A Practical Approach to New (5Z) 2-Alkylthio-5-arylmethylene-1-methyl-1,5-dihydro-4H-imidazol-4-one Derivatives

Abstract
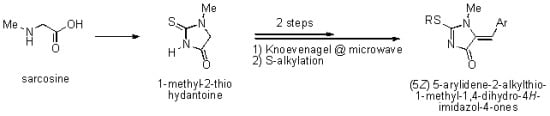
:
1. Introduction

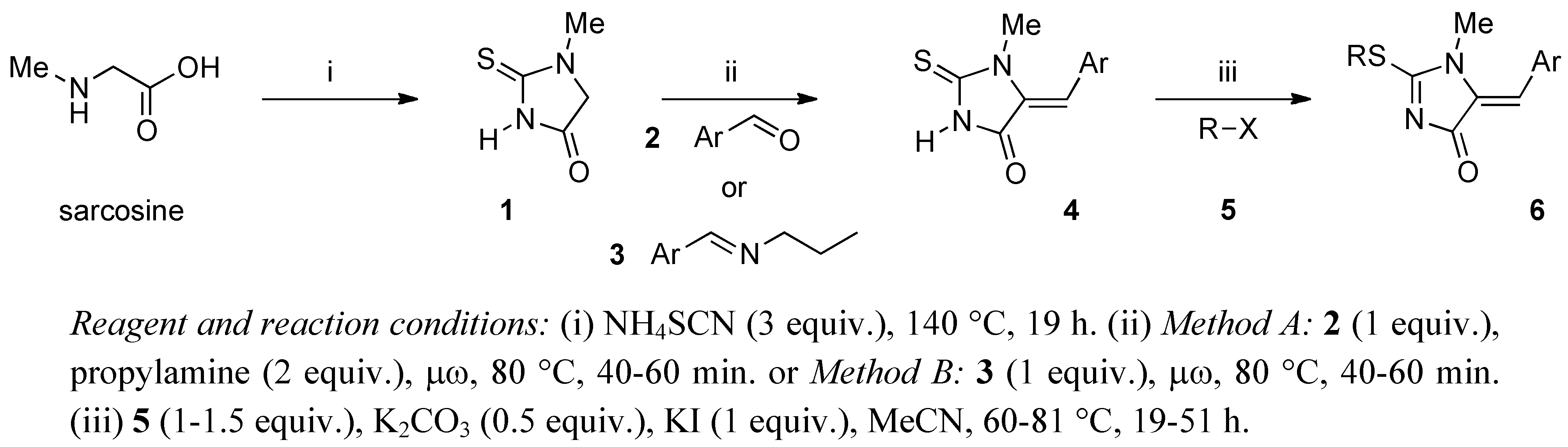
2. Results and Discussion



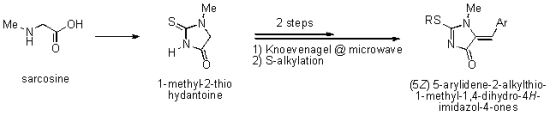{kind=link}
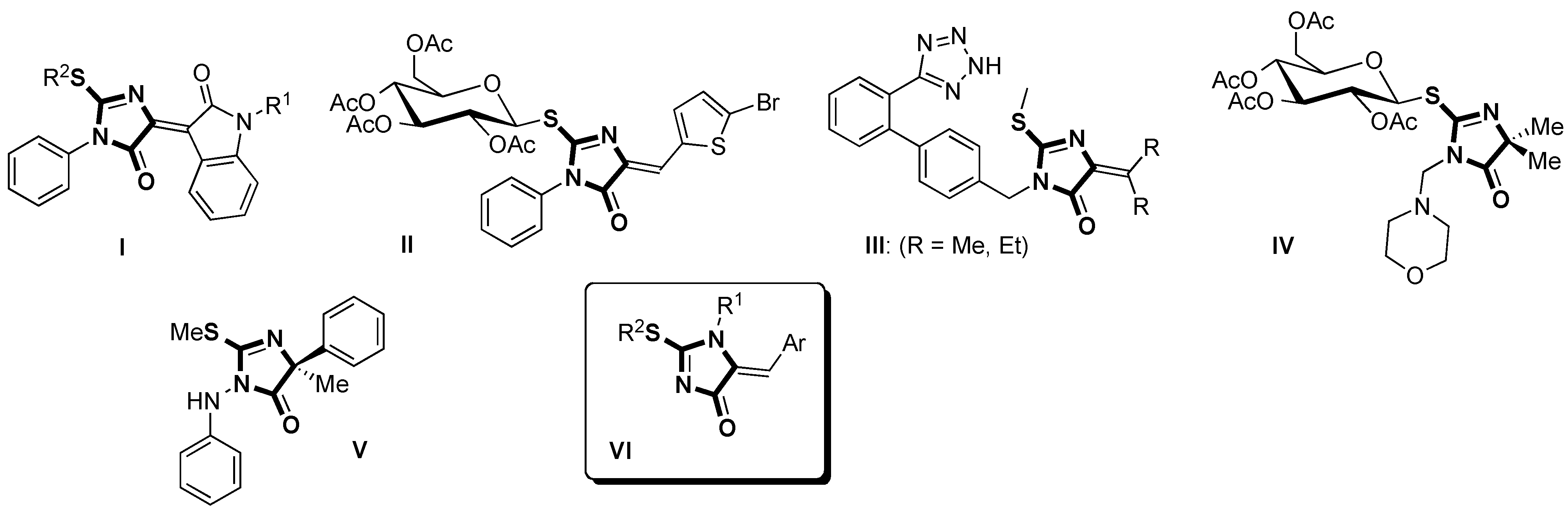{kind=link}
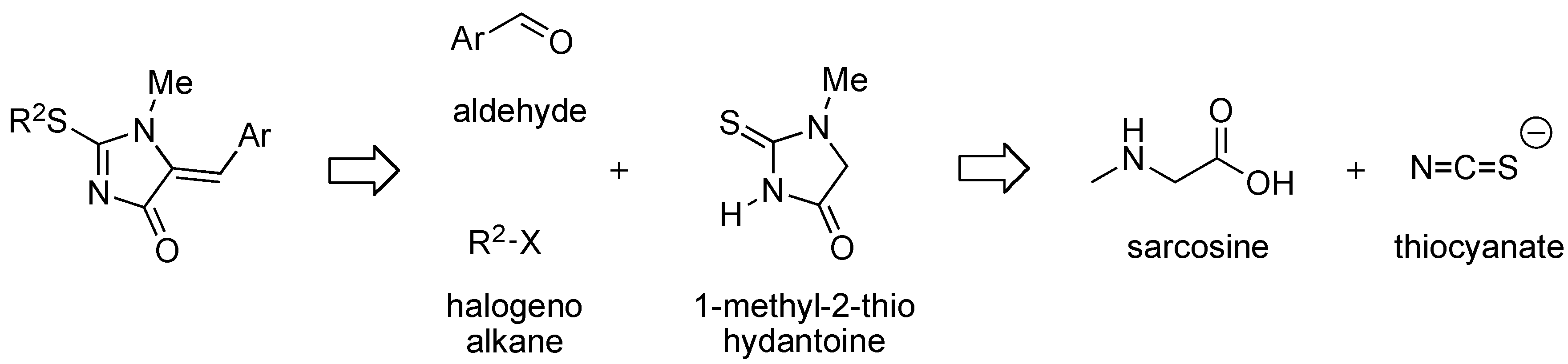{kind=link}
{kind=link}
| Product | Method A | Method B | ||
|---|---|---|---|---|
| Starting reagent | Yield a of 4 (%) | Starting reagent | Yield a of 4 (%) | |
| 4a | 2a | 96 | 3a | 88 |
| 4b | 2b | 96 | 3b | 92 |
| 4c | 2c | 90 | 3c | 90 |
| 4d | 2d | 86 | 3d | -b |
| 4e | 2e | 98 | 3e | 92 |
 | ||||
| Product 6 | R | Starting product 4 | Reagent 5 | Reaction conditions: temperature a and reaction time | Yield b of 6 (%) |
|---|---|---|---|---|---|
| 6a | CH3CH2 | 4a | 5a | 60 °C, 51 h | 60 |
| 6b | CH2=CH-CH2 | 4a | 5b | 66 °C, 48 h | 65 |
| 6c | CH3CH2CH2 | 4a | 5c | 65 °C, 24 h | 62 |
| 6d | NC-CH2 | 4a | 5d | 81 °C, 19h | 66 |
| 6e | EtO2C-CH2 | 4a | 5e | 60 °C, 14 h | 68 |
| 6f | EtO2C-CH2 | 4b | 5e | 60 °C, 19 h | 62 |
| 6g | HOCH2CH2CH2 | 4a | 5f | 70 °C, 46 h | 42 |
 | |||||
3. Experimental
3.1. General
3.2. General Procedure for the Solventless Synthesis of (5Z) 5-Arylmethylene-1-methyl-2-thioxo-imidazolin-4-ones 4 under Microwave Dielectric Heating according to Method A and Method B:
3.3. General Procedure for the Synthesis of Compounds 6 by S-Alkylation of 5-Arylmethylene-1-methyl-2-thioxo imidazolin-4-ones 4(a-g) with Halogeno Compounds 5
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Khodair, A.I.A.; El-Ashry, E.S.H.; Al-Masoudi, N.A.L. Synthesis of 5-spirocyclohexyl-2,4-dithiohydantoin derivatives: A potential anti-leishmaniasis agent. Monat. Chem. 2004, 135, 1061–1079. [Google Scholar]
- Ware, E. The chemistry of hydantoins. Chem. Rev. 1950, 46, 403–470. [Google Scholar] [CrossRef]
- Chazeau, V.; Cussac, M.; Boucherle, A. Synthesis and immunomodulating activity of 1-amino-2-thiohydantoin derivatives. Eur. J. Med. Chem. 1992, 27, 839–843. [Google Scholar] [CrossRef]
- Al-Obaid, A.M.; El-Subagh, H.I.; Khodair, A.I.A.; Elmazar, M.M.A. 5-substituted-2-thiohydantoin analogs as a novel class of antitumor agents. Anticancer Drugs 1996, 7, 873–880. [Google Scholar] [CrossRef]
- Okasaki, T.; Kikuchi, K.; Watanabe, T.; Suga, A.; Shibasaki, M.; Fujimori, A.; Inagaki, O.; Yanagisawa, I. Studies on Nonpeptide Angiotensin II Receptor Antagonists. III. Synthesis and Biological Evaluation of 5-Alkylidene-3, 5-dihydro-4H-imidazol-4-one-Derivatives. Chem. Pharm. Bull. 1998, 46, 777–781. [Google Scholar] [CrossRef]
- Aly, Y.L.; El-Barbary, A.A.; Hashem, A.F.M.; El-Shehawy, A.A. Alkylation of Thiohydantoins Including Synthesis, Conformational and Configurational Studies of Some Acetylated S-Pyranosides. Phosph.Sulfur Silicon 2004, 179, 185–202. [Google Scholar] [CrossRef]
- Morton, J.; Enisz, J.; Hasztafi, S.; Timar, T. Preparation and fungicidal activity of 5-substituted hydantoins and their 2-thio analogs. J. Agric. Food Chem. 1993, 41, 148–152. [Google Scholar] [CrossRef]
- Bascou, J.P.; Lacroix, G.; Gadras, A.; Perez, J. Dérivés optiquement actifs de 2-imidazoline-5-ones et 2-imidazoline-5-thiones fongicides. EP Patent 0 629 616 A2, 1994. [Google Scholar]
- Ding, M-W.; Fu, B-Q.; Yuan, J.-Z. Facile synthesis of 2-alkylthio-3-amino-4 H-imidazol-4-ones and 2H-imidazo[2,1-b]-1,3,4-thiadiazin-6(7H)-ones via N-vinylic iminophosphorane. Heteroatom Chem. 2005, 16, 76–80. [Google Scholar] [CrossRef]
- Hital, K.; Fuchikami, T.; Fujita, A.; Hirose, H.; Yokota, M.; Nakato, S. Preparation of phenylhydantoin derivatives as herbicides. EP Patent 262 428, 1988. [Google Scholar]
- Han, J.; Wang, J.; Dong, H.; Lei, J.; Wang, M.; Fang, J. Synthesis and herbicidal activity of 5-(4-hydroxybenzyl)-2-thioxoimidazolidin-4-one esters. Molecules 2011, 16, 2833–2845. [Google Scholar] [CrossRef]
- Unangst, P.C.; Connor, D.T.; Cotenko, W.A.; Sorenson, R.J.; Kostlan, C.R.; Sircor, J.C.; Wright, C.D.; Schrier, D.J.; Dyer, R.D. Synthesis and biological evaluation of 5-[[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]methylene]oxazoles, -thiazoles, and -imidazoles: novel dual 5-lipoxygenase and cyclooxygenase inhibitors with antiinflammatory activity. J. Med. Chem. 1994, 37, 322–328. [Google Scholar] [CrossRef]
- Belleau, B.; Brasili, L.; Chan, L.; Di Marco, M.P.; Zacharie, B.; Nguyen-Ba, N.; Jenkinson, H.J.; Coates, J.A.V.; Cameron, J.M. A novel class of 1,3-oxathiolane nucleoside analogues having potent anti-HIV activity. Bioorg. Med. Chem. Lett. 1993, 3, 1723–1728. [Google Scholar] [CrossRef]
- Attardo, G.; Tripathy, S.; Gagnon, M. Preparation of arylmethylidene heterocycles as novel analgesics. Patent WO 2009/097 695 A1, 2009. [Google Scholar]
- Bazureau, J.P.; Carreaux, F.; Renault, S.; Meijer, L.; Lozach, O. Imidazolone derivatives, preparation method thereof and biological use of same. Patent WO 2009/05032 A2, 2009. [Google Scholar]
- Roué, N.; Bergman, J. Synthesis of the marine alkaloid leucettamine B. Tetrahedron 1999, 55, 14729–14738. [Google Scholar] [CrossRef]
- Molina, P.; Torroga, A.; Lidon, M.J. Preparation and thermal ring-closure of β-aryl vinyl carbodi-imides: synthesis of isoquinoline derivatives. J. Chem. Soc. Perkin Trans 1 1990, 1727–1731. [Google Scholar]
- Staudinger, H.; Hauser, E. Über neue organische phosphorverbindungen IV phosphinimine. Helv. Chim. Acta 1921, 4, 861–886. [Google Scholar] [CrossRef]
- Ding, M.-W.; Cheng, L.; Fu, B.-Q. New facile synthesis of imidazo[2,1-b]-1,3,4-thiadiazol-5(6H)-ones via aza-Wittig reaction. Synthesis 2004, 1067–1071. [Google Scholar]
- Ding, M.-W.; Cheng, L.; Fu, B.-Q. An efficient synthesis of 2-alkylthio- 5-phenylmethylidene-4 H -imidazolin-4-ones. Synth. Commun. 2003, 33, 1267–1284. [Google Scholar] [CrossRef]
- Rowley, G.L.; Greenleaf, A.L.; Kenyon, G.L. On the specificity of creatine kinase. New glycocyamines and glycocyamine analogs related to creatine. J. Am. Chem. Soc. 1971, 93, 5542–5551. [Google Scholar] [CrossRef]
- Reddick, R.E.; Kenyon, G.L. Syntheses and NMR studies of specifically labeled [2-15N]phosphocreatine, [2-15N]creatinine, and related 15N-labeled compounds. J. Am. Chem. Soc. 1987, 109, 4380–4387. [Google Scholar] [CrossRef]
- Porwal, S.; Chauhan, S.; Chauhan, P.M.S.; Shakya, N.; Verma, A.; Gupta, S. Discovery of novel antileishmanial agents in an attempt to synthesize pentamidine-aplysinopsin hybrid molecule. J. Med. Chem. 2009, 52, 5793–5802. [Google Scholar] [CrossRef]
- Majouga, A.G.; Beloglazkina, E.K.; Vatsadze, S.Z.; Frolova, N.A.; Zyk, N.K. Synthesis of isomeric 3-phenyl-5-(pyridylmethylene)-2-thiohydantoins and their S-methylated derivatives. Molecular and crystal structures of (5Z)-3-phenyl-5-(pyridylmethylene)-2-thiohydantoin and (5Z)-2-methylthio-3-phenyl-5-(pyridin-2-ylmethylene)3,5-dihydro-4H-imidazol-4-one. Russ. Chem. Bull. Int. Ed. 2004, 2850–2855. [Google Scholar]
- El-Barbary, A.A.; Khodair, A.I.; Pedersen, E.B.; Nielsen, C. S-Glycosylated Hydantoins as New Potential Antiviral Agents. J. Med. Chem. 1994, 37, 73–77. [Google Scholar] [CrossRef]
- Krstenansky, J.L.; Cotteril, I. Recent advances in microwave-assisted organic syntheses. Curr. Opin. Drug Discov. Dev. 2000, 3, 454–461. [Google Scholar]
- Larhed, M.; Hallberg, A. Microwave-assisted high speed chemistry: A new technique in drug disvovery. Drug Discov. Today 2001, 6, 406–416. [Google Scholar] [CrossRef]
- Ben Alloum, A.; Villemin, D. Potassium fluoride on alumina: Condensation of 3-methyl-2-thiono-4-thiazolidinone with aldehydes. Synthesis of α-thioacrylic acids and phosphonothionothiazolidinones. Phosph. Sulfur Silicon Relat. Elem. 1993, 79, 33–37. [Google Scholar] [CrossRef]
- Ben Alloum, A.; Labiad, B.; Villemin, D. Application of microwave heating techniques for dry organic reactions. J. Chem. Soc., Chem. Commun. 1989, 386–387. [Google Scholar]
- Commarmot, R.; Didenot, R.; Gardais, J.F. Temperatures are measured by an IR captor. Prolabo. Fr. Demande 1985. [Chem. Abstr. 1986,105, 17442]. [Google Scholar]
- Renault, S.; Bertrand, S.; Carreaux, F.; Bazureau, J.P. Parallel solution phase synthesis of 2-alkylthio-3,5-dihydro-4H-imidazol-4-one by one-pot three component domino reaction. J. Comb. Chem. 2007, 9, 935–942. [Google Scholar] [CrossRef]
- Lerestif, J.M.; Perrocheau, J.; Tonnard, F.; Bazureau, J.P.; Hamelin, J. 1,3-Dipolar cycloaddition of imidate ylides on imino-alcohols : synthesis of new imidazolones using solvent free conditions. Tetrahedron 1995, 51, 6757–6774. [Google Scholar] [CrossRef]
- Martin, J.; Villemin, D. Dry Condensation of Creatinine with Aldehydes Under Focused Microwave Irradiation. Synth. Commun. 1995, 25, 3135–3140. [Google Scholar] [CrossRef]
- Primot, A.; Baratte, B.; Gompel, M.; Borgne, A.; Liabeuf, S.; Romette, J.L.; F. Costantini, F.; Meijer, L. Purification of GSK-3 by affinity chromatography on immobilized axin. Protein Expr. Purif. 2000, 20, 394–404. [Google Scholar] [CrossRef]
- Borgne, A.; Meijer, L. Sequential dephosphorylation of p34cdc2 on Thr-14 and Tyr-15 at the prophase/metaphase transition. J. Biol. Chem. 1996, 271, 27847–27854. [Google Scholar] [CrossRef]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S.H. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bourahla, K.; Paquin, L.; Lozach, O.; Meijer, L.; Carreaux, F.; Bazureau, J.P. A Practical Approach to New (5Z) 2-Alkylthio-5-arylmethylene-1-methyl-1,5-dihydro-4H-imidazol-4-one Derivatives. Molecules 2011, 16, 7377-7390. https://doi.org/10.3390/molecules16097377
Bourahla K, Paquin L, Lozach O, Meijer L, Carreaux F, Bazureau JP. A Practical Approach to New (5Z) 2-Alkylthio-5-arylmethylene-1-methyl-1,5-dihydro-4H-imidazol-4-one Derivatives. Molecules. 2011; 16(9):7377-7390. https://doi.org/10.3390/molecules16097377
Chicago/Turabian StyleBourahla, Khadidja, Ludovic Paquin, Olivier Lozach, Laurent Meijer, François Carreaux, and Jean Pierre Bazureau. 2011. "A Practical Approach to New (5Z) 2-Alkylthio-5-arylmethylene-1-methyl-1,5-dihydro-4H-imidazol-4-one Derivatives" Molecules 16, no. 9: 7377-7390. https://doi.org/10.3390/molecules16097377