Recent Developments in Molecular Dynamics Simulations of Fluorescent Membrane Probes


Abstract
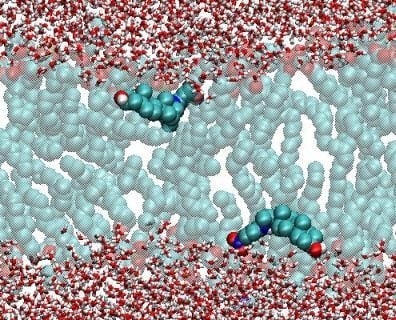
:
1. Introduction

2. Atomistic MD Simulations of Fluorophores in Lipid Bilayers
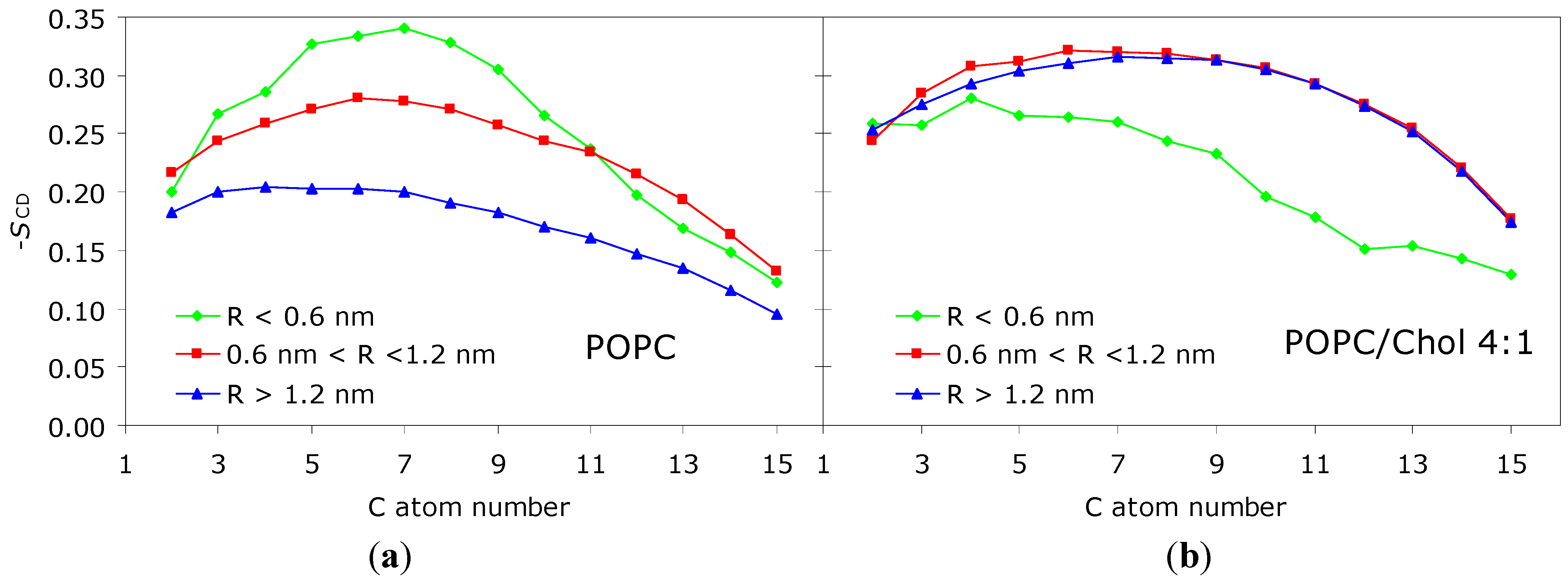
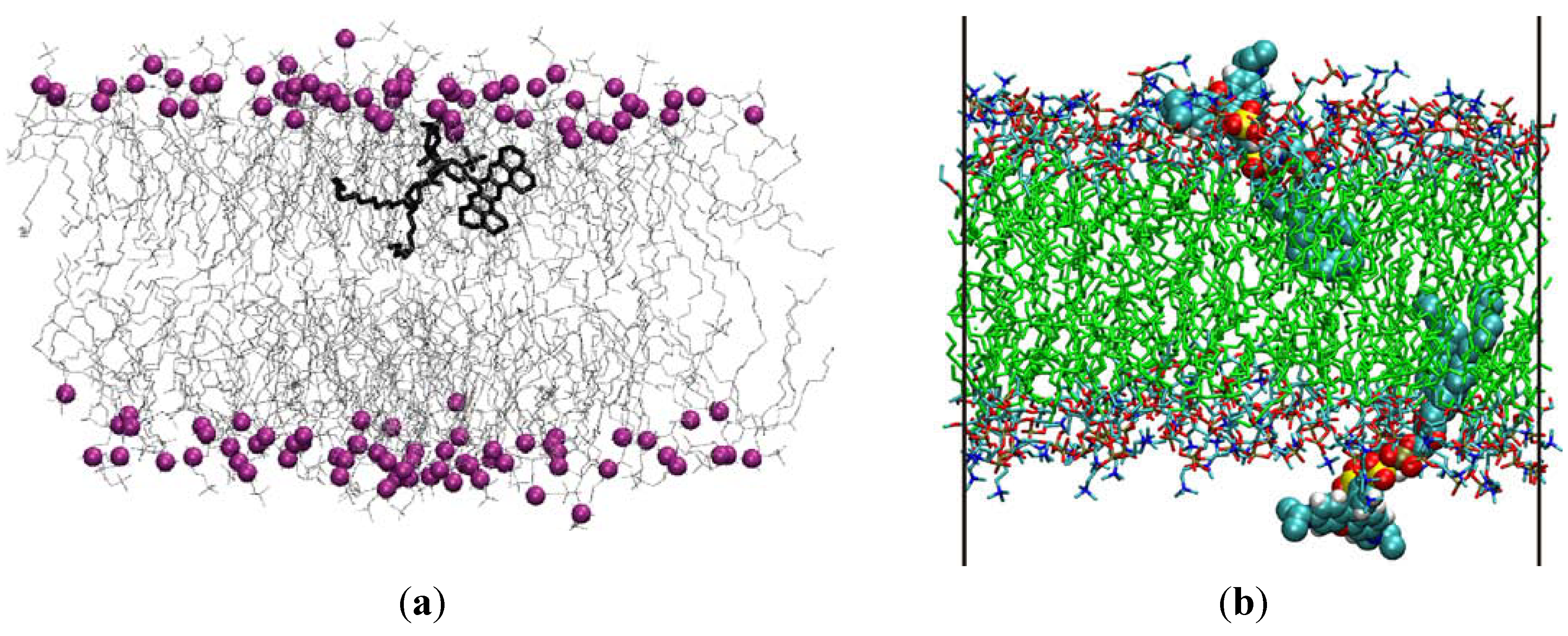
2.1. Apolar Fluorophores

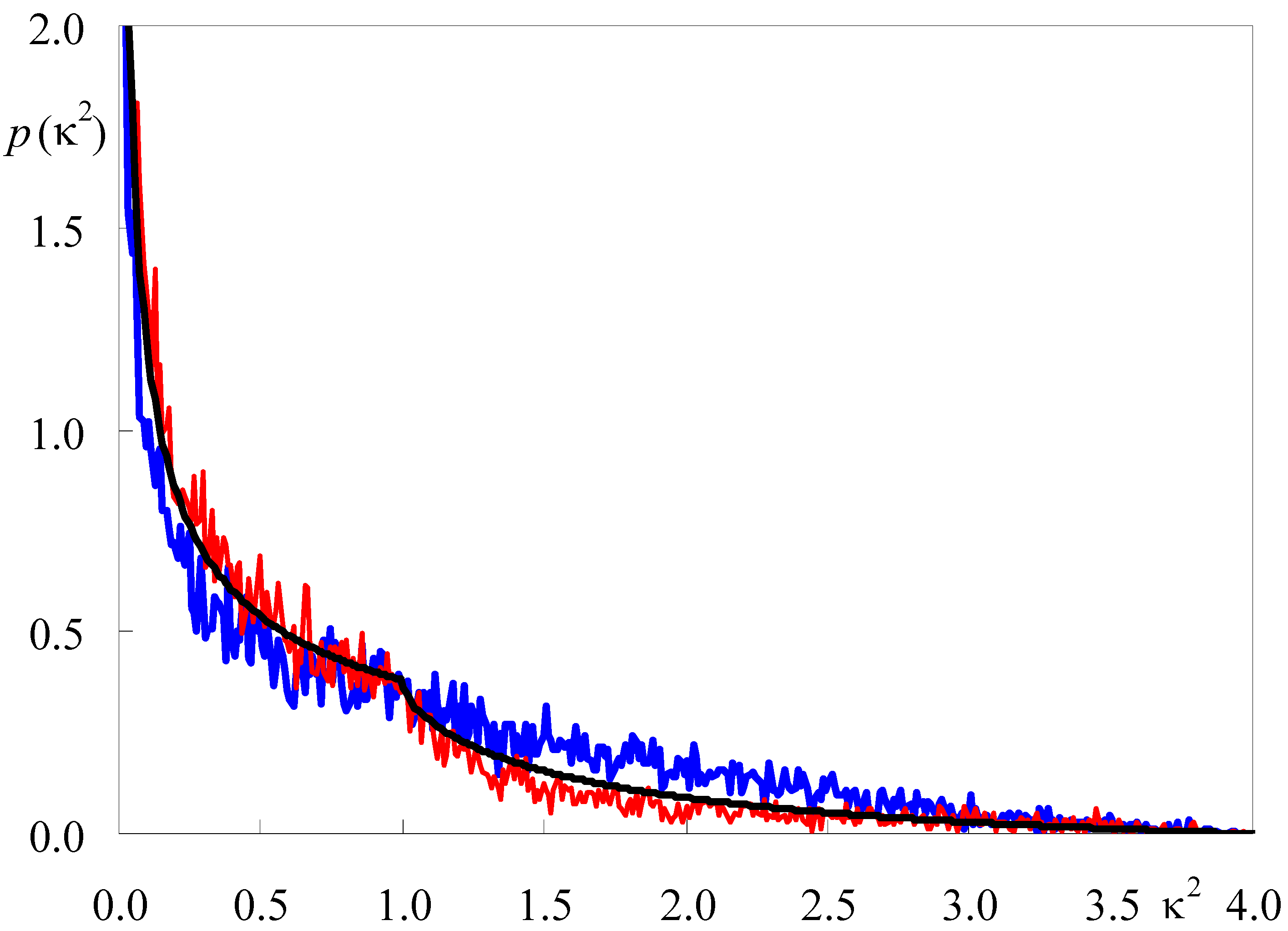
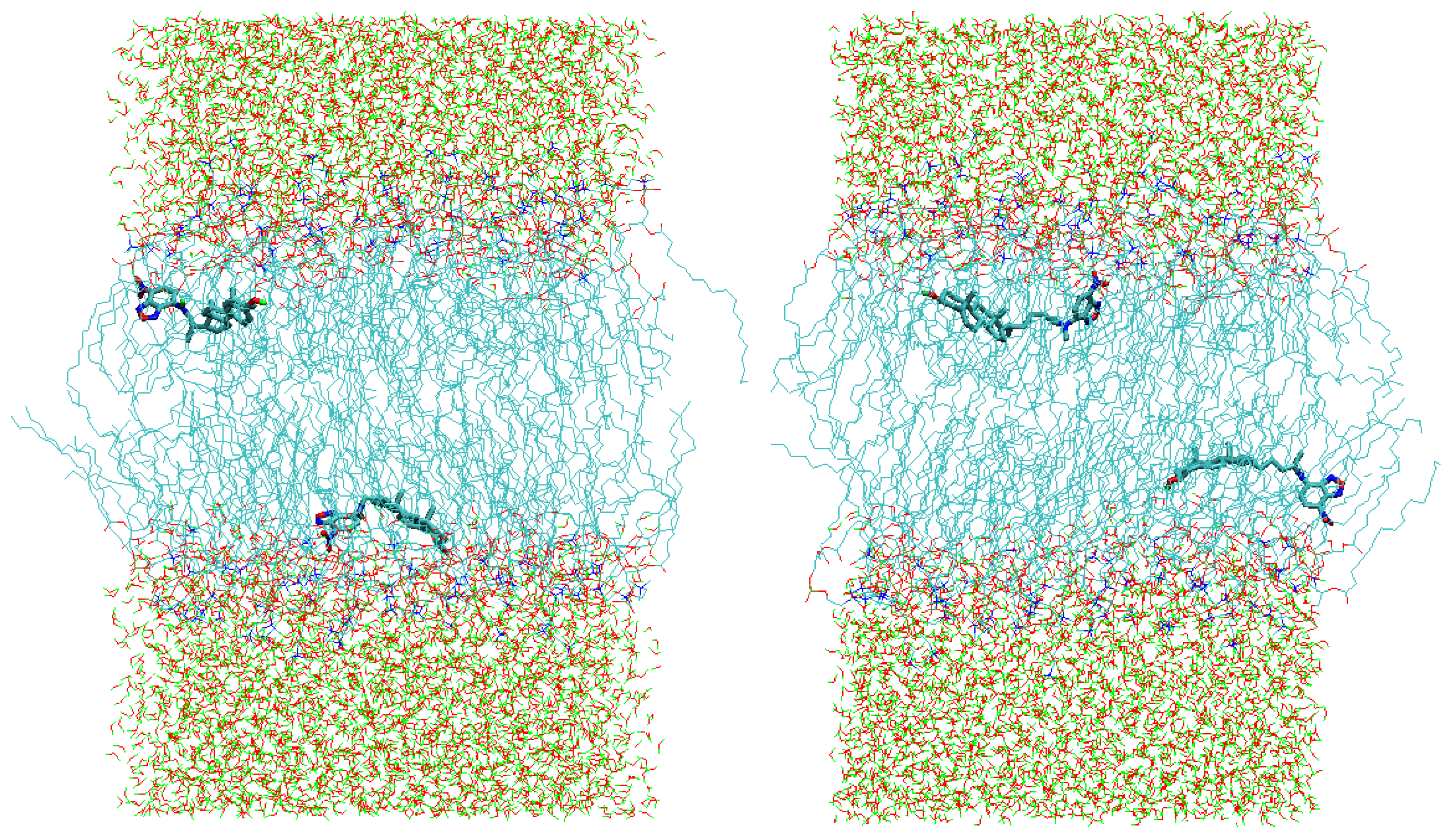
2.2. Polar Fluorophores



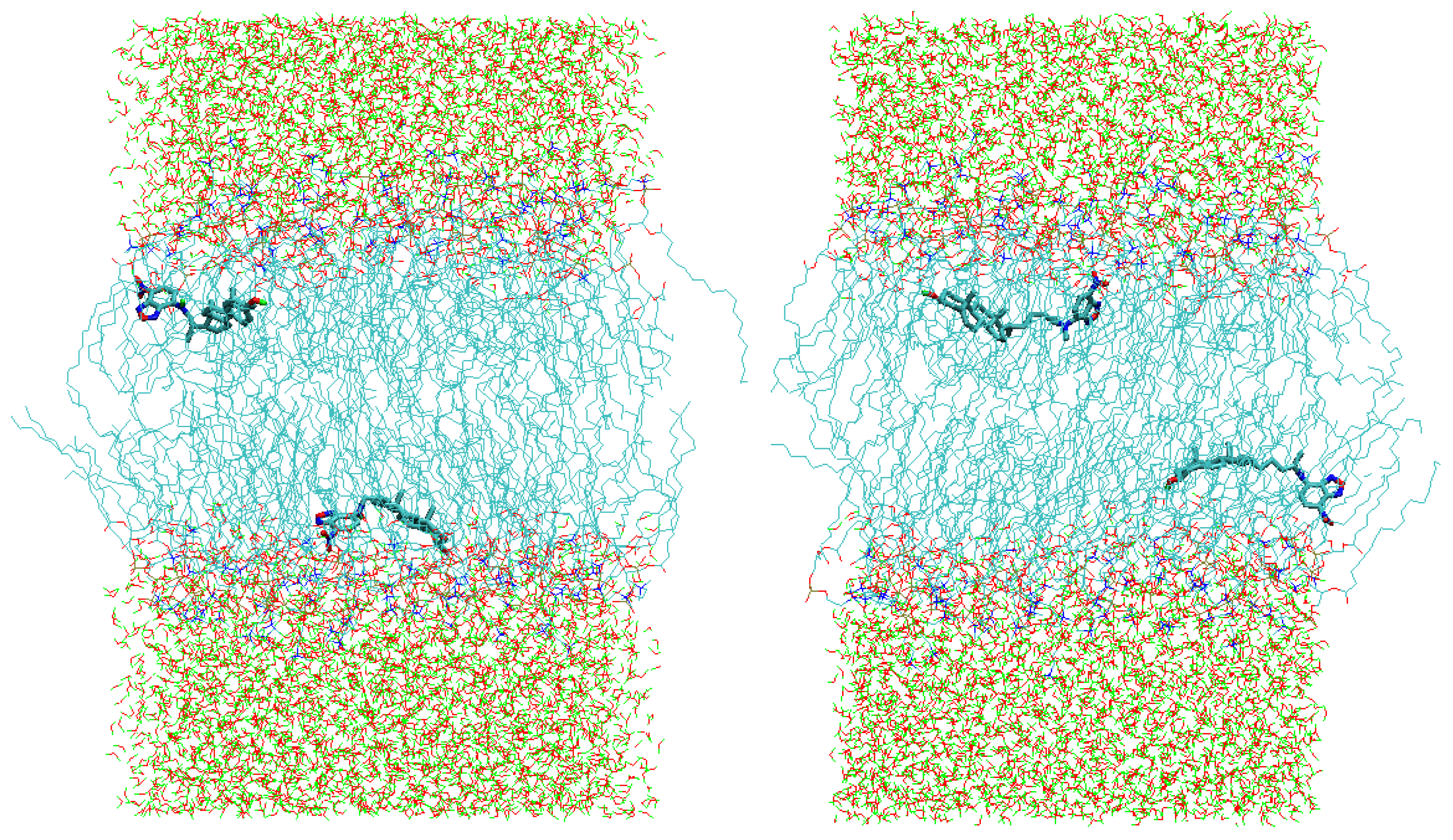

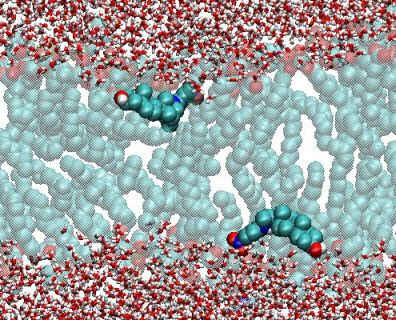{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
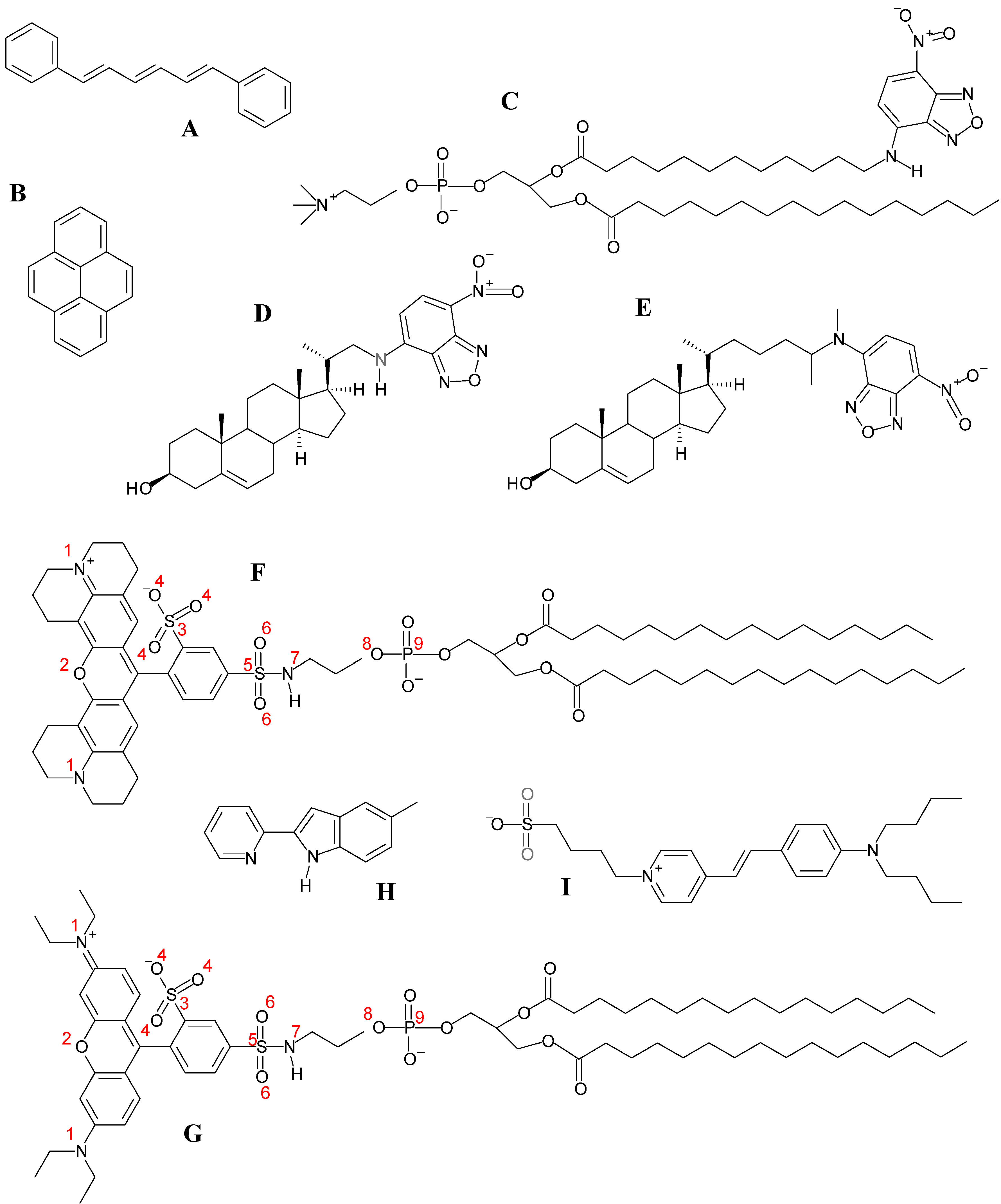
| Atom number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| TR-DHPE | −0.035, | −0.368 | 0.950 | −0.519, | 0.979 | −0.537, | −0.174 | −0.421 | 1.202 |
| 0.018 | −0.591, | −0.558 | |||||||
| −0.600 | |||||||||
| Rhod-DPPE | −0.8 | −0.6 | 1.6 | −0.8 | 1.6 | −0.7 | −0.5 | −0.8 | 1.7 |

3. Coarse-Grained Simulations of Dyes in Lipid Bilayers
4. Conclusions
Acknowledgements
References
- Gennis, R.B. Biomembranes: Molecular Structure and Function; Springer-Verlag: New York, NY, USA, 1989. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed; Springer: New York, NY, USA, 2006. [Google Scholar]
- Royer, C.A.; Scarlata, S.F. Fluorescence approaches to quantifying biomolecular interactions. Methods Enzymol. 2008, 450, 79–106. [Google Scholar]
- de Almeida, R.F.; Loura, L.M.S.; Prieto, M. Membrane lipid domains and rafts: current applications of fluorescence lifetime spectroscopy and imaging. Chem. Phys. Lipids 2009, 157, 61–77. [Google Scholar] [CrossRef]
- Bouvrais, H.; Pott, T.; Bagatolli, L.A.; Ipsen, J.H.; Méléard, P. Impact of membrane-anchored fluorescent probes on the mechanical properties of lipid bilayers. Biochim. Biophys. Acta 2010, 1798, 1333–1337. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Prates Ramalho, J.P. Fluorescent membrane probes’ behaviour in lipid bilayers: Insights from molecular dynamics simulations. Biophys. Rev. 2009, 1, 141–148. [Google Scholar] [CrossRef]
- Lentz, B.R. Membrane “fluidity” as detected by diphenylhexatriene probes. Chem. Phys. Lipids 1989, 50, 171–190. [Google Scholar] [CrossRef]
- Lentz, B.R. Use of fluorescence probes to monitor molecular order and motions within liposome bilayers. Chem. Phys. Lipids 1993, 64, 99–116. [Google Scholar] [CrossRef]
- Straume, M.; Litman, B.J. Equilibrium and dynamic structure of large, unilamellar, unsaturated acyl chain phosphatidylcholine vesicles. Higher order analysis of 1,6-diphenyl-1,3,5-hexatriene and 1-[4-(trimethylammonio)phenyl]-6-phenyl-1,3,5-hexatriene anisotropy decay. Biochemistry 1987, 26, 5113–5120, Erratum in: Biochemistry 1987, 26, 8030. [Google Scholar] [CrossRef]
- Mitchell, D.C.; Litman, B.J. Molecular order and dynamics in bilayers consisting of highly polyunsaturated phospholipids. Biophys. J. 1998, 74, 879–891. [Google Scholar] [CrossRef]
- López Cascales, J.J.; Huertas, M.L.; García de la Torre, J. Molecular dynamics simulation of a dye molecule in the interior of a bilayer: 1,6-diphenyl-1,3,5-hexatriene in dipalmitoylphosphatidylcholine. Biophys. Chem. 1997, 69, 1–8. [Google Scholar] [CrossRef]
- Repáková, J.; Čapková, P.; Holopainen, J.M.; Vattulainen, I. Distribution, orientation, and dynamics of DPH probes in DPPC bilayer. J. Phys. Chem. B 2004, 108, 13438–13448. [Google Scholar]
- Kaiser, R.D.; London, E. Location of diphenylhexatriene (DPH) and its derivatives within membranes: comparison of different fluorescence quenching analyses of membrane depth. Biochemistry 1998, 37, 8180–8190. [Google Scholar] [CrossRef]
- Repáková, J.; Holopainen, J.M.; Morrow, M.R.; McDonald, M.C.; Čapková, P.; Vattulainen, I. Influence of DPH on the structure and dynamics of a DPPC bilayer. Biophys. J. 2005, 88, 3398–3410. [Google Scholar] [CrossRef]
- Franová, M.; Repáková, J.; Čapková, P.; Holopainen, J.M.; Vattulainen, I. Effects of DPH on DPPC-cholesterol membranes with varying concentrations of cholesterol: from local perturbations to limitations in fluorescence anisotropy experiments. J. Phys. Chem. B 2010, 114, 2704–2711. [Google Scholar] [CrossRef]
- Somerharju, P. Pyrene-labeled lipids as tools in membrane biophysics and cell biology. Chem. Phys. Lipids 2002, 116, 57–74. [Google Scholar] [CrossRef]
- Hoff, B.; Strandberg, E; Ulrich, A.S.; Tieleman, D.P.; Posten, C. 2H-NMR study and molecular dynamics simulation of the location, alignment, and mobility of pyrene in POPC bilayers. Biophys. J. 2005, 88, 1818–1827. [Google Scholar] [CrossRef]
- Čurdová, J.; Čapková, P.; Plášek, J.; Repáková, J.; Vattulainen, I. Free pyrene probes in gel and fluid membranes: perspective through atomistic simulations. J. Phys. Chem. B 2007, 111, 3640–3650. [Google Scholar] [CrossRef]
- Repáková, J.; Holopainen, J.M.; Karttunen, M.; Vattulainen, I. Influence of pyrene-labeling on fluid lipid membranes. J. Phys. Chem. B 2006, 110, 15403–15410. [Google Scholar]
- Loura, L.M.S.; Prates Ramalho, J.P. Location and dynamics of acyl chain NBD-labeled phosphatidylcholine (NBD-PC) in DPPC bilayers. A molecular dynamics and time-resolved fluorescence anisotropy study. Biochim. Biophys. Acta 2007, 1768, 467–478. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Fernandes, F.; Fernandes, A.C.; Prates Ramalho, J.P. Effects of fluorescent probe NBD-PC on the structure, dynamics and phase transition of DPPC. A molecular dynamics and differential scanning calorimetry study. Biochim. Biophys. Acta 2008, 1778, 491–501. [Google Scholar] [CrossRef]
- Gullapalli, R.R.; Demirel, M.C.; Butler, P.J. Molecular dynamics simulations of DiI-C18(3) in a DPPC lipid bilayer. Phys. Chem. Chem. Phys. 2008, 10, 3548–3560. [Google Scholar]
- Muddana, H.S.; Gullapalli, R.R.; Manias, E.; Butler, P.J. Atomistic simulation of lipid and DiI dynamics in membrane bilayers under tension. Phys. Chem. Chem. Phys. 2011, 13, 1368–1378. [Google Scholar]
- Hölttä-Vuori, M.; Uronen, R.L.; Repáková, J.; Salonen, E.; Vattulainen, I.; Panula, P.; Li, Z.; Bittman, R.; Ikonen, E. BODIPY-cholesterol: A new tool to visualize sterol trafficking in living cells and organisms. Traffic 2008, 9, 1839–1849. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Palace Carvalho, A.J.; Prates Ramalho, J.P. Direct calculation of Förster orientation factor of membrane probes by molecular simulation. J. Mol. Struct. (Theochem) 2010, 946, 107–112. [Google Scholar] [CrossRef]
- Van Der Meer, B.W.; Coker, G., III; Chen, S.-Y.S. Resonance Energy Transfer: Theory and Data; VCH: New York, NY, USA, 1994. [Google Scholar]
- Loura, L.M.S.; Prieto, M.; Fernandes, F. Quantification of protein-lipid selectivity using FRET. Eur. Biophys. J. 2010, 39, 565–578. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Fernandes, F.; Prieto, M. Membrane microheterogeneity: Förster resonance energy transfer characterization of lateral membrane domains. Eur. Biophys. J. 2010, 39, 565–578. [Google Scholar] [CrossRef]
- Stryer, L. Fluorescence energy transfer as a spectroscopic ruler. Ann. Rev. Biochem. 1978, 47, 829–846. [Google Scholar]
- Dale, R.E.; Eisinger, J.; Blumberg, W.E. The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys. J. 1979, 26, 161–193, Erratum in: Biophys. J. 1980, 30, 365. [Google Scholar] [CrossRef]
- Harriman, A.; Izzet, G.; Ziessel, R. Rapid energy transfer in cascade-type bodipy dyes. J. Am. Chem. Soc. 2006, 128, 10868–10875. [Google Scholar] [CrossRef]
- Cardoso, R.M.; Filipe, H.A.; Gomes, F.; Moreira, N.D.; Vaz, W.L.; Moreno, M.J. Chain length effect on the binding of amphiphiles to serum albumin and to POPC bilayers. J. Phys. Chem. B 2010, 114, 16337–16346. [Google Scholar]
- Gimpl, G.; Gehrig-Burger, K. Probes for studying cholesterol binding and cell biology. Steroids 2011, 76, 216–231. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Fedorov, A.; Prieto, M. Exclusion of a cholesterol analog from the cholesterol-rich phase in model membranes. Biochim. Biophys. Acta 2001, 1511, 236–243. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; London, E. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 1987, 26, 39–45. [Google Scholar] [CrossRef]
- Scheidt, H.A.; Müller, P.; Herrmann, A; Huster, D. The potential of fluorescent and spin-labeled steroid analogs to mimic natural cholesterol. J. Biol. Chem. 2003, 278, 45563–45569. [Google Scholar]
- Franks, N.P. Structural analysis of hydrated egg lecithin and cholesterol bilayers. I. X-ray diffraction. J. Mol. Biol. 1976, 100, 345–358. [Google Scholar] [CrossRef]
- Worcester, D.L.; Franks, N.P. Structural analysis of hydrated egg lecithin and cholesterol bilayers. II. Neutrol diffraction. J. Mol. Biol. 1976, 100, 359–378. [Google Scholar] [CrossRef]
- Skaug, M.J.; Longo, M.L.; Faller, R. Computational studies of Texas Red-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine - model building and applications. J. Phys. Chem. B 2009, 113, 8758–8766. [Google Scholar] [CrossRef]
- Kyrychenko, A. A molecular dynamics model of rhodamine-labeled phospholipid incorporated into a lipid bilayer. Chem. Phys. Lett. 2010, 485, 95–99. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz, K.M.; Kollman, P.A. Atomic Charges Derived from Semiempirical Methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Kachel, K.; Asuncion-Punzalan, E.; London, E. The location of fluorescence probes with charged groups in model membranes. Biochim. Biophys. Acta 1998, 1374, 63–76. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Wu, F.; Thummel, R.P.; Waluk, J.; Ladokhin, A.S. Partitioning and localization of environment-sensitive 2-(2'-pyridyl)- and 2-(2'-pyrimidyl)-indoles in lipid membranes: A joint refinement using fluorescence measurements and molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 13574–13584. [Google Scholar] [CrossRef]
- Kyrychenko, A.; Sevriukov, I.Y.; Syzova, Z.A.; Ladokhin, A.S.; Doroshenko, A.O. Partitioning of 2,6-Bis(1H-Benzimidazol-2-yl)pyridine fluorophore into a phospholipid bilayer: Complementary use of fluorescence quenching studies and molecular dynamics simulations. Biophys. Chem. 2011, 154, 8–17. [Google Scholar] [CrossRef]
- Norman, K.E.; Nymeyer, H. Indole localization in lipid membranes revealed by molecular simulation. Biophys. J. 2006, 91, 2046–2054. [Google Scholar] [CrossRef]
- MacCallum, J.L.; Bennett, W.F.D.; Tieleman, D.P. Partitioning of amino acid side chains into lipid bilayers: results from computer simulations and comparison to experiment. J. Gen. Physiol. 2007, 129, 371–377. [Google Scholar] [CrossRef]
- MacCallum, J.L.; Bennett, W.F.D.; Tieleman, D.P. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys. J. 2006, 94, 3393–3404. [Google Scholar]
- Hinner, M.J.; Marrink, S.J.; de Vries, A.H. Location, tilt, and binding: a molecular dynamics study of voltage-sensitive dyes in biomembrane. J. Phys. Chem. B 2009, 113, 15807–15819. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI forcefield: coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Loura, L.M.S.; Ramalho, J.P.P. Recent Developments in Molecular Dynamics Simulations of Fluorescent Membrane Probes. Molecules 2011, 16, 5437-5452. https://doi.org/10.3390/molecules16075437
Loura LMS, Ramalho JPP. Recent Developments in Molecular Dynamics Simulations of Fluorescent Membrane Probes. Molecules. 2011; 16(7):5437-5452. https://doi.org/10.3390/molecules16075437
Chicago/Turabian StyleLoura, Luís M. S., and J. P. Prates Ramalho. 2011. "Recent Developments in Molecular Dynamics Simulations of Fluorescent Membrane Probes" Molecules 16, no. 7: 5437-5452. https://doi.org/10.3390/molecules16075437
APA StyleLoura, L. M. S., & Ramalho, J. P. P. (2011). Recent Developments in Molecular Dynamics Simulations of Fluorescent Membrane Probes. Molecules, 16(7), 5437-5452. https://doi.org/10.3390/molecules16075437