2. Results and Discussion
Our designed dual linker also has an amino end, but our approach differs from that of a previous report [
5], by directly attaching to the resin through an acid sensitive linker (linker 2,
Figure 2). Cleavage at site B with a weak protonic acid will release the products bearing linker 1 and a hydrophilic spacer. This spacer could not only improve the resin’s swelling properties and the loading capacity as well as the overall yield of the solid phase reaction [
9], but also exhibit more desirable physical properties, provide a friendly environment for target interaction and reduce the binding to nonspecific proteins in affinity chromatography for target identification [
10]. In this initial study, we employed diethylene glycol to simply demonstrate the feasibility of the method. On the other hand, an alkoxymethyl ester fragment was chosen as linker 1 to protect the carboxyl group of the triterpene. It was supposed that the alkoxymethylene group would be sensitive to a Lewis acid like a 2-methoxy-ethoxymethyl ether (MEM), a commonly used alcohol protective group, so that facile cleavage at site A will yield the unlabeled product.
Figure 2.
Diagram of the dual linker for OA solid phase synthesis.
Figure 2.
Diagram of the dual linker for OA solid phase synthesis.
The dual linker was synthesized as illustrated in
Scheme 1. Monosulfonation of diethyleneglycol (
2) with tosyl chloride (TsCl) in CH
2Cl
2 in the presence of pyridine and 4-dimethylaminopyridine (DMAP) showed poor selectivity towards the symmetrical diol, and yielded only a 48.9% of the monotosylate
3a, along with 17.8% of the ditosylate
3b, respectively. In contrast, use of a stoichiometric amount of TsCl in the presence of Ag
2O and a catalytic amount of KI under neutral condition [
11] improved the yield of the desired product
3a to 90.6%. Then,
3a was treated with NaN
3 under reflux conditions in MeCN, giving the corresponding azide
4 in almost quantitative yield. A portion of DMF should be added to the system to accelerate the reaction process. Because of the high yield, compound
4 was hydrogenated in MeOH at atmospheric pressure (catalyzed by Pd/C) to give 2-(2-aminoethoxy)-ethan-1-ol (
5) in 98.3% yield without further purification.
Scheme 1.
Synthesis of chloromethyl ether 9.
Scheme 1.
Synthesis of chloromethyl ether 9.
We initially chose benzyloxycarbonyl chloride (Cbz) as the amino protective group before chloromethylation of the hydroxyl, but the yield is not satisfactory due to the poor selectivity of Cbz for the two nucleophiles, OH and NH
2. Finally, we found that phthalimide group was the best choice to meet our needs, giving the protected compound
7 in a yield of 92.3%. Chloromethylation of
7 with trioxane and dry HCl (dissolved in dry MeOH) did not afford the wanted product but rather gave the dimer of the molecule itself by connecting with a methylene. Thus an
O,
S-acetal
8 was prepared in 83.7% yield by reacting of the alcohol
7 with DMSO in the presence of AcOH and Ac
2O [
12]. Then chloromethyl ether
9 was obtained by cleavage of
8 with a stoichiometric amount of SOCl
2. It should be mentioned that
9 may be unstable and decompose on heating or during storage, so that it should be used immediately without any purification after evaporation of the reaction solvent at low temperature.
Before attempting the solid-phase experiment, preliminary liquid-phase tests were first performed on the assessment of dual cleavage ability of the linker (
Scheme 2). Treatment of
1a with
9 (~2 equiv) and
N,
N-diisopropylethylamine (DIPEA) (3 equiv) in CH
2Cl
2, furnished
10a in 97.2% yield. The C-3 hydroxyl of
1a is inert under this reaction condition. After hydrolysis of the phthalimide group in an ethanol solution of methylamine (33 wt. %), the amino product was obtained in 78.2% yield. Trityl chloride was chosen to simulate the resin, and we first attempted to couple it with
11a through a carbamate, but were unsuccessful. Alternatively, a direct connection of the amine and trityl chloride in the presence of DIPEA proved effective, affording
12a in a yield of 85.4%. Then, the cleavage conditions for the two different sites in
12a were explored. It was found that a solution of 1% (v/v) trifluoroacetic acid (TFA) in CH
2Cl
2 could be used for removing the trityl group to afford
11a, albeit accompanied with the formation of
1a. Little improvement was achieved by reducing the concentration of TFA to 0.5%. Thus, a mixture of AcOH, 2,2,2-trifluoroethanol (TFE) and CH
2Cl
2 (1:2:7 v/v/v) was employed, giving solely the desired product
11a. On the other hand, several Lewis acids, such as MgBr
2, ZnBr
2, AlCl
3/
N,
N-dimethylaniline,
etc, were utilized for the cleavage of the MEM-like moiety. Ultimately, anhydrous TiCl
4 could smoothly release the free carboxyl group of OA without any side reactions after carefully controlling the reaction time. We also repeated the same reaction sequence starting from 3β-acetoxyl-OA
1b. From the results, it was found that the finely tuned cleavage conditions of our designed dual linker did not affect the integrity of the triterpene skeleton and C-3 acetate.
Scheme 2.
Dual cleavage property of the designed linker.
Scheme 2.
Dual cleavage property of the designed linker.
Compound
11a was then loaded onto trityl chloride resin (Nankai HECHENG, 0.97 mmol/g) by procedures similar to those in solution described above. As indicated in
Scheme 3,
11a was incubated with pre-swelled resin (~2 equiv) in CH
2Cl
2 at room temperature in the presence of DIPEA. The reaction was quenched by MeOH as soon as the substrate disappeared in solution, as monitored by TLC. The loading of the substrate was calculated as 0.34 mmol/g (substrate/resin) based on the weight gain of the dried resin. Treatment of
13 with TiCl
4 in CH
2Cl
2 easily recovered the free OA in 94.6%, while the treatment with AcOH/TFE could release the amino end from the resin in 93.5% smoothly. To show the viability of the dual linker for solid phase organic synthesis of derivatives of OA, a model reaction has been performed. The swelled resin
13 was mixed with excess
E-(2-thiophene)acrylic acid,
N,
N’-diisopropylcarbodiimide (DIC) and DMAP in DMF. After 1 day, the reaction was terminated and the modified resin was filtered, dried and subjected to cleavage reactions under the conditions described above. The final products
15 and
16 were obtained in yields of 92.0% and 89.9%, respectively. The structures of the cleaved compounds
1a,
11a,
15, and
16 were confirmed by MS,
1H- and
13C-NMR analysis.
Scheme 3.
Dual cleavage property of the designed linker on the resin.
Scheme 3.
Dual cleavage property of the designed linker on the resin.
Next, we measured and compared the cytotoxicity of two compounds, 1a and 10a, on non-small cell lung cancer cell line A549 (IC50 51 μM and 50 μM, respectively), and their inhibition activity of human umbilical vein endothelial cells ECV304 (IC50 20 μM and 10.3 μM, respectively). The results showed that both activity of these two compounds were on the same level, and indicated that the long and flexible alkyl ether functionality in the linker is less likely to be critical for the binding event, and the major binding interaction will come from the compound scaffold. This demonstrates its ability for application in the subsequent target identification program.
Since Merrifield and Tam first introduced the concept of “multidetachable” resins as early as 1979 [
13,
14], dozens of similar studies have been published [
15,
16,
17,
18,
19]. Among these, a technique, named “analytical construct”, for solid phase reaction monitoring has been developed by Geysen
et al. [
20] and attracted much attention [
21,
22,
23,
24,
25,
26,
27,
28,
29,
30]. The construct typically comprises two selective-cleavable in-line linkers, involving a MS sensitizer (usually an amine) which can greatly improve the ionization properties of the materials and thus allowing easy detection by electrospray mass spectrometry [
31] and a MS splitter which is an isotope label giving rise to a characteristic split-peak pattern in MS [
20,
32,
33]. Accordingly, the amino-terminated dual linker designed by us may act as an analytical construct too, whose reaction monitoring ability is worthy of study in the near future.
3. Experimental
3.1. General
1H- and 13C-NMR spectra were measured on a Varian Mercury-400 or Varian Mercury-300 spectrometer. The mass spectra (MS) were measured on Agilent 1100 LC/MSD high performance ion trap mass spectrometer or LCQ ESI mass spectrometer.
3.2. Materials
Oleanolic acid was a reference compound (purity > 98%) purchased from the Division of Chinese Materia Medica and Natural Products, National Institute for the Control of Pharmaceutical and Biological Products (NICPBP), Ministry of Public Health, China. Trityl chloride resin (loading capacity 0.97 mmol/g) was purchased from Nankai HECHENG S&T Co. (Tianjing, China). All other reagents were of standard quality and used without further purification. All solvents were dried before use through standard procedures.
3.3. Tosylation of Diethyleneglycol (2)
To a stirred solution of diethyleneglycol (2, 0.1 mL, 1.0 mmol) in CH2Cl2 (10 mL) was added fresh Ag2O (350 mg, 1.5 mmol), TsCl (210 mg, 1.1 mmol), and KI (33 mg, 0.2 mmol). The reaction mixture was stirred at 0 °C for 4 h, then filtered through a small pad of silica gel, and washed with EtOAc. Evaporation of the solvent in vacuo, followed by column chromatography on silica gel (hexane-EtOAc = 1:1), gave the desired monotosylate product 3a (as a colorless oil) and a spot of ditosylate 3b (also a colorless oil) as a by-product.
2-(2-{[(4-Methylbenzene)sulfonyl]oxy}ethoxy)ethan-1-ol (3a) (242.5 mg, 90.6%): 1H-NMR (300 MHz, CDCl3): δ 7.78 (2H, d, J = 8.1 Hz, o-H of Ph), 7.33 (2H, d, J = 8.1 Hz, m-H of Ph), 4.17 (2H, t, J = 4.5 Hz, TsOCH2CH2O-), 3.66 (4H, m, -CH2CH2OCH2CH2-), 3.51 (2H, t, J = 4.5 Hz, -OCH2CH2OH), 2.43 (3H, s, -CH3), 2.18 (1H, s, -OH). 13C-NMR (75 MHz, CDCl3): δ 144.89, 132.87, 129.78 (2C), 127.86 (2C), 72.41, 69.12, 68.47, 61.52, 21.55. ESI-MS: m/z [M + H]+ 261.2, [M + Na]+ 283.1.
2-(2-{[(4-Methylbenzene)sulfonyl]oxy}ethoxy)ethyl4-methylbenzene-1-sulfonate (3b) (18.2 mg, 4.3%): 1H-NMR (300 MHz, CDCl3): δ 7.77 (4H, d, J = 8.1Hz, o-H of Ph), 7.34 (4H, d, J = 7.8 Hz, m-H of Ph), 4.08 (4H, t, J = 4.5 Hz, TsOCH2CH2O-), 3.59 (4H, t, J = 4.5 Hz, TsOCH2CH2O-), 2.44 (6H, s, -CH3). 13C-NMR (75 MHz, CDCl3): δ 144.91 (2C), 132.74 (2C), 129.83 (4C), 127.86 (4C), 68.96 (2C), 68.64 (2C), 21.56 (2C). ESI-MS: m/z [M + H]+ 415.1, [M + NH4]+ 432.1, [M + Na]+ 437.1, [M + K]+ 453.1.
3.4. Synthesis of 2-(2-azidoethoxy)ethan-1-ol (4)
To a stirred solution of 3a (75.0 mg, 0.29 mmol) in dry CH3CN (2 mL) was added NaN3 (56.2 mg, 0.87 mmol) and DMF (200 μL). The reaction mixture was heated to reflux for 9 h, then another batch of NaN3 (56.2 mg, 0.87 mmol) was added to the system. After another 20 h, the reaction was quenched with water (2 mL), and extracted with CH2Cl2. The organic layer was dried over Na2SO4 and concentrated in vacuo to afford product 4 (36.4 mg, 96.2%) as a colorless oil. This compound was used in the next step without further purification. 1H-NMR (300 MHz, CDCl3): δ 3.76 (2H, t, J = 4.2 Hz,-OCH2CH2OH), 3.70 (2H, t, J = 4.8Hz, N3CH2CH2O-), 3.62 (2H, t, J = 4.2Hz, -OCH2CH2OH), 3.41 (2H, t, J = 4.8 Hz, N3CH2CH2O-), 2.05 (1H, br s, -OH). 13C-NMR (75 MHz, CDCl3): δ 72.38, 70.09, 61.78, 50.70. ESI-MS: m/z [M + NH4]+ 149.0.
3.5. Synthesis of 2-(2-aminoethoxy)ethan-1-ol (5)
A suspension of compound 4 (30 mg, 0.23 mmol), 10% Pd/C (10 mg) in MeOH (1 mL) was stirred under 1 atm of hydrogen pressure for 8 h, and then filtered. The filtrate was concentrated in vacuo to afford product 5 (23.6 mg, 98.3%) as a colorless oil. This compound was used in the next step without further purification. 1H-NMR (300 MHz, CDCl3): δ 3.64 (2H, t, J = 4.5Hz, -OCH2CH2OH), 3.49 (2H, t, J = 4.5 Hz, -OCH2CH2OH), 3.46 (2H, t, J = 5.1 Hz, H2NCH2CH2O-), 2.87 (3H, br s, -NH2 and -OH), 2.81 (2H, t, J = 5.1 Hz, H2NCH2CH2O-). 13C-NMR (75 MHz, CDCl3): δ 72.45 (2C), 61.16, 41.34. ESI-MS: m/z [M + H]+ 106.1, [M + Na]+ 128.1.
3.6. Synthesis of 2-[2-(2-hydroxyethoxy)ethyl]-2,3-dihydro-1H-isoindole-1,3-dione (7)
Compound 5 (1.05 g, 10 mmol) and phthalic anhydride (1.53 g, 10.1 mmol) were mixed together and stirred until the system become clear. Then the mixture was heated to 100 °C and maintained for 1.5 h. After cooling to room temperature, CHCl3 (15 mL) and water (6 mL) were added. The organic layer was dried over Na2SO4 and concentrated in vacuo to afford product 7 (2.17 g, 92.3%) as a white solid. This compound was used in the next step without further purification. 1H-NMR (300 MHz, CDCl3): δ 7.81 (2H, m, Ph), 7.69 (2H, m, Ph), 3.88 (2H, t, J = 5.4Hz, -NCH2CH2O-), 3.72 (2H, t, J = 5.4 Hz, -NCH2CH2O-), 3.66 (2H, t, J = 4.2Hz, -OCH2CH2OH), 3.57 (2H, t, J = 4.2Hz, -OCH2CH2OH), 2.51 (1H, br s, -OH). 13C-NMR (75 MHz, CDCl3): δ 168.37 (2C), 133.95 (2C), 131.93 (2C), 123.23 (2C), 72.13, 68.27, 61.63, 37.47. ESI-MS: m/z [M + H]+ 236.2, [M + Na]+ 258.1, [M + K]+ 274.1.
3.7. Synthesis of 2-(2-{2-[(methylsulfanyl)methoxy]ethoxy}ethyl)-2,3-dihydro-1H-isoindole-1,3-dione (8)
To a stirred solution of 7 (2.17 g, 9.2 mmol) in DMSO (27.1 mL) was added AcOH (5.4 mL,94.4 mmol) and Ac2O (17.9 mL, 191.4 mmol). The reaction mixture was stirred at room temperature for 20 h, then poured into a cold water (271 mL) solution of Na2CO3 (27.1 g). The mixture was extracted with CH2Cl2. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane-EtOAc = 5:1) to give the product 8 (2.28 g, 83.7%) as a viscous oil. 1H-NMR (300 MHz, CDCl3): δ 7.82 (2H, m, Ph), 7.69 (2H, m, Ph), 4.58 (2H, s, -SCH2O-), 3.89 (2H, t, J = 6.0 Hz, -NCH2CH2O-), 3.73 (2H, t, J = 6.0 Hz, -NCH2CH2O-), 3.63 (4H, br s, -OCH2CH2O-), 2.07 (3H, s, -SCH3). 13C-NMR (75 MHz, CDCl3): δ 168.17 (2C), 133.84 (2C), 132.07 (2C), 123.16 (2C), 75.32, 69.74, 67.80, 66.91, 37.16, 13.64. ESI-MS: m/z [M + H]+ 296.4, [M + Na]+ 318.4.
3.8. Synthesis of {2-[2-(1,3-dione-2,3-dihydro-1H-isoindol-2-yl)ethoxy]ethoxy}methyl olean-12-en-28-oate (10a)
To a stirred solution of 8 (51 mg, 0.17 mmol) in dry CH2Cl2 (1.0 mL) was added SOCl2 (12.3 μL, 0.17 mmol) at 0 °C. The reaction was warmed to room temperature and stirred for 2 h before it was evaporated under reduced pressure at 30 °C to remove the excess SOCl2. The crude product 2-{2-[2-(chloromethoxy)ethoxy]ethyl}-2,3-dihydro-1H-isoindole-1,3-dione (9) could be used in the next step without further purification. The residue mentioned above dissolved in dry CH2Cl2 (0.5 mL) was added to a solution of the mixture of oleanolic acid (1a, 21.2 mg, 0.046 mmol) and DIPEA (21.3 μL, 0.13 mmol) in dry CH2Cl2 (0.5 mL). The mixture was stirred at room temperature for 2 h before it was diluted with CH2Cl2 (10 mL) and washed with a saturated solution of NaHCO3. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane-EtOAc = 3:1) to give the product 10a (31.8 mg, 97.2%) as a white solid. 1H-NMR (400 MHz, CDCl3): δ 7.84 (2H, m, Ph), 7.72 (2H, m, Ph), 5.28 (1H, s, H-12), 5.24–5.18 (2H, ABq, J = 6.0 Hz, -COOCH2O-), 3.90 (2H, t, J = 6.0 Hz, -NCH2CH2O-), 3.74 (4H, m, -NCH2-, -OCH2CH2OCH2O-), 3.65 (2H, m,-OCH2CH2OCH2O-), 3.21 (1H, dd, J = 10.8, 4.4Hz, H-3α), 2.85 (1H, d, J = 10.8 Hz, H-18), 1.12 (3H, s, CH3), 0.98 (3H, s, CH3), 0.91 (3H, s, CH3), 0.89 (6H, s, CH3), 0.78 (3H, s, CH3), 0.72 (3H, s, CH3). 13C-NMR (100 MHz, CDCl3): δ 177.07, 168.20 (2C), 143.61, 133.91 (2C), 132.12 (2C), 123.22 (2C), 122.47, 89.37, 78.98, 69.62, 69.47, 68.00, 55.21, 47.59, 46.85, 45.83, 41.69, 41.17, 39.32, 38.73, 38.43, 37.15, 37.00, 33.81, 33.06, 32.74, 32.27, 30.66, 28.09, 27.58, 27.19, 25.81, 23.59, 23.39, 22.91, 18.31, 16.99, 15.56, 15.30. ESI-MS: m/z [M + Na]+ 726.4, [M + K]+ 742.4, [M + Cl]− 738.6.
3.9. Synthesis of [2-(2-aminoethoxy)ethoxy]methyl olean-12-en-28oate (11a)
A mixture of 10a (13.5 mg, 0.020 mmol) and a solution of methylamine in ethanol (0.7 mL, 33 wt. %) was stirred at room temperature for 0.5 h before it was concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3-MeOH = 10:1) to give the product 11a (8.6 mg, 78.2%) as a white solid. 1H-NMR (400 MHz, CDCl3): δ 5.32–5.26 (3H, m, H-12, -COOCH2O-), 3.78 (2H, t, J = 4.4 Hz, -NCH2CH2O-), 3.62 (2H, t, J = 4.8 Hz, -OCH2CH2OCH2O-), 3.52 (2H, t, J = 5.2 Hz, -OCH2CH2OCH2O-), 3.21 (1H, dd, J = 12.4, 4.0 Hz, H-3α), 2.89-2.85 (3H, m, H-18,-NCH2CH2O-), 1.14 (3H, s, CH3), 0.98 (3H, s, CH3), 0.92 (3H, s, CH3), 0.90 (6H, s, CH3), 0.78 (3H, s, CH3), 0.74 (3H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ 177.13, 143.49, 122.44, 89.21, 78.74, 72.21, 69.84, 69.29, 55.11, 47.48, 46.81, 45.71, 41.62, 41.22, 41.10, 39.24, 38.64, 38.37, 36.90, 33.71, 32.95, 32.66, 32.23, 30.57, 28.02, 27.50, 27.04, 25.73, 23.50, 23.30, 22.83, 18.22, 16.94, 15.53, 15.23. ESI-MS: m/z [M + H]+ 574.5, [M + Na]+ 596.7.
3.10. Synthesis of (2-{2-[(triphenylmethyl)amino]ethoxy}methyl olean-12-en-28-oate (12a)
To a solution of 11a (63.4 mg, 0.11 mmol) in CH2Cl2 (2.0 mL) was added DIPEA (91.3 μL, 0.55 mmol) and trityl chloride (92.1 mg, 0.33 mmol). The reaction mixture was stirred at room temperature for 3 h before it was concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane-EtOAc = 6:1) to afford product 12a (77.0 mg, 85.4%) as a white solid. 1H-NMR (400 MHz, CDCl3): δ 7.47 (6H, d, J = 7.6Hz, o-H of Ph), 7.27 (6H, t, J = 7.6 Hz, m-H of Ph), 7.18 (3H, t, J = 7.2 Hz, p-H of Ph), 5.30-5.22 (3H, m, H-12, -COOCH2O-), 3.73 (2H, t, J = 4.8 Hz, -NCH2CH2O-), 3.60 (2H, t, J = 5.2 Hz, -OCH2CH2OCH2O-), 3.53 (2H, t, J = 4.8 Hz,-OCH2CH2OCH2O-), 3.21 (1H, dd, J = 10.8, 4.4 Hz, H-3α), 2.86 (1H, dd, J = 13.6, 4.0 Hz, H-18), 2.36 (2H, br s, -NCH2CH2O-), 1.13 (3H, s, CH3), 0.98 (3H, s, CH3), 0.91 (3H, s, CH3), 0.90 (3H, s, CH3), 0.89 (3H, s, CH3), 0.77 (3H, s, CH3), 0.72 (3H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ 177.04, 145.99 (3C), 143.53, 128.58 (6C), 127.69 (6C), 126.13 (3C), 122.41, 89.33, 78.83, 71.31, 70.51, 69.66, 69.31, 55.11, 47.50, 46.78, 45.74, 42.95, 41.62, 41.10, 39.24, 38.64, 38.34, 36.92, 33.71, 32.98, 32.68, 32.20, 30.57, 28.02, 27.52, 27.09, 25.75, 23.50, 23.30, 22.83, 18.22, 16.94, 15.50, 15.23. ESI-MS: m/z [M + H]+ 816.7.
3.11. Cleavage at Site A of 12a with TiCl4
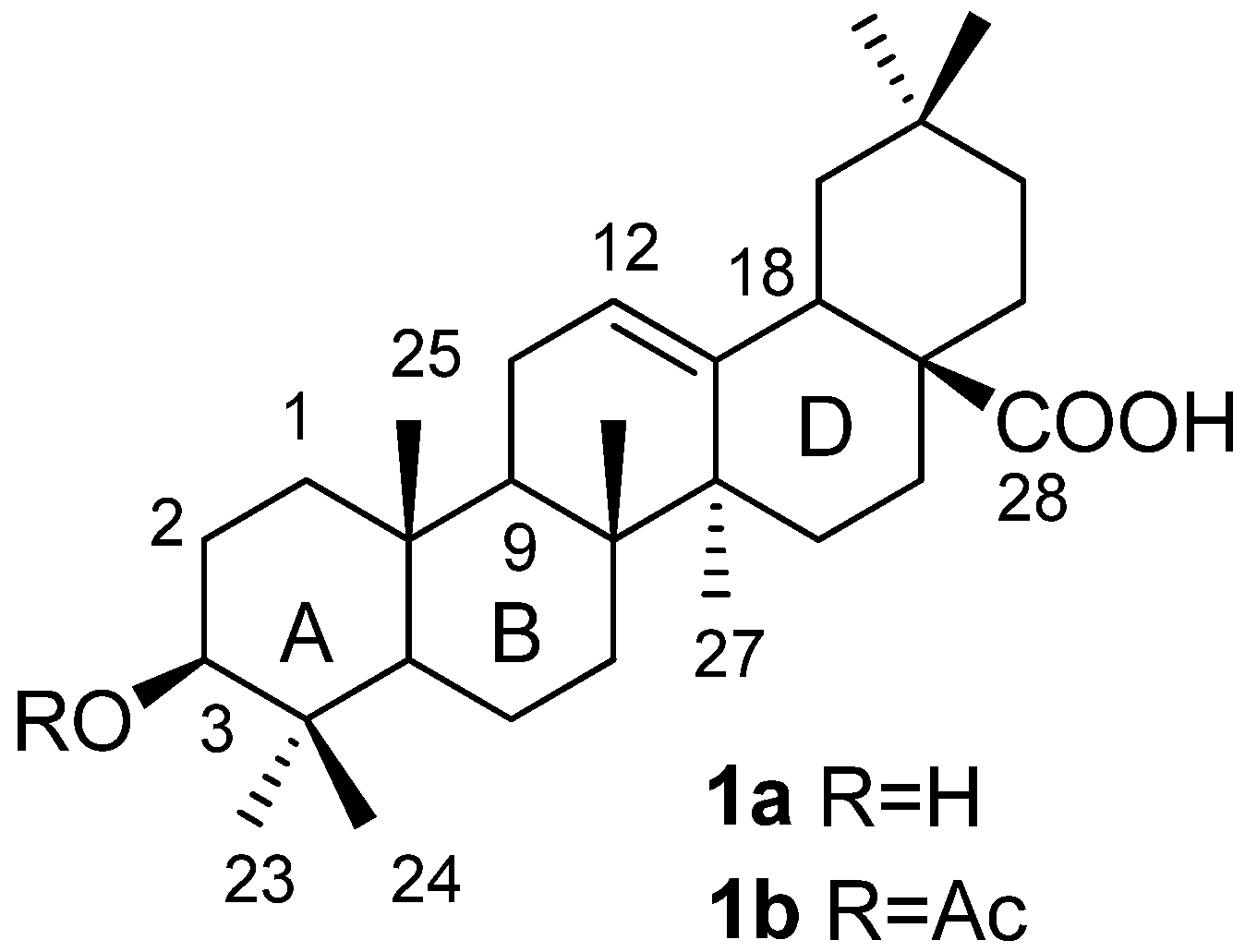
To a solution of 12a (10.6 mg, 0.013 mmol) in CH2Cl2 (1.0 mL) was added TiCl4 (5 μL, 0.045 mmol) dropwise. The reaction mixture was stirred at room temperature for 0.5 h before it was poured into water (1 mL) and extracted with CH2Cl2. The organic layer was washed with a saturated solution of NaHCO3, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane-acetone = 5:1) to afford product 1a (5.3 mg, 89.6%) as a white solid. 1H-NMR (300 MHz, pyridine-d5): δ 5.49 (1H, br s, H-12), 3.44 (1H, m, H-3α), 3.29 (1H, dd, J = 13.5, 3.6 Hz, H-18), 1.29 (3H, s, CH3), 1.24 (3H, s, CH3), 1.02 (9H, s, CH3), 0.95 (3H, s, CH3), 0.90 (3H, s, CH3). 13C-NMR (75 MHz, pyridine-d5): δ 180.20, 144.88, 122.61, 78.13, 55.88, 48.18, 46.72, 46.55, 42.23, 42.06, 39.82, 39.44, 39.01, 37.44, 34.29, 33.35 (2C), 33.24, 31.03, 28.85, 28.39, 28.16, 26.25, 23.89, 23.85, 23.76, 18.86, 17.49, 16.62, 15.63. ESI-MS: m/z [M−H]− 455.3.
3.12. Cleavage at Site B of 12a with TFE
A mixture of 12a (25.0 mg, 0.031 mmol) and a solution of AcOH/TFE/CH2Cl2 (1.0 mL, v/v/v = 1:2:7) was stirred at room temperature for 2 h before it was quenched with a saturated solution of NaHCO3 (0.4 mL). The water layer was extracted with CH2Cl2, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3-MeOH = 10:1) to afford product 11a (16.5 mg, 94.0%) as a white solid.
3.13. Synthesis of {2-[2-(1,3-dione-2,3-dihydro-1H-isoindol-2-yl)ethoxy]ethoxy}methyl 3β-acetoxylolean-12-en-28-oate (10b)
To a stirred solution of 8 (59 mg, 0.20 mmol) in dry CH2Cl2 (1.0 mL) was added SOCl2 (14.4 μL, 0.20 mmol) at 0 °C. The reaction was warmed to room temperature and stirred for 2 h before it was evaporated under reduced pressure at 30 °C to remove the excess SOCl2. The crude product 9 could be used in the next step without further purification. The crude product dissolved in dry CH2Cl2 (0.5 mL) was added to a solution of the mixture of 3β-acetoxyl-OA (1b, 22.9 mg, 0.046 mmol) and DIPEA (21.3 μL, 0.13 mmol) in dry CH2Cl2 (0.5 mL). The mixture was stirred at room temperature for 2 h before it was diluted with CH2Cl2 (10 mL) and washed with a saturated solution of NaHCO3. The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane-EtOAc = 6:1) to give the product 10b (32.7 mg, 95.4%) as a white solid. 1H-NMR (300 MHz, CDCl3): δ 7.82 (2H, m, Ph), 7.69 (2H, m, Ph), 5.25 (1H, br s, H-12), 5.23–5.15 (2H, ABq, J = 6.0 Hz, -COOCH2O-), 4.46 (1H, t, J = 8.1 Hz, H-3α), 3.88 (2H, t, J = 5.7 Hz, -NCH2CH2O-), 3.70 (4H, m, -NCH2-,-OCH2CH2OCH2O-), 3.62 (2H, t, J = 4.5 Hz, -OCH2CH2OCH2O-), 2.82 (1H, m, H-18), 2.02 (3H, s, CH3 of acetyl), 1.09 (3H, s, CH3), 0.89 (6H, s, CH3), 0.87 (3H, s, CH3), 0.84 (3H, s, CH3), 0.83 (3H, s, CH3), 0.70 (3H, s, CH3). 13C-NMR (75MHz, CDCl3): δ 176.99, 170.90, 168.12 (2C), 143.56, 133.86 (2C), 132.06 (2C), 123.16 (2C), 122.33, 89.27, 80.80, 69.55, 69.40, 67.93, 55.22, 47.43, 46.76, 45.73, 41.60, 41.10, 39.27, 38.03, 37.59, 37.07, 36.83, 33.74, 33.00, 32.60, 32.19, 30.58, 27.96, 27.49, 25.70, 23.53, 23.44, 23.32, 22.83, 21.23, 18.13, 16.91, 16.62, 15.29. ESI-MS: m/z [M + NH4]+ 763.5, [M + Na]+ 768.5.
3.14. Synthesis of [2-(2-aminoethoxy)ethoxy]methyl 3β-acetoxylolean-12-en-28-oate (11b)
A mixture of 10b (129 mg, 0.17 mmol) and a solution of methylamine in ethanol (6.0 mL, 33 wt. %) was stirred at room temperature for 0.5 h before it was concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3-MeOH = 10:1) to give the product 11b (86.9 mg, 82.0%) as a white solid. 1H-NMR (300 MHz, CDCl3): δ 5.28 (3H, m, H-12, -COOCH2O-), 4.48 (1H, t, J = 7.8 Hz, H-3α), 4.00 (2H, br s, -NH2), 3.77 (2H, m, -NCH2CH2O-), 3.63 (4H, m, -OCH2CH2O-), 3.02 (2H, m, -NCH2CH2O-), 2.86 (1H, m, H-18), 2.04 (3H, s, CH3 of acetyl), 1.13 (3H, s, CH3), 0.92 (6H, s, CH3), 0.90 (3H, s, CH3), 0.86 (3H, s, CH3), 0.85 (3H, s, CH3), 0.73 (3H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ 177.05, 170.87, 143.47, 122.29, 89.15, 80.71, 72.56, 69.80, 69.26, 55.11, 47.34, 46.75, 45.62, 41.54, 41.31, 41.02, 39.18, 37.94, 37.51, 36.75, 33.67, 32.93, 32.54, 32.17, 30.54, 27.90, 27.42, 25.65, 23.47, 23.35, 23.26, 22.77, 21.20, 18.05, 16.88, 16.56, 15.26. ESI-MS: m/z [M + H]+ 616.4.
3.15. Synthesis of (2-{2-[(triphenylmethyl)amino]ethoxy}methyl 3β-acetoxylolean-12-en-28-oate (12b)
To a solution of 11b (82.8 mg, 0.14 mmol) in CH2Cl2 (2.0 mL) was added DIPEA (155.5 μL, 0.95 mmol) and trityl chloride (188.3 mg, 0.68 mmol). The reaction mixture was stirred at room temperature for 3 h before it was concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane-EtOAc = 12:1) to afford product 12b (82.3 mg, 71.3%) as a white solid. 1H-NMR (300 MHz, CDCl3): δ 7.49 (6H, d, J = 7.5 Hz, o-H of Ph), 7.27 (6H, t, J = 7.2 Hz, m-H of Ph), 7.18 (3H, t, J = 7.2 Hz, p-H of Ph), 5.26 (3H, m, H-12, -COOCH2O-), 4.51 (1H, t, J = 7.8 Hz, H-3α), 3.74 (2H, t, J = 4.5 Hz, -NCH2CH2O-), 3.61 (2H, t, J = 4.8 Hz, -OCH2CH2OCH2O-), 3.53 (2H, t, J = 4.8 Hz, -OCH2CH2OCH2O-), 2.88 (1H, dd, J = 13.5, 3.3 Hz, H-18), 2.37 (2H, t, J = 4.8 Hz, -NCH2CH2O-), 2.05 (3H, s, CH3 of acetyl), 1.15 (3H, s, CH3), 0.93 (3H, s, CH3), 0.92 (3H, s, CH3), 0.89 (3H, s, CH3), 0.88 (3H, s, CH3), 0.87 (3H, s, CH3), 0.75 (3H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ 177.05, 170.90, 146.04 (3C), 143.58, 128.61 (6C), 127.72 (6C), 126.18 (3C), 122.38, 89.36, 80.82, 71.35, 70.57, 69.70, 69.34, 55.23, 47.47, 46.82, 45.74, 42.99, 41.65, 41.15, 39.30, 38.06, 37.62, 36.86, 33.76, 33.01, 32.64, 32.25, 30.61, 27.99, 27.53, 25.73, 23.55, 23.46, 23.35, 22.86, 21.23, 18.16, 16.97, 16.63, 15.32. ESI-MS: m/z [M + H]+ 858.5, [M + Na]+ 880.5.
3.16. Cleavage at Site A of 12b with TiCl4
To a solution of 12b (25.0 mg, 0.030 mmol) in CH2Cl2 (1.0 mL) was added TiCl4 (22 μL, 0.20 mmol) dropwise. The reaction mixture was stirred at room temperature for 0.5 h before it was poured into water (1 mL) and extracted with CH2Cl2. The organic layer was washed with a saturated solution of NaHCO3, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane-EtOAc = 4:1) to afford product 1b (12.0 mg, 82.8%) as a white solid. 1H-NMR (300 MHz, CDCl3): δ 5.27 (1H, br s, H-12), 4.49 (1H, t, J = 7.8 Hz, H-3α), 2.81 (1H, dd, J = 13.5, 4.2 Hz, H-18), 2.04 (3H, s, CH3 of acetyl), 1.12 (3H, s, CH3), 0.94 (3H, s, CH3), 0.92 (3H, s, CH3), 0.90 (3H, s, CH3), 0.86 (3H, s, CH3), 0.85 (3H, s, CH3), 0.74 (3H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ 184.15, 171.01, 143.58, 122.53, 80.89, 55.27, 47.53, 46.52, 45.80, 41.51, 40.87, 39.25, 38.03, 37.67, 36.96, 33.76, 33.04, 32.49, 32.42, 30.65, 28.02, 27.64, 25.88, 23.56, 23.50, 23.36, 22.85, 21.29, 18.14, 17.15, 16.63, 15.37. ESI-MS: m/z [M−H]− 497.3.
3.17. Cleavage at Site B of 12b with TFE
A mixture of 12b (26.6 mg, 0.031 mmol) with a solution of AcOH/TFE/CH2Cl2 (1.0 mL, v/v/v = 1:2:7) was stirred at room temperature for 2 h before it was quenched with a saturated solution of NaHCO3 (0.4 mL). The water layer was extracted with CH2Cl2, dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CHCl3-MeOH = 10:1) to afford product 11b (14.3 mg, 92.7%) as a white solid.
3.18. Loading of 11a onto Trityl Chloride Resin
The trityl chloride resin (332.4 mg, 0.32 mmol) was swelled in CH2Cl2 (1 mL) for 1 h. Then a solution of 11a (98.8 mg, 0.17 mmol) was added to the system, followed by DIPEA (177.8 μL, 1.21 mmol) dropwise. The reaction mixture was stirred at room temperature for 16 h. The system was treated with MeOH (1 mL) to cap unloaded resin. The resin was filtered and washed sequentially with CH2Cl2, DMF, CH2Cl2, diethyl ether and dried in vacuo for 24 h, affording resin 13 (406.2 mg). The loading of the substrate was calculated as about 0.34 mmol/g (substrate/resin) based on the weight gain of the resin.
3.19. Acidic Cleavage at Site A of Resin 13 with TiCl4
Resin 13 (36.0 mg, 0.012 mmol) was swelled in CH2Cl2 (1 mL) for 1 h. TiCl4 (8.2 μL, 0.060 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 0.5 h before the resin was filtered and washed with MeOH and CH2Cl2. The filtrate was concentrated in vacuo to afford product 1a (5.3 mg, 94.6%) without any by-products.
3.20. Acidic Cleavage at Site B of Resin 13 with TFE
Resin 13 (39.5 mg, 0.013 mmol) was mixed with a solution of AcOH/TFE/CH2Cl2 (1.0 mL, v/v/v = 1:2:7) and the mixture was stirred at room temperature for 2 h before the resin was filtered and washed with MeOH and CH2Cl2. The filtrate was concentrated in vacuo, and the residue was purified by column chromatography on silica gel (CHCl3-MeOH = 10:1) to afford product 11a (7.2 mg, 93.5%).
3.21. Coupling Reaction of (E)-(2-thiophene)acrylic Acid with Resin 13
Resin 13 (88.8 mg, 0.030 mmol) was swelled in DMF (1 mL) for 3 h. Then, (E)-(2-thiophene)acrylic acid (268.7 mg, 1.74 mmol), DMAP (21.3 mg, 0.17 mmol) and DIC (381.9 μL, 2.44 mmol) were added to the system. The reaction mixture was stirred at room temperature for 24 h before the resin was filtered, washed sequentially with DMF, MeOH, CH2Cl2, diethyl ether and dried in vacuo for 24 h, affording resin 14 (92.6 mg). The conversion of the substrate was calculated as 93.3% based on the weight gain of the resin.
3.22. Acidic Cleavage at Site A of Resin 14 with TiCl4
Resin 14 (42.3 mg, 0.0137 mmol) was swelled in CH2Cl2 (1 mL) for 1 h. TiCl4 (9.6 μL, 0.085 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 0.5 h before the resin was filtered and washed with MeOH and CH2Cl2. The filtrate was concentrated in vacuo, and the residue was purified by column chromatography on silica gel (hexane-acetone = 10:1) to afford product 15 (6.4 mg, 92.0%) and unreacted substrate 1a (0.5 mg).
3β-[(E)-(2-Thiophene)acryloxyl]-12-en-28-oate (15): 1H-NMR (400 MHz, CDCl3): δ 7.76 (1H, d, J = 15.6 Hz, -CH = CHCO-), 7.36 (1H, d, J = 4.8 Hz, H-3 of thiophene), 7.25 (1H, d, J = 3.2 Hz, H-5 of thiophene), 7.05 (1H, t, J = 4.0 Hz, H-4 of thiophene), 6.23 (1H, d, J = 16.0 Hz, -CH = CHCO-), 5.29 (1H, br s, H-12), 4.63 (1H, t, J = 8.0 Hz, H-3α), 2.83 (1H, dd, J = 10.4, 3.2 Hz, H-18), 1.14 (3H, s, CH3), 0.97 (3H, s, CH3), 0.93 (6H, s, CH3), 0.91 (6H, s, CH3), 0.77 (3H, s, CH3). 13C-NMR (100 MHz, CDCl3): δ 183.78, 166.67, 143.59, 139.68, 136.75, 130.70, 128.23, 128.01, 122.57, 117.60, 80.98, 55.33, 47.55, 46.53, 45.83, 41.56, 40.92, 39.28, 38.09, 37.94, 37.00, 33.78, 33.05, 32.53, 32.43, 30.66, 28.09, 27.66, 25.91, 23.63, 23.57, 23.40, 22.88, 18.19, 17.17, 16.82, 15.40. ESI-MS: m/z [M + Cl]− 591.4.
3.23. Acidic Cleavage at Site B of Resin 14 with TFE
Resin 14 (42.6 mg, 0.0138 mmol) was mixed with a solution of AcOH/TFE/CH2Cl2 (1.0 mL, v/v/v = 1:2:7). And the mixture was stirred at room temperature for 2 h before the resin was filtered and washed with MeOH and CH2Cl2. The filtrate was concentrated in vacuo, and the residue was purified by column chromatography on silica gel (CHCl3-MeOH = 10:1) to afford product 16 (7.7 mg, 89.1%) and unreacted substrate 11a (0.8 mg).
[2-(2-Aminoethoxy)ethoxy]methyl 3β-[(E)-(2-thiophene)acryloxyl]-12-en-28-oate (16): 1H-NMR (300 MHz, CDCl3): δ 7.75 (1H, d, J = 15.6Hz, -CH = CHCO-), 7.35 (1H, d, J = 4.8 Hz, H-3 of thiophene), 7.23 (1H, d, J = 2.7 Hz, H-5 of thiophene), 7.03 (1H, t, J = 3.9 Hz, H-4 of thiophene), 6.23 (1H, d, J = 15.9 Hz, -CH = CHCO-), 5.28 (3H, m, H-12, -COOCH2O-), 4.61 (1H, t, J = 7.8 Hz, H-3α), 4.32 (2H, br s, -NH2), 3.78 (2H, br s, -NCH2CH2O-), 3.63 (4H, br s, -OCH2CH2O-), 2.99 (2H, br s, -NCH2CH2O-), 2.86 (1H, d, J = 11.1 Hz, H-18), 1.14 (3H, s, CH3), 0.94 (3H, s, CH3), 0.92 (6H, s, CH3), 0.90 (6H, s, CH3), 0.74 (3H, s, CH3). 13C-NMR (75 MHz, CDCl3): δ 177.21, 166.63, 143.58, 139.64, 136.73, 130.68, 128.22, 128.00, 122.44, 117.57, 89.24, 80.91, 71.03, 69.95, 69.35, 55.30, 47.48, 46.89, 45.76, 41.70, 41.18, 39.33, 38.11, 37.90, 36.90, 33.79, 33.03, 32.68, 32.31, 30.65 (2C), 28.07, 27.58, 25.76, 23.58 (2C), 23.39, 22.91, 18.21, 17.03, 16.83, 15.38. ESI-MS: m/z [M + H]+ 710.4.
3.24. Cytotoxicity Assay
Cells grown in 96-well plates were treated with gradient concentrations of each compound for 48 h. Cells were fixed with 50% trichloroacetic acid and stained with 0.4% sulforhodamine B dissolved in 1% acetic acid. Cells were then washed with 1% acetic acid to remove unbound dye. The protein-bound dye was extracted with 10 mM Tris base to determine the optical density at 564 nm wavelength using a SPECTRAmax PLUS 384 microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). The percentage of cell survival as a function of drug concentration was plotted to determine the IC50 value, which stands for the drug concentration needed to kill cells by 50%.
3.25. Cell Migration Assay
Human umbilical vein endothelial cells grown in 24-well plates as confluent monolayers were mechanically scratched using a 20 µL pipette tip to create the wound. Cells were washed with phosphate-buffered saline to remove the debris, and complete culture media were then added to allow wound healing. Phase contrast images of the wound were taken at three random locations first immediately after wounding and then at the same location after 24 h to examine wound closure by migrating cells. Cells migrated into the wound area were then counted, and the extent of wound closure was determined.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




