z 671.3779 [M + Na]+ (calc. C36H56O10Na, 671.3771), 649.3953 [M + H]+ (calc. C36H57O10, 649.3952), 666.4221 [M + NH4]+ (calc. C36H60O10N, 666.4217); 1H- and 13C-NMR (pyridine-d5) data, see Table 1.
+ 16.5° (c 1.05, MeOH). IR (KBr): 3419.6, 1735.7, 1711.3, 1640.8, 1070.4, 1029.9, 993.3 cm-1. HR-ESI-MS m/z 671.3779 [M + Na]+ (calc. C36H56O10Na, 671.3771), 649.3953 [M + H]+ (calc. C36H57O10, 649.3952), 666.4221 [M + NH4]+ (calc. C36H60O10N, 666.4217); 1H- and 13C-NMR (pyridine-d5) data, see Table 1.