Recent Advances in Microflow Photochemistry



Abstract
:
1. Introduction

2. Reactor Comparison




3. Photochemical Reactions in Microreactors
3.1. Homogeneous Reactions
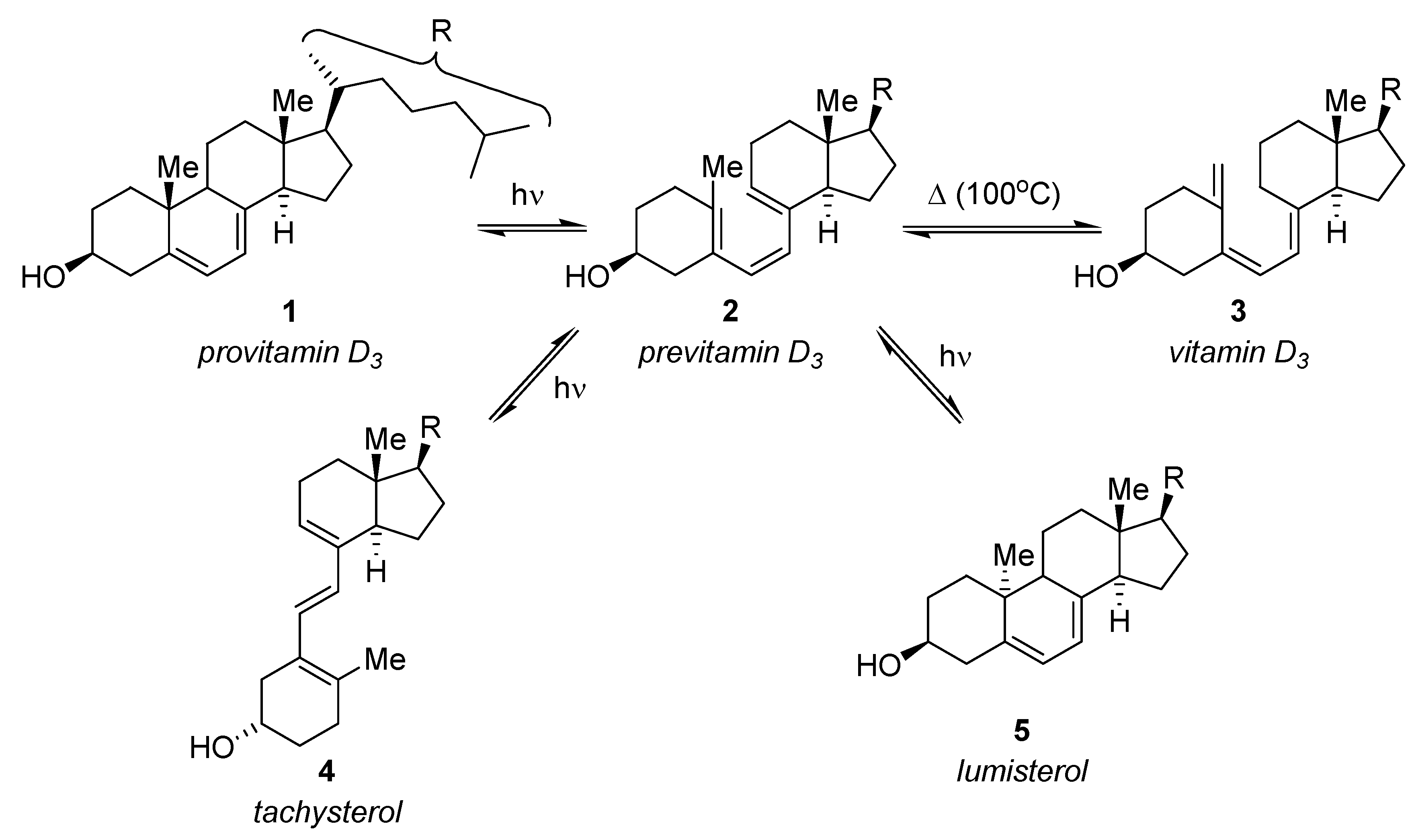
3.1.1. Synthesis of Vitamin D3


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
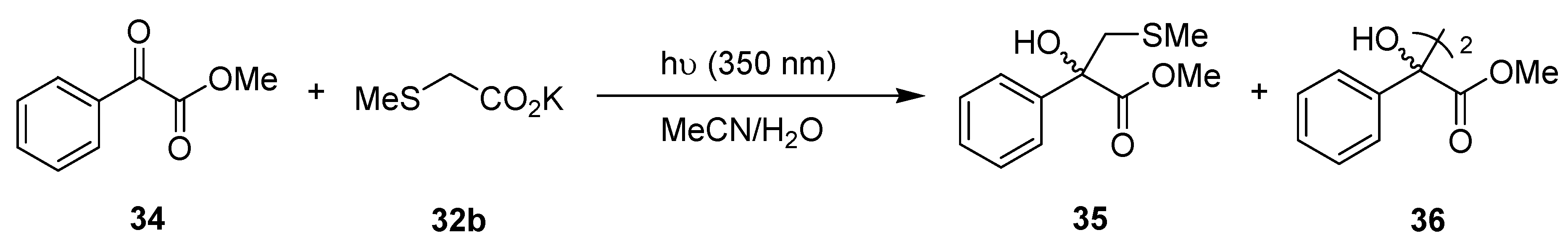{kind=link}
{kind=link}
{kind=link}
{kind=link}
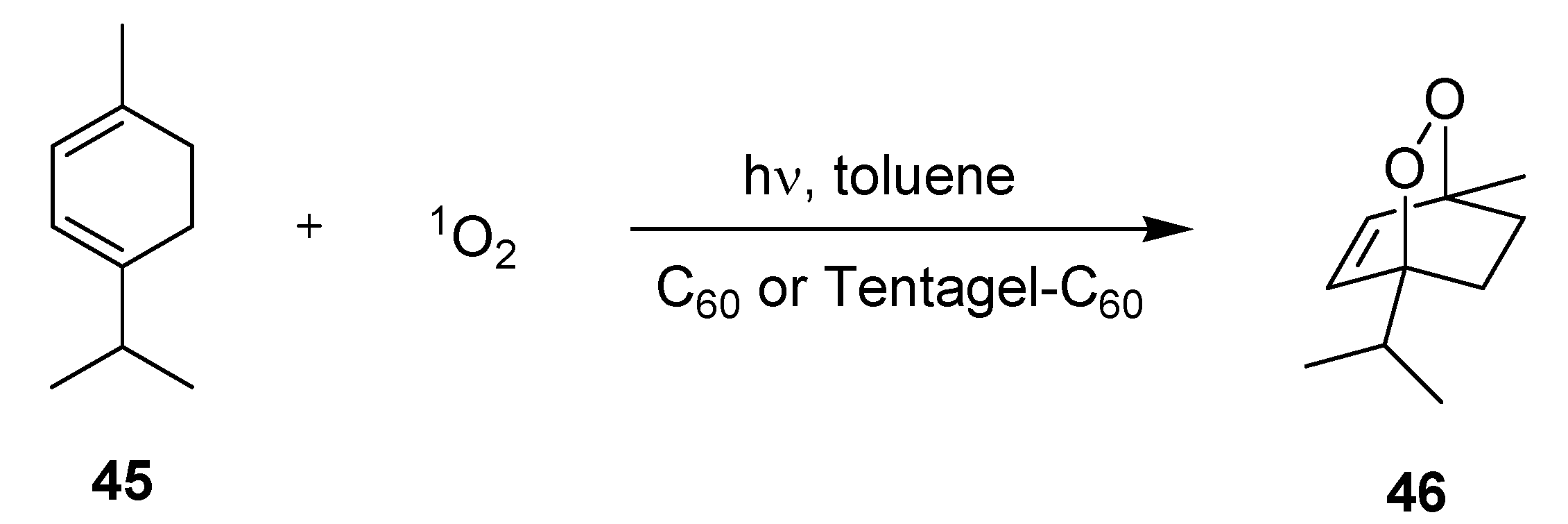{kind=link}
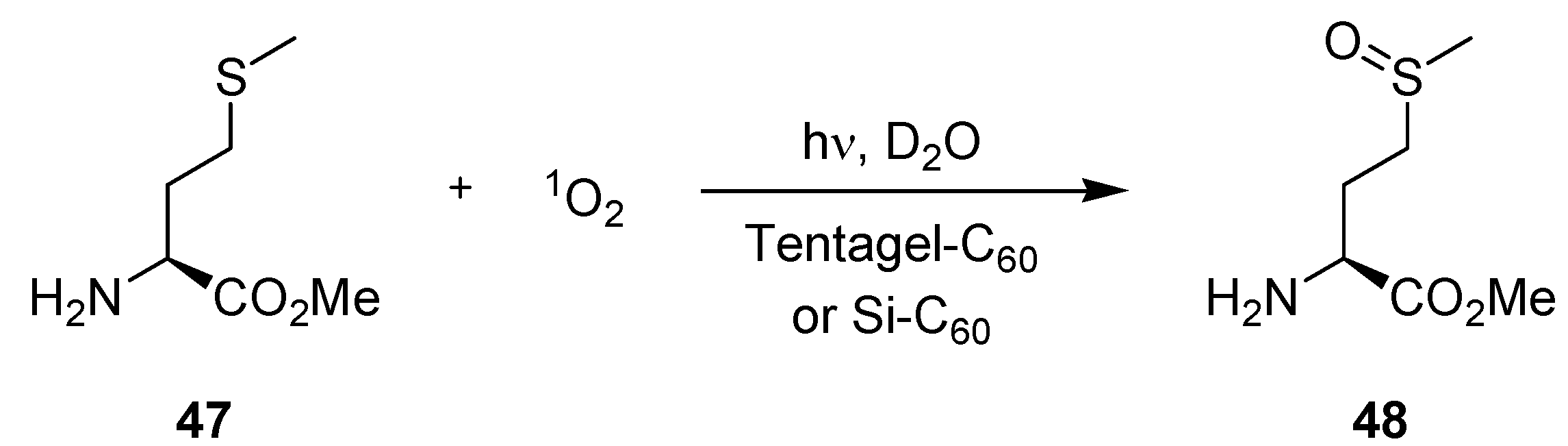{kind=link}
{kind=link}
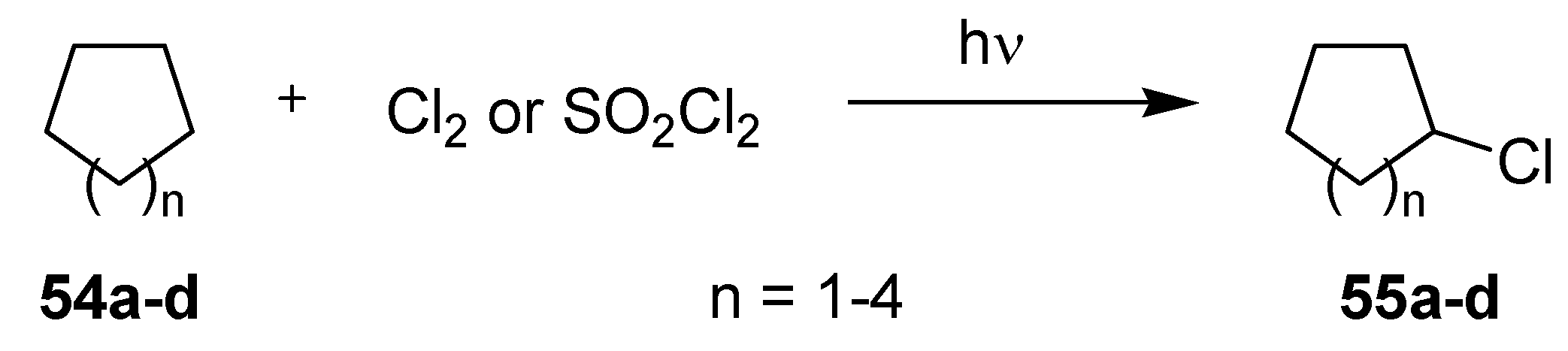{kind=link}
{kind=link}
{kind=link}
| Photo-μ-reactor | Photo/thermal-μ-reactor |
|---|---|
| Custom-made (quartz) 1,000 μm × 200 μm × 25 cm (W × D × L) 50 μL (Vchannel) | Custom-made (quartz) 1,000 μm × 200 μm × 50 cm (W × D × L) 100 μL (Vchannel) Submerged in oil bath (100 °C) |
| 400 W high pressure mercury lamp (Vycor filter) | 400 W high pressure mercury lamp (Vycor and glass UV filter) |
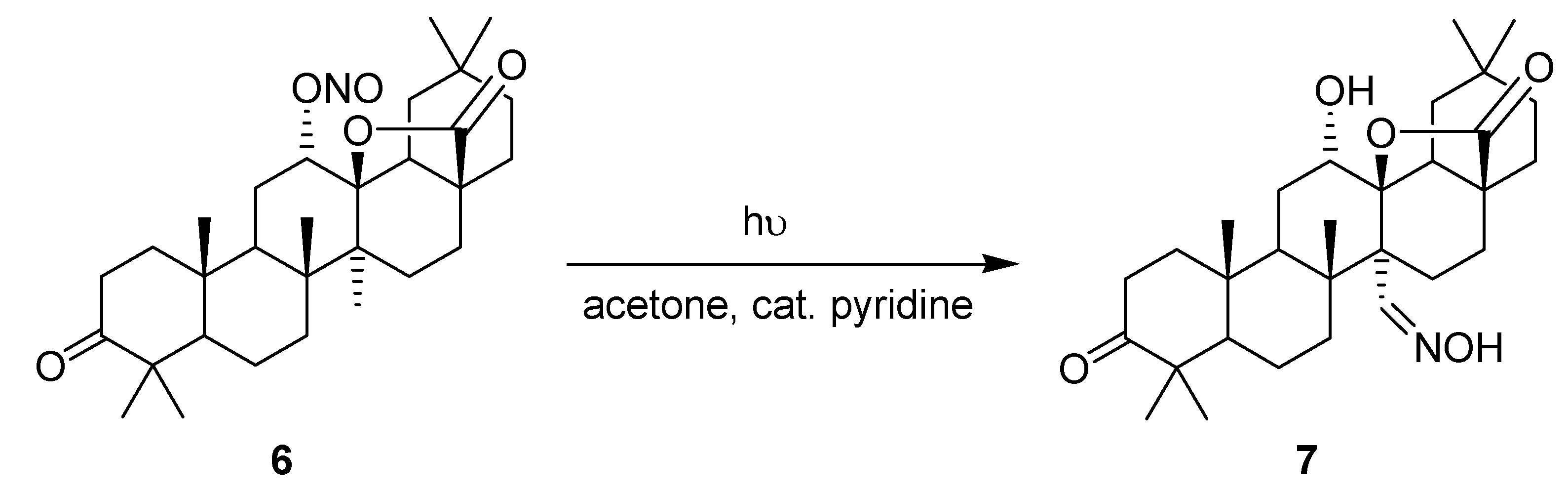
3.1.2. Barton Reaction

| Batch | Single-lane μ-reactor |
|---|---|
| 20 mL Pyrex round bottom flask 10 mL (Vsolution) | Dainippon Screen (stainless steel, glass window) 1,000 μm × 107 μm × 2.2 m (W × D × L) 0.2 mL (Vchannel) |
| 15 W black light | 15 W black light or 1.7 W UV-LED array |
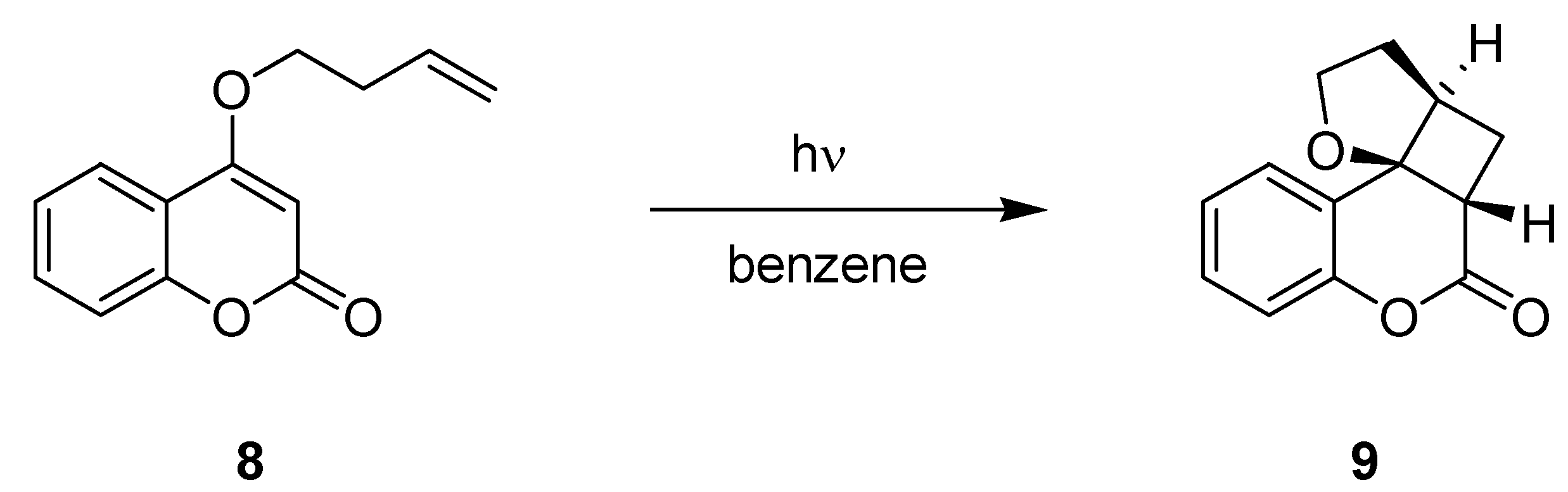
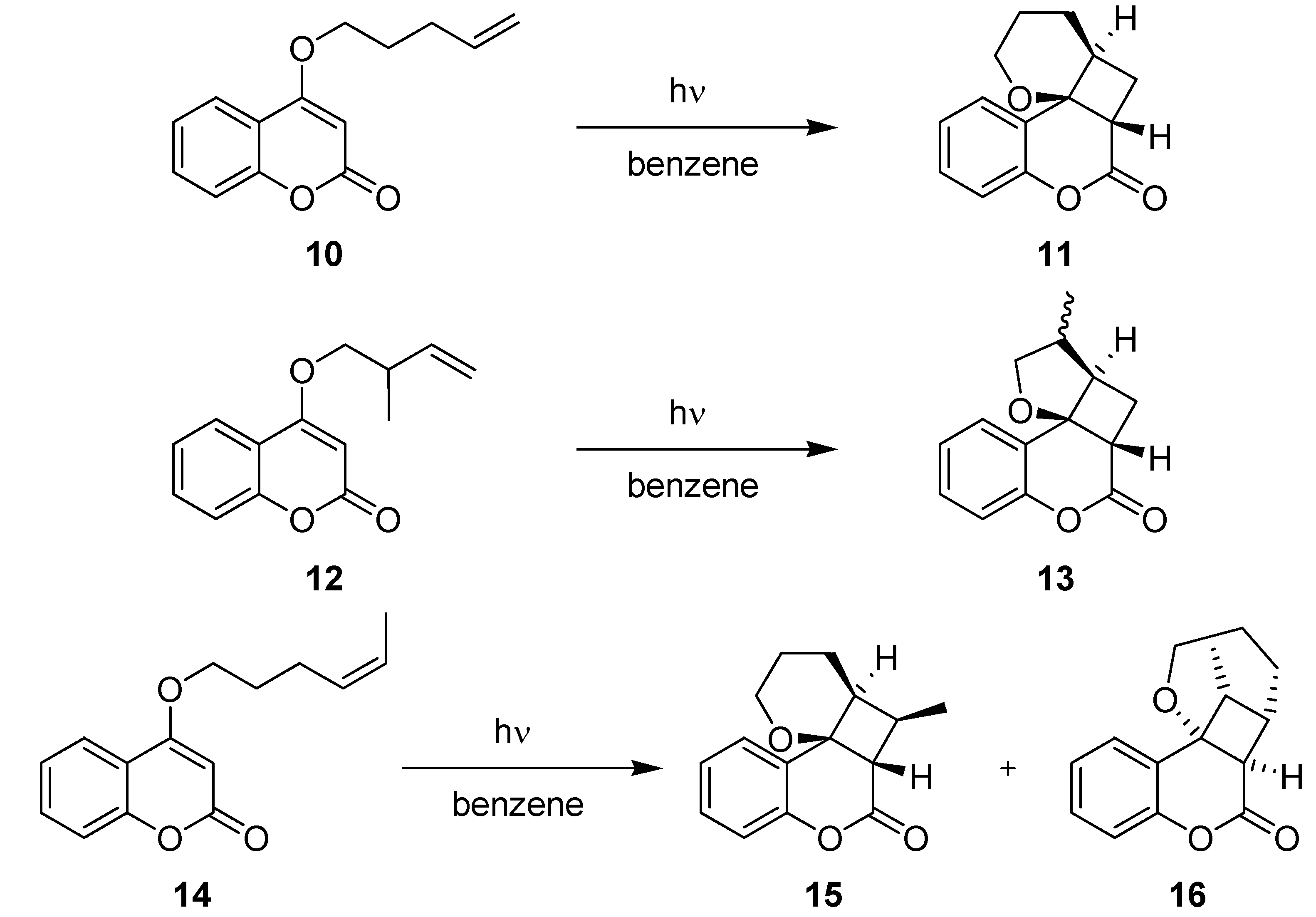
3.1.3. [2+2]-Cycloadditions of Coumarin Derivatives

| Batch | μ-reactor |
|---|---|
| 10 mL quartz Round bottom flask 1 mL (Vsolution) | LOPHTOR (stainless steel with FEP membrane cover, quartz window) 1,000 μm × 250 μm × 3.93 m (W × D × L) 0.98 mL (Vchannel) |
| 450 W medium pressure mercury lamp | 450 W medium pressure mercury lamp (with elliptical concentrator and integral UVEXS cold mirror) |

| Transformation | LOPHTOR | Batch | ||
|---|---|---|---|---|
| Time [h] | Yield [%] | Time [h] | Yield [%] | |
| 10 → 11 | 4 | 99 | 48 | 67 |
| 12 → 13 | 7 | 50 (1:1 a) | 24 | 30 (1:1 a) |
| 14 → 15 + 16 | 5 | 40 (7:1 b) | 24 | 25 (8:1 b) |
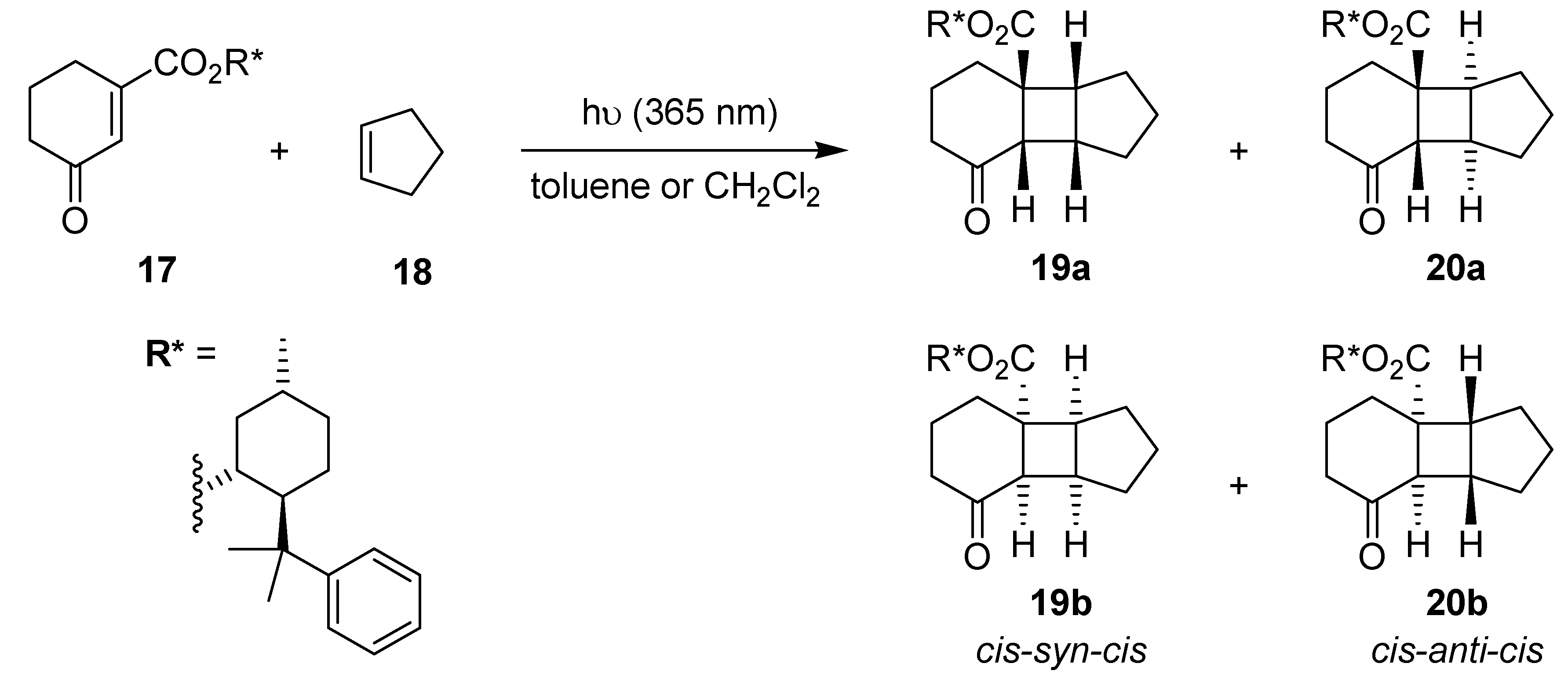
3.1.4. Diastereoselective [2+2]-Cycloaddition of a Chiral Cyclohexenone

| Batch | μ-Reactor |
|---|---|
| Pyrex test tube 13 mm (ID) / 17 mm (OD) 2 mL (Vsolution) | Dainippon Screen (stainless steel, Pyrex window) 1,000 μm × 100 μm × 2.2 m (W × D × L) 0.2 mL (Vchannel) |
| 500 W high pressure mercury lamp (inside a quartz immersion well) | 500 W high pressure mercury lamp (inside a quartz immersion well) |
| Reactor | Temp. [°C] | Solvent | Time [h] | Ratio19:20 | d.e. [%] | |
|---|---|---|---|---|---|---|
| 19 | 20 | |||||
| μ-reactor | 0 | toluene | 0.5 | 39:61 | 71 | 53 |
| ‑20 | 41:59 | 72 | 53 | |||
| ‑40 | 50:50 | 82 | 54 | |||
| batch | 0 | 1 | 38:62 | 60 | 37 | |
| ‑20 | 41:59 | 70 | 42 | |||
| ‑40 | 50:50 | 72 | 44 | |||
| μ-reactor | 0 | CH2Cl2 | 0.5 | 38:62 | 65 | 30 |
| ‑20 | 50:50 | 70 | 32 | |||
| ‑40 | 51:49 | 71 | 34 | |||
| batch | 0 | 1 | 35:65 | 57 | 27 | |
| ‑20 | 46:54 | 60 | 30 | |||
| ‑40 | 50:50 | 67 | 33 | |||
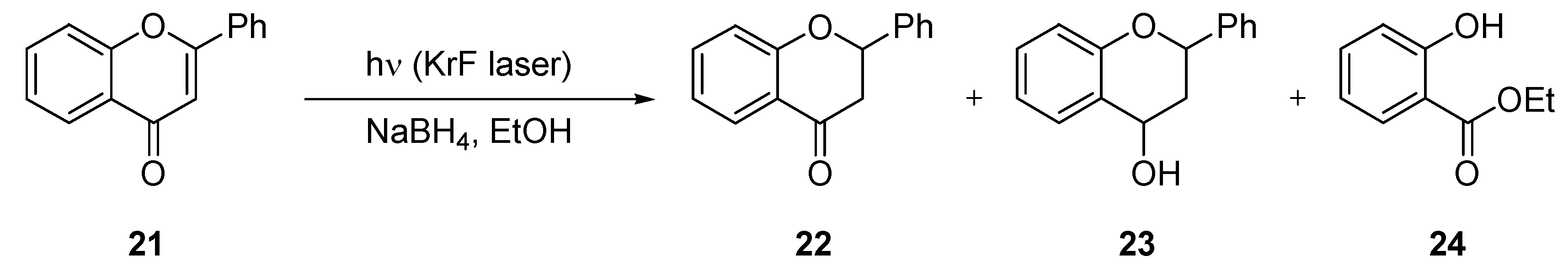
3.1.5. Photochemical Reduction of Flavone

| Batch | μ-Reactor |
|---|---|
| Synthetic quartz cell (10 mm width and 1 mm optical path); 50 μL (Vsolution) | IMT Co. Ltd. (quartz) 100 μm × 40 μm × 16.5 mm (W × D × L a) |
| KrF (248 nm) or XeCl (308 nm) excimer laser | KrF (248 nm) excimer laser |
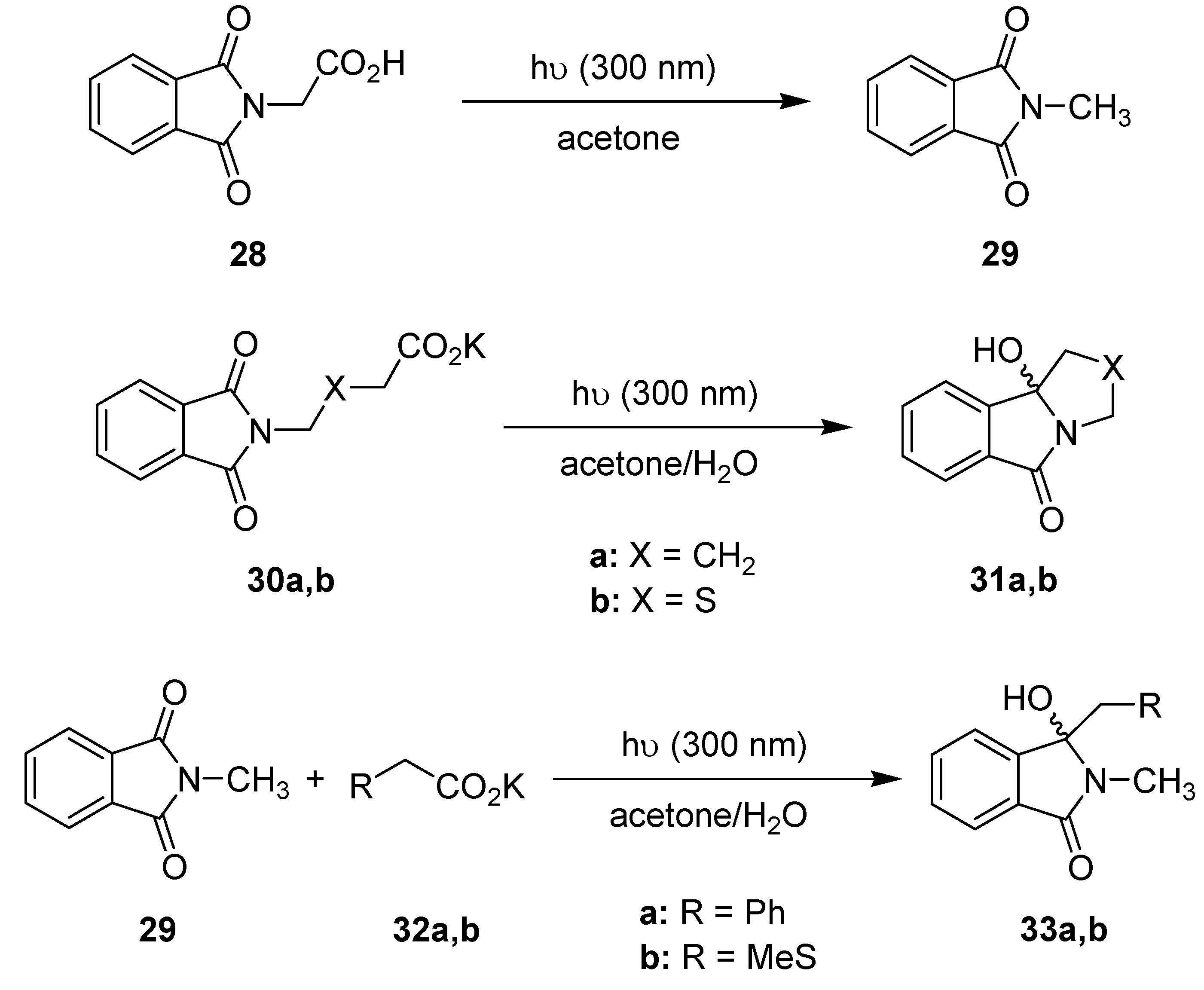
3.1.6. Photodecarboxylations Involving Phthalimides

| Batch | μ-Reactor |
|---|---|
| Pyrex Schlenk flask (32 mm ID) with cold finger (24 mm OD) 100 mL (Vsolution) | mikroglas chemtech dwell device (Foturan™) 2,000 μm × 500 μm × 1.15 m (W × D × L) 1.68 mL (Vchannel) |
| Rayonet chamber reactor (RPR-200) equipped with 16 × 8 W UVB lamps | Luzchem UV panel equipped with 5 × 8 W UVB lamps |

| Transformation | Time [min] | Dwell device | Batch |
|---|---|---|---|
| Yields/Conv. [%] a | Yields/Conv. [%] a | ||
| 28 → 29 | 3.4 | 5 | 2 |
| 11 | 23 | 7 | |
| 21 | 44 | 19 | |
| 40 | 74 | 39 | |
| 60 | 92 | 59 (22 b) | |
| 30a → 31a | 21 | 33 | 46 |
| 40 | 69 | 53 | |
| 60 | 77 | 69 (19 b) | |
| 30b → 31b | 21 | 39 | 36 |
| 40 | 70 | 59 | |
| 60 | 80 | 72 (21 b) | |
| 29 + 32a → 33a | 14 | 83 | 46 |
| 21 | 97 | 93 | |
| 40 | 100 | 100 | |
| 60 | 100 | 100 (29 b) | |
| 29 + 32b → 33b | 14 | 4 | 2 |
| 21 | 34 | 20 | |
| 40 | 66 | 44 | |
| 60 | 100 | 86 (17 b) |
3.1.7. Photodecarboxylative Addition to Methyl Phenylglyoxolate

| Batch | μ-Reactor |
|---|---|
| Pyrex Schlenk flask (32 mm ID) with cold finger (24 mm OD) 50 mL (Vsolution) | mikroglas chemtech dwell device (Foturan™) 2,000 μm × 500 μm × 1.15 m (W × D × L) 1.68 mL (Vchannel) |
| Rayonet chamber reactor (RPR-200) equipped with 16 × 8 W UVA lamps | Luzchem UV panel equipped with 5 × 8 W UVA lamps |
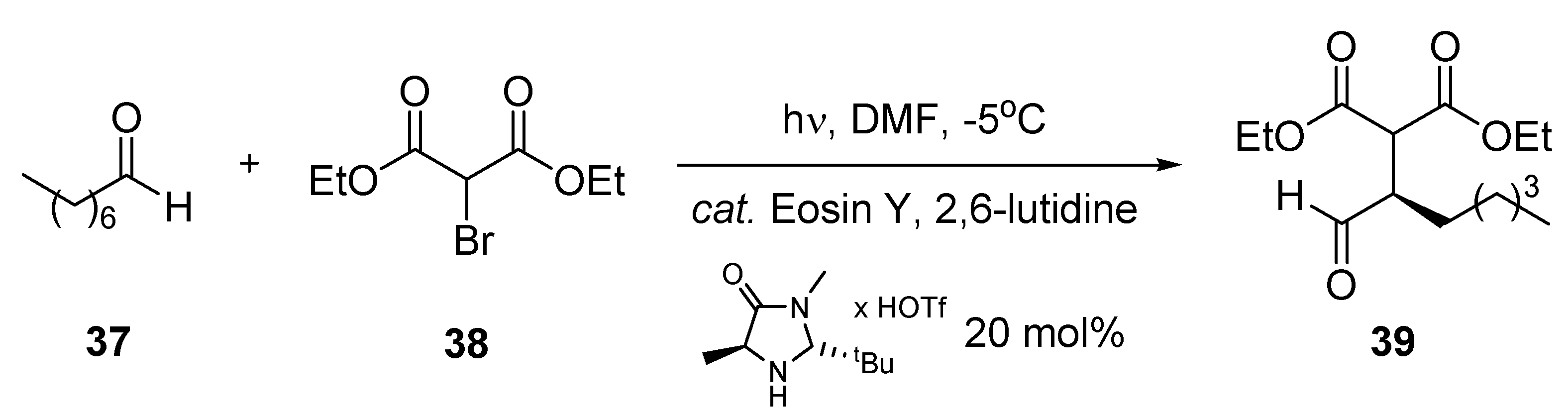
3.1.8. Organo-Photocatalytic Addition to Diethyl 2-Bromomalonate

| Batch | μ-reactor |
|---|---|
| Borosilicate vial (18 mm ID) 5 mL (Vsolution) | Future Chemistry photochemistry module with M-111 basic μ-reactor (Borosilicate) 600 μm × 500 μm (Wmax × D) 100 μL (Vreaction) |
| 1 × 1 W 530 nm LED | 2 × 1 W 530 nm LEDs |
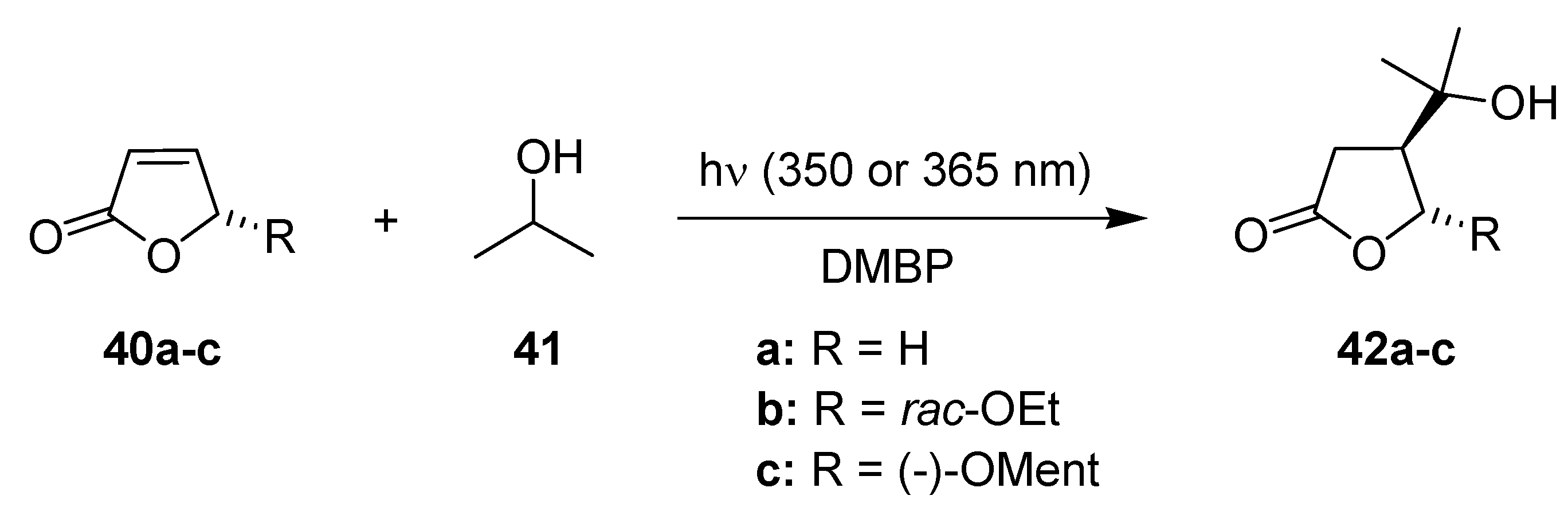
3.1.9. Isopropanol Addition to Furanones

| batch | μ-Reactor | μ-Chip | μ-Capillary tower |
|---|---|---|---|
| Pyrex test tube 9 mm (ID) / 10 mm (OD) 15 mL (Vsolution) | mikroglas chemtech dwell device (Foturan™) 2,000 μm × 500 μm × 1.15 m (W × D × L) 1.68 mL (Vchannel) | Micronit Microfluidics (Borofloat®) 150 μm × 150 μm × 757 mm (W × D × L) 13 μL (Vchannel) | PTFE capillary (wrapped around a Pyrex cylinder) 558 μm × 460 cm (ID × L) 2 capillaries 2 × 1.12 mL (Vcapillary) |
| Rayonet chamber reactor (RPR-200) equipped with 16 × 8 W UVA lamps | Luzchem UV panel equipped with 5 × 8 W UVA lamps | 6 × 75 mW 365 nm UV-LED array | 1 × 8 W UVA lamp (inside the Pyrex cylinder) |
| Batch | μ-Reactor | μ-Chip | μ-Capillary tower | |||||
|---|---|---|---|---|---|---|---|---|
| Time [min] | Conv. [%] | Time [min] | Conv. [%] | Time [min] | Conv. [%] | Time [min] | Conv. [%] | |
| 42a | 5 | 90 | 5 | 81 | 1 | 58 | 2.5 | 35 |
| 20 | 100 | 10 | 100 | 2.5 | 89 | 5 | 75 | |
| 5 | 100 | 7.5 | 95 | |||||
| 42b | 5 | 90 | 5 | 99 | 1 | 60 | 2.5 | 50 |
| 20 | 100 | 10 | 100 | 2.5 | 100 | 5 | 96 | |
| 5 | 100 | 7.5 | 99 | |||||
| 42c | 5 | 87 | 5 | 98 | 1 | 59 | 2.5 | 64 |
| 20 | 100 | 10 | 100 | 2.5 | 100 | 5 | 99 | |
| 5 | 100 | 7.5 | 100 | |||||
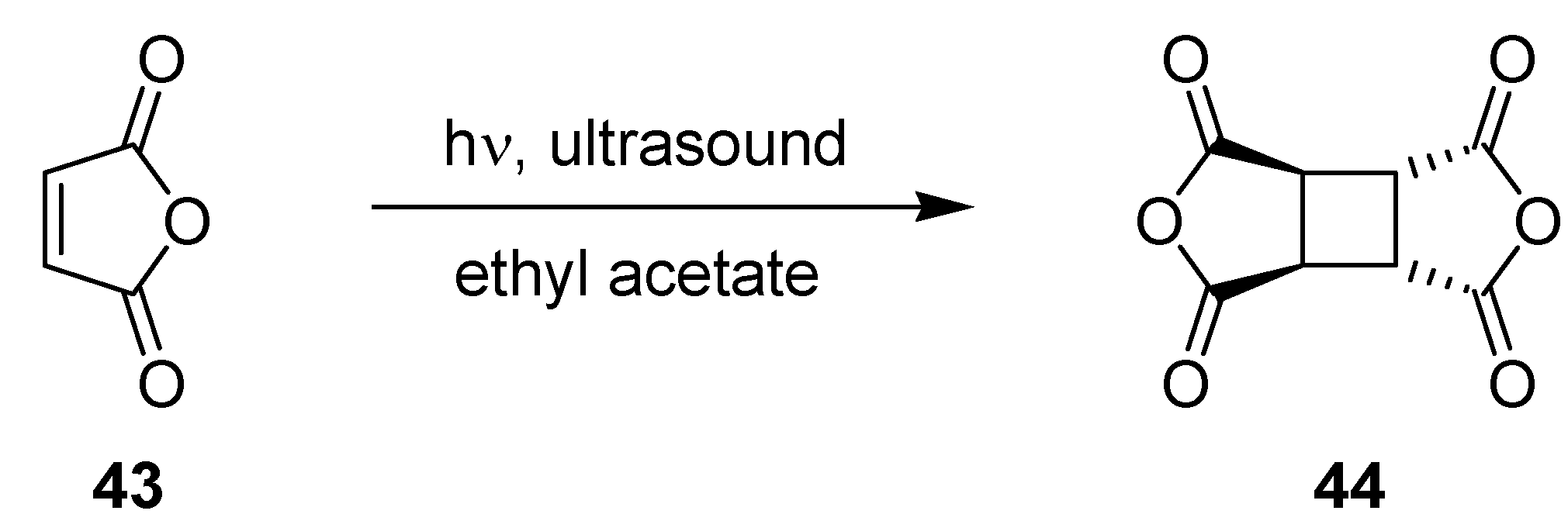
3.1.10. Photodimerization of Maleic Anhydride
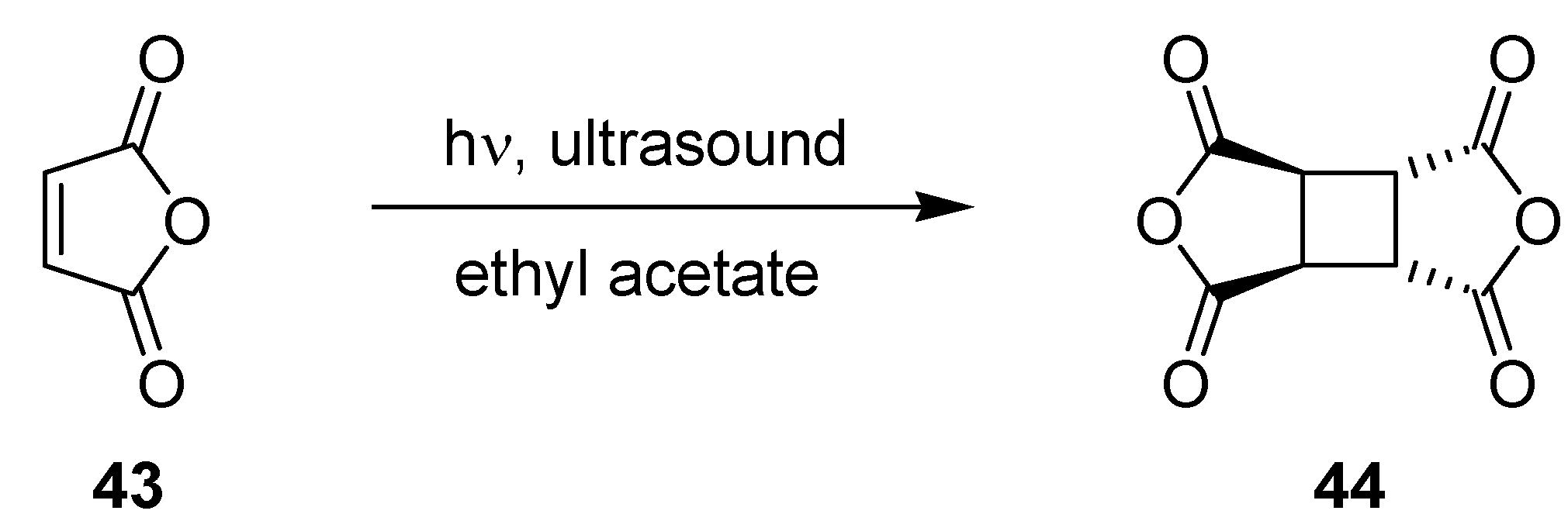
| Batch | μ-Reactor |
|---|---|
| Cylindrical glass vessel 75 × 300 mm (ID × H) 600 g (msolution) | FEP tube (wrapped around a quartz beaker) 0.5–1.6 mm × 6.4–19.2 m (ID × L) 0.8–12.9 mL (Vcapillary) |
| 400 W high pressure mercury lamp (inside a Pyrex immersion well; immersed in glass vessel) | 400 W high pressure mercury lamp (inside a Pyrex immersion well; placed inside the quartz beaker) |
3.2. Heterogeneous Reactions
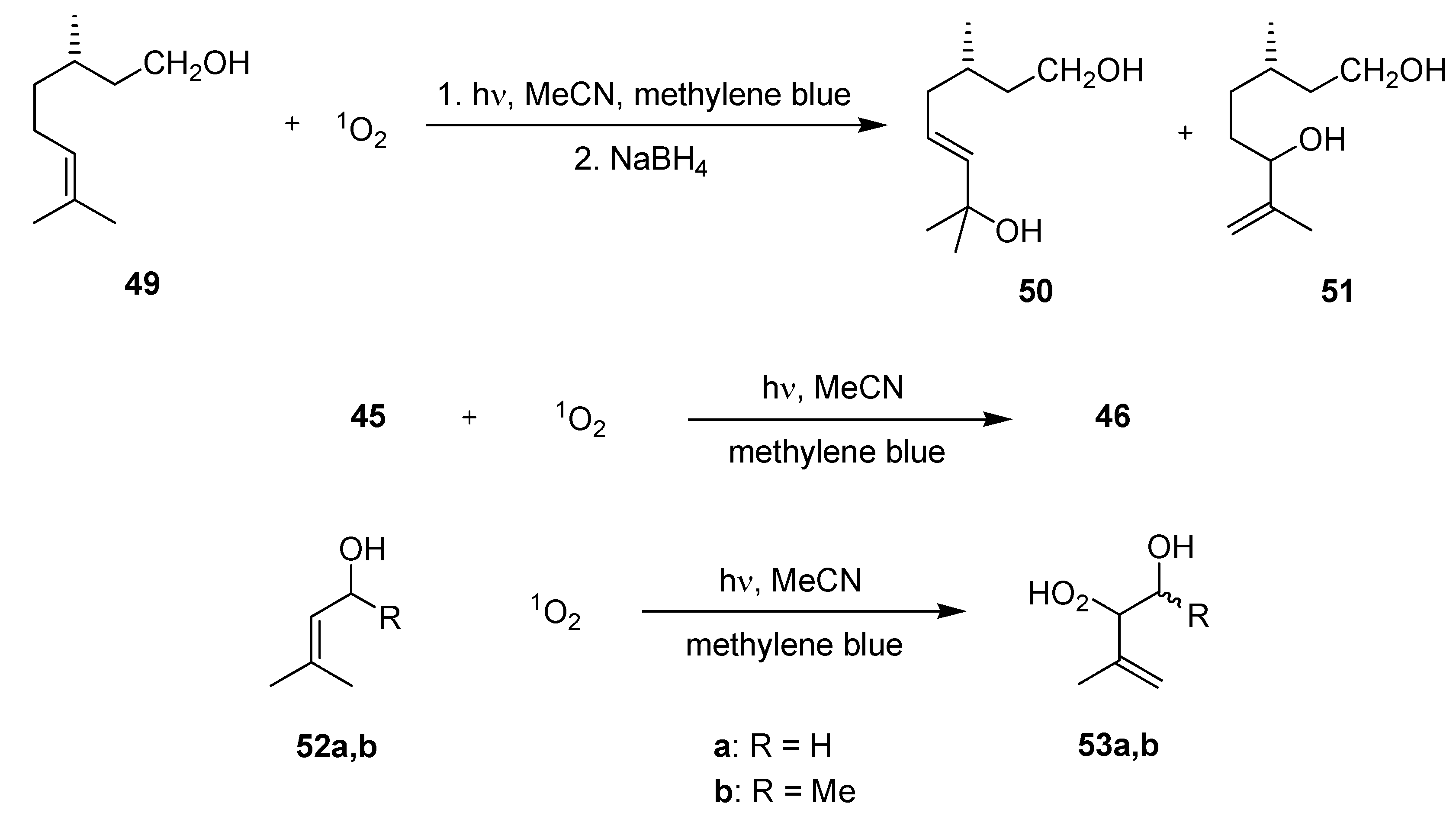
3.2.1. Photooxygenation Reactions
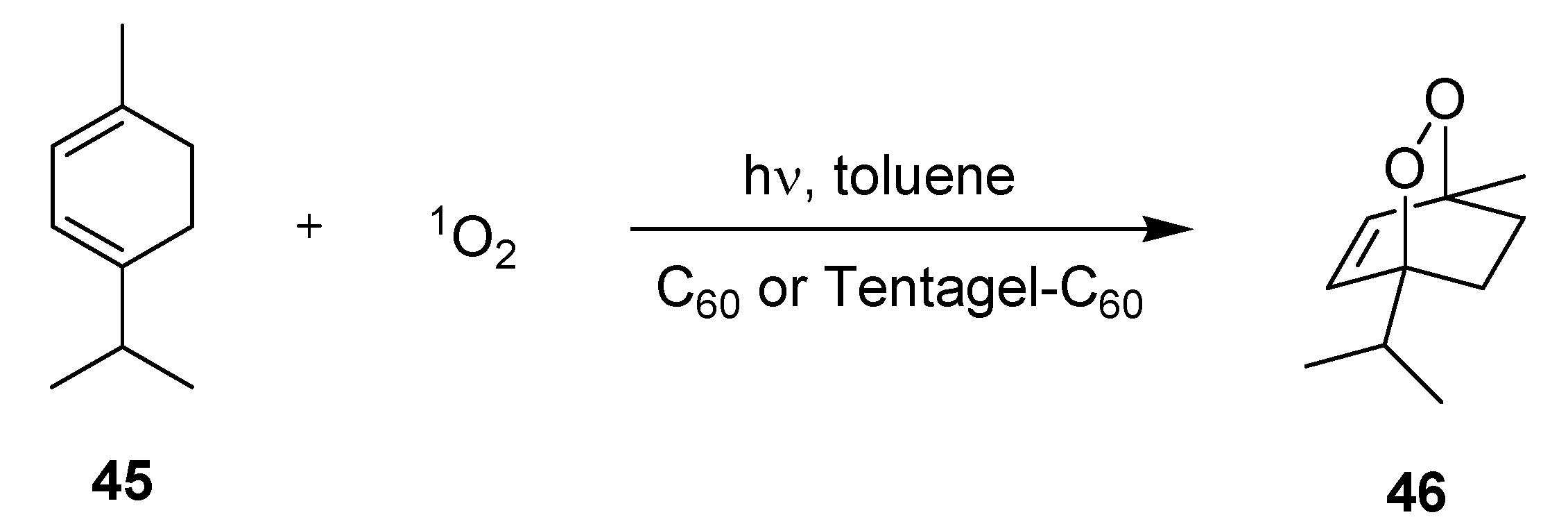

| Batch | Serpentine μ-reactor | Chamber μ-reactor |
|---|---|---|
| Glass photochemical reactor 10 mL (Vsolution) | In-house (glass-thiolene) no channel dimensions given 0.152 mL (Vreactor) | In-house (glass-thiolene) serpentine oxygenation zone500 μm (L) with chamber 3 mm × 30 mm (W × L) 0.152 mL (Vreactor) |
| 300 W tungsten halogen lamp | 300 W tungsten halogen lamp or 15 × 110 mW white LED array | |

| Batch | Mono-channel μ-reactor | Dual-channel μ-reactor |
|---|---|---|
| 50 ml round bottom flask 4.4 cm (ODmax) 20 mL (Vsolution) | In-house (PDMS, PVSZ coating in reaction channel) concave reaction channel 220 μm × 0.9 m (W × L) 38.9 μL (Vchannel) segment flow | In-house (PDMS with PDMS membrane, PVSZ coating in reaction channel) concave reaction channel 220 μm × 0.9 m (W × L) 38.9 μL (Vchannel) |
| 16 W white LED spot light | 16 W white LED spot light | 16 W white LED spot light |
3.2.2. Photochlorinations of Cycloalkanes
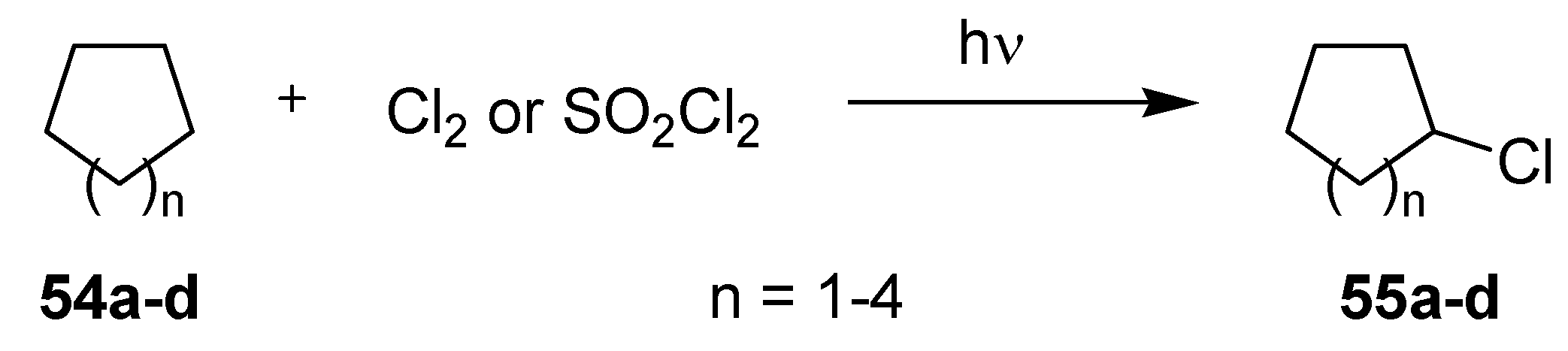
| Steel based μ-reactor | Glass based μ-reactor 2 |
|---|---|
| Dainippon Screen (stainless steel, Pyrex window) 1,000 μm × 300 μm × 2.35 m (W × D × L) 0.7 mL (Vchannel) | mikroglas chemtech dwell device (Foturan™) 1,000 μm × 500 μm × 1.9 m (W × D × L) 0.95 mL (Vchannel) |
| ambient light | 15 W black light |
3.3. Scale-Up and Industrial Applications
3.3.1. Barton Reaction
| Multi-lane μ-reactor | Automated Multi-lane μ-reactor |
|---|---|
| Dainippon Screen (stainless steel, Pyrex window) 1,000 μm × 500 μm × 0.5 m (W × D × L) 16 lanes, 2 reactors in series8 mL (Vchannel) | Dainippon Screen DS-AMS-1 (stainless steel, Pyrex window) 1,000 μm × 500 μm × 0.5 m (W × D × L) 16 lanes 4 mL (Vchannel) |
| 8 × 20 W black light | 6 × 15 W black light |
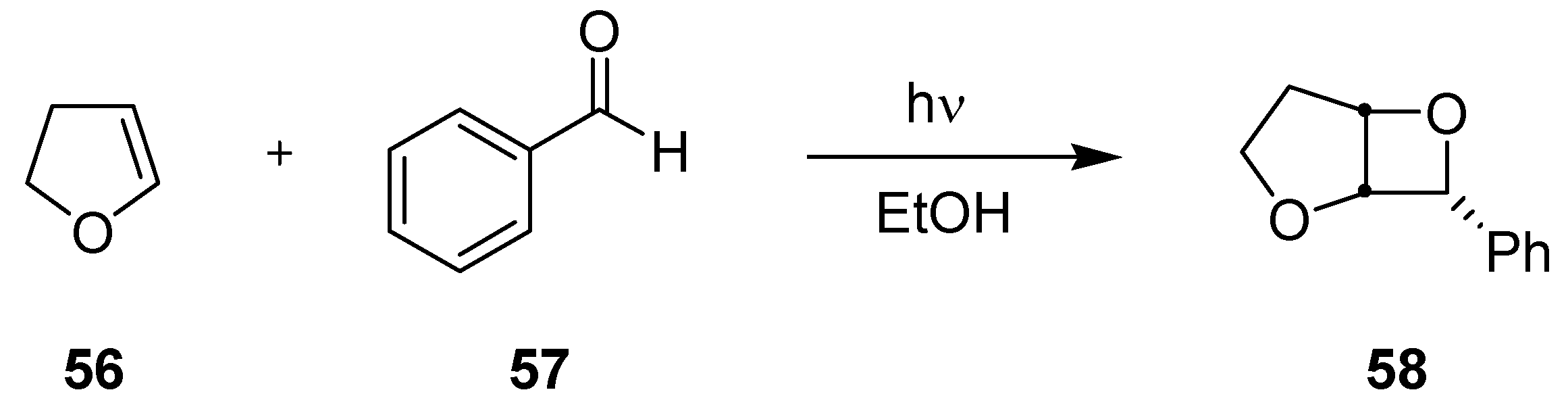
3.3.2. Paternò-Büchi Reaction


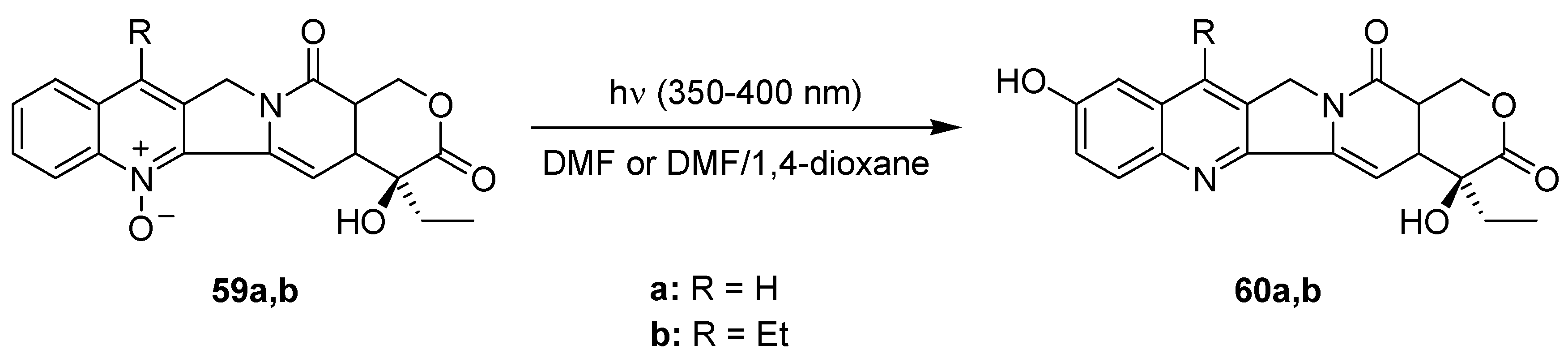
3.3.3. Photoisomerization of Camptothecin-N-oxides


4. Conclusions
Acknowledgments
References and Notes
- Hoffmann, N. Photochemical reactions as key steps in organic synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar]
- Bach, T.; Hehn, J.P. Photochemical reactions as key steps in natural product synthesis. Angew. Chem. Int. Ed. 2011, 50, 1000–1045. [Google Scholar] [CrossRef]
- Iriondo-Alberdi, J.; Greaney, M.F. Photocycloaddition in natural product synthesis. Eur. J. Org. Chem. 2007, 4801–4815. [Google Scholar] [CrossRef]
- Ciana, C.L.; Bochet, C.G. Clean and easy photochemistry. Chimia 2007, 61, 650–654. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Jung, C.; Mattay, J. Green photochemistry: Production of fine chemicals with sunlight. Pure Appl. Chem. 2007, 79, 1939–1947. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Healy, N.; de Oliveira, L.; Jung, C.; Mattay, J. Green photochemistry: Solar-chemical synthesis of juglone with medium concentrated sunlight. Green Chem. 2006, 8, 831–834. [Google Scholar] [CrossRef]
- Braun, A.M.; Maurette, M.; Oliveros, E. Photochemical Technology; Wiley: Chichester, UK, 1991. [Google Scholar]
- Fischer, M. Industrial applications of photochemical syntheses. Angew. Chem. Int. Ed. 1978, 17, 16–26. [Google Scholar] [CrossRef]
- Wegner, J.; Ceylan, S.; Kirschning, A. Ten key issues in modern flow chemistry. Chem. Commun. 2011, 47, 4583–4592. [Google Scholar]
- Pohar, A.; Plazl, I. Process intensification through microreactor application. Chem. Biochem. Eng. Q. 2009, 23, 537–544. [Google Scholar]
- Wiles, C.; Watts, P. Recent advances in micro reaction technology. Chem. Commun. 2011, 47, 6512–6535. [Google Scholar] [CrossRef]
- Webb, D.; Jamison, T.F. Continuous flow multi-step organic synthesis. Chem. Sci. 2010, 1, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Hartman, R.L; Jensen, K.F. Microchemical systems for continuous-flow synthesis. Lab Chip 2009, 9, 2495–2507. [Google Scholar]
- Rubin, A.E.; Tummala, S.; Both, D.A.; Wang, C.C.; Delaney, E.J. Emerging technologies supporting chemical process R&D and their increasing impact on productivity in the pharmaceutical industry. Chem. Rev. 2006, 106, 2794–2810. [Google Scholar] [CrossRef]
- Chin, P.; Barney, W.S.; Pindzola, B.A. Microstructured reactors as tools for the intensification of pharmaceutical reactions and processes. Curr. Opin. Drug Discov. Dev. 2009, 12, 848–861. [Google Scholar]
- Mason, B.P.; Price, K.E.; Steinbacher, J.L.; Bogdan, A.R.; McQuade, D.T. Greener approaches to organic synthesis using microreactor technology. Chem. Rev. 2007, 107, 2300–2318. [Google Scholar]
- Yoshida, J.I.; Kim, H.; Nagaki, A. Green and sustainable chemical synthesis using flow microreactors. ChemSusChem 2011, 4, 331–340. [Google Scholar] [CrossRef]
- Hessel, V.; Kralisch, D.; Krtschil, U. Sustainability through green processing—Novel process windows intensify micro and milli process technologies. Energy Environ. Sci. 2008, 1, 467–478. [Google Scholar] [CrossRef]
- Lerou, J.J.; Tonkovich, A.L.; Silva, L.; Perry, S.; McDaniel, J. Microchannel reactor architecture enables greener processes. Chem. Eng. Sci. 2010, 65, 380–385. [Google Scholar] [CrossRef]
- Coyle, E.E.; Oelgemöller, M. Photochemistry goes micro. Chem. Technol. 2008, 5, T95. [Google Scholar]
- Débieux, J.-L.; Cosandey, A.; Helgen, C.; Bochet, C.G. Photoacylation of alcohols in neutral medium. Eur. J. Org. Chem. 2007, 2073–2077. [Google Scholar]
- Carney, J.M.; Hammer, R.J.; Hulce, M.; Lomas, C.M.; Miyashiro, D. Hight-efficient microphotooxidation using milliwatt LED sources. Tetrahedron Lett. 2011, 52, 352–355. [Google Scholar]
- Gano, J.E.; Gano, A.J.; Garry, P.; Sekher, P. Small scale reactor for ultraviolet photochemistry. J. Chem. Educ. 2002, 79, 1361. [Google Scholar] [CrossRef]
- Penn, J.H.; Orr, R.D. A microscale immersion well for photochemical reactions. J. Chem. Educ. 1989, 66, 86–88. [Google Scholar] [CrossRef]
- Rubin, M.B.; Braslavsky, S.E. Quantum yield: The term and the symbol. A historical search. Photochem. Photobiol. Sci. 2010, 9, 670–674. [Google Scholar] [CrossRef]
- Sosnin, E.A.; Oppenländer, T.; Tarasenko, V.F. Applications of capacitive and barrier discharge exilamps in photoscience. J. Photochem. Photobiol. C-Photo. 2006, 7, 145–163. [Google Scholar]
- Hackett, P.A. Making light work—Applications of lasers to chemical processes. Laser Chem. 1988, 10, 75–106. [Google Scholar] [CrossRef]
- Kreisel, G.; Meyer, S.; Tietze, D.; Fidler, T.; Gorges, R.; Kirsch, A.; Schäfer, B.; Rau, S. Leuchtdioden in der Chemie - Eine Hochzeit verschiedener Technologien. Chem. Ing. Tech. 2007, 79, 153–159. [Google Scholar] [CrossRef]
- André, J.-C.; Viriot, M.-L.; Villermaux, J. New developments in photochemical technology. Pure Appl. Chem. 1986, 58, 907–916. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Kramer, W.; Oelgemöller, M. Photoinduced decarboxylation reactions. Radical chemistry in water. Green Chem. 1999, 1, 205–207. [Google Scholar]
- Griesbeck, A.G.; Maptue, N.; Bondock, S.; Oelgemöller, M. The excimer radiation system: A powerful tool for preparative organic photochemistry. A technical note. Photochem. Photobiol. Sci. 2003, 2, 450–451. [Google Scholar] [CrossRef]
- Shama, G.; Peppiatt, C.; Biguzzi, M. A novel thin film photoreactor. J. Chem. Tech. Biotechnol. 1996, 65, 56–64. [Google Scholar] [CrossRef]
- Dunk, B.; Jachuck, R. A novel reactor for UV irradiated reactions. Green Chem. 2000, 2, G13–G14. [Google Scholar] [CrossRef]
- Yatmaz, H.C.; Wallis, C.; Howarth, C.R. The spinning disc reactor—Studies on a novel TiO2 photocatalytic reactor. Chemosphere 2001, 42, 397–403. [Google Scholar] [CrossRef]
- Lu, H.; Schmidt, M.A.; Jensen, K.F. Photochemical reactions and on-line UV detection in microfabricated reactors. Lab Chip 2001, 1, 22–28. [Google Scholar] [CrossRef]
- Benito-Lopex, F.; Verboom, W.; Kakuta, M.; Gardeniers, J.G.E.; Egberink, R.J.M.; Oosterbroek, E.R.; van der Berg, A.; Reinhoudt, D.N. Optical fiber-based on-line UV/Vis spectroscopic monitoring of chemical reaction kinetics under high pressure in a capillary microreactor. Chem. Commun. 2005, 2857–2859. [Google Scholar]
- Bentrup, U.; Küpper, L.; Budde, U.; Lovis, K.; Jähnisch, K. Mid-Infrared monitoring of gas-liquid reactions in Vitamin D analogue synthesis with a novel fiber optical diamond ATR sensor. Chem. Eng. Technol. 2006, 29, 1216–1220. [Google Scholar] [CrossRef]
- Marre, S.; Baek, J.; Park, J.; Bawendi, M.G.; Jensen, K.F. High-pressure/high-temperature microreactors for nanostructure synthesis. J. Assoc. Lab. Autom. 2009, 14, 367–373. [Google Scholar] [CrossRef]
- Jähnisch, K.; Baerns, M.; Hessel, V.; Ehrfeld, W.; Haverkamp, V.; Löwe, H.; Wille, C.; Cuber, A. Direct fluorination of toluene using elemental fluorine in gas/liquid microreactors. J. Fluorine Chem. 2000, 105, 117–128. [Google Scholar] [CrossRef]
- Hornung, C.H.; Hallmark, B.; Baumann, M.; Baxendale, I.R.; Ley, S.V.; Hester, P.; Clayton, P.; Mackley, M.R. Multiple microcapillary reactor for organic synthesis. Ind. Eng. Chem. Res. 2010, 49, 4576–4582. [Google Scholar]
- Coyle, E.E.; Oelgemöller, M. Micro-photochemistry: Photochemistry in microstructured reactors. The new photochemistry of the future? Photochem. Photobiol. Sci. 2008, 7, 1313–1322. [Google Scholar]
- Matsushita, Y.; Ichimura, T.; Ohba, N.; Kumada, S.; Sakeda, K.; Suzuki, T.; Tanibata, H.; Murata, T. Recent progress on photoreactions in microreactors. Pure Appl. Chem. 2007, 79, 1959–1968. [Google Scholar] [CrossRef]
- Ichimura, T.; Matsushita, Y.; Sakeda, K.; Suzuki, T. Photoreactions. In Microchemical Engineering in Practice; Dietrich, T.R., Ed.; John Wiley & Sons: Hoboken, New Jersey, NJ, USA, 2009; pp. 385–402, Chapter 17. [Google Scholar]
- Vesborg, P.C.K.; In, S.-I.; Olsen, J.L.; Henriksen, T.R.; Abrams, B.L.; Hou, Y.; Kleiman-Shwarsctein, A.; Hansen, O.; Chorkendorff, I. Quantitative measurements of photocatalytic CO-oxidation as a function of light intensity and wavelength over TiO2 nanotube thin films in μ-reactors. J. Phys. Chem. C 2010, 114, 11162–11168. [Google Scholar]
- Vesborg, P.C.K.; Olsen, J.L.; Henriksen, T.R.; Chorkendorff, I.; Hansen, O. Gas-phase photocatalysis in μ-reactors. Chem. Eng. J. 2010, 160, 738–741. [Google Scholar] [CrossRef]
- Ye, M.-Y.; Li, B.-X.; Ye, R.-M.; Liu, J.-H. Photodegradation of organics with titanium oxide/hydrogen peroxide catalyst system in microreactor and its application in environmental analysis. Chin. J. Anal. Chem. 2010, 38, 643–647. [Google Scholar]
- He, Z.; Li, Y.; Zhang, Q.; Wang, H. Capillary microchannel-based microreactors with highly durable ZnO/TiO2 nanorod arrays for rapid, high efficiency and continuous-flow photocatalysis. Appl. Catal. B-Environ. 2010, 93, 376–382. [Google Scholar]
- Matsushita, Y.; Iwasawa, M.; Suzuki, T.; Ichimura, T. Multiphase photocatalytic oxidation in a microreactor. Chem. Lett. 2009, 38, 846–847. [Google Scholar] [CrossRef]
- Charles, G.; Corbel, S.; Carré, M.-C.; Roques-Carmes, T.; Zahraa, O. Design of a micro-channel reactor for decomposition of organic pollutants in waste water treatment. In Innovative Developments in Design and Manufacturing. Advanced Research in Virtual and Rapid Prototyping; Bartolo, P.J.d.S., Jorge, M.A., Batista, F.d.C., Almeida, H.A., Matias, J.M., Vasco, J.C., Gaspar, J.B., Correia, M.A., Andre, N.C., Alves, N.F., Novo, P.P., Martinho, P.G., Carvalho, R.A., Eds.; Taylor & Francis Group: London, UK, 2010; Leiria Portugal; Chapter 66. [Google Scholar]
- Fuse, S.; Tanabe, N.; Yoshida, M.; Yoshida, H.; Doi, T.; Takahashi, T. Continuous-flow synthesis of vitamin D3. Chem. Commun. 2010, 46, 8722–8724. [Google Scholar]
- Sugimoto, A.; Sumino, Y.; Takagi, M.; Fukuyama, T.; Ryu, H. The Barton reaction using a microreactor and black light. Continuous-flow synthesis of a key steroid intermediate for an endothelin receptor antagonist. Tetrahedron Lett. 2006, 47, 6197–6200. [Google Scholar]
- Sugimoto, A.; Fukuyama, T.; Sumino, Y.; Takagi, M.; Ryu, I. Microflow photo-radical reaction using a compact light source: Application to the Barton reaction leading to a key intermediate for myriceric acid A. Tetrahedron 2009, 65, 1593–1598. [Google Scholar] [CrossRef]
- Vasudevan, A.; Villamil, C.; Trumbull, J.; Olson, J.; Sutherland, D.; Pan, J.; Djuric, S. LOPHTOR: A convenient flow-based photochemical reactor. Tetrahedron Lett. 2010, 51, 4007–4009. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Terao, K.; Yamaguchi, H.; Yoshimura, S.; Morimoto, T.; Kakiuchi, K.; Fukuyama, T.; Ryu, I. Diastereoselective [2+2] photocycloaddition of chiral cyclic enone and cyclopentene using a microflow reactor system. Chem. Lett. 2010, 39, 828–829. [Google Scholar] [CrossRef]
- Ouchi, A.; Sakai, H.; Oishi, T.; Kaneda, M.; Suzuki, T.; Saruwatari, A.; Obata, T. Photochemical reduction of flavone with NaBH4 in batch and micro-channel reactors using excimer lasers. J. Photochem. Photobiol. A-Chem. 2008, 199, 261–266. [Google Scholar]
- Nakayama, T.; Shimizu, T.; Torii, Y.; Miki, S.; Hamanoue, K. A comparison of the photochemistry of flavanone with that of flavone originating from their lowest excited triplet states in ethanol. J. Photochem. Photobiol. A-Chem. 1997, 111, 35–39. [Google Scholar]
- Griesbeck, A.G.; Kramer, W.; Oelgemöller, M. Synthetic applications of photoinduced electron transfer decarboxylation reactions. Synlett 1999, 1169–1178. [Google Scholar]
- McDermott, G.; Yoo, D.J.; Oelgemöller, M. Photochemical addition reactions involving phthalimides. Heterocycles 2005, 65, 2221–2257. [Google Scholar] [CrossRef]
- Belluau, V.; Noeureuil, P.; Ratzke, E.; Skvortsov, A.; Gallagher, S.; Motti, C.A.; Oelgemöller, M. Photodecarboxylative benzylations of phthalimide in pH 7 buffer: A simple access to 3-arylmethyleneisoindolin-1-ones. Tetrahedron Lett. 2010, 51, 4738–4741. [Google Scholar]
- Shvydkiv, O.; Gallagher, S.; Nolan, K.; Oelgemöller, M. From conventional to microphotochemistry: Photodecarboxylation reactions involving phthalimides. Org. Lett. 2010, 12, 5170–5173. [Google Scholar] [CrossRef]
- Shvydkiv, O.; Nolan, K.; Oelgemöller, M. Microphotochemistry—4,4’-dimethoxybenzophenone mediated photodecarboxylation reactions involving phthalimides. Beilstein J. Org. Chem. 2011, 7, 1055–1063. [Google Scholar] [CrossRef]
- Tan, S.B.; Shvydkiv, O.; Fiedler, J.; Hatoum, F.; Nolan, K.; Oelgemöller, M. Photodecarboxylative additions of α-thioalkyl-substituted carboxylates to alkyl phenylglyoxolates. Synlett 2010, 2240–2243. [Google Scholar]
- Neumann, M.; Zeitler, K. Organocatalysis meets photoredox chemistry. In Book of Abstracts, Organocatalysis. New Methodologies for Sustainable Chemistry; CATAFLU.OR Symposium: Bologna, Italy, 2011; p. 23. [Google Scholar]
- Neumann, M.; Füldner, S.; König, B.; Zeitler, K. Metal-free, cooperative asymmetric organophotoredox catalysis with visible light. Angew. Chem. Int. Ed. 2011, 50, 951–954. [Google Scholar]
- Shvydkiv, O.; Yavorskyy, A.; Nolan, K.; Youssef, A.; Riguet, E.; Hoffmann, N.; Oelgemöller, M. Photosensitized addition of isopropanol to furanones in a 365 nm UV-LED microchip. Photochem. Photobiol. Sci. 2010, 9, 1601–1603. [Google Scholar] [CrossRef]
- Yavorskyy, A.; Shvydkiv, O.; Nolan, K.; Hoffmann, N.; Oelgemöller, M. Photosensitized addition of isopropanol to furanones in a continuous-flow dual capillary microreactor. Tetrahedron Lett. 2011, 52, 278–280. [Google Scholar] [CrossRef]
- Shvydkiv, O.; Yavorskyy, A.; Tan, S.B.; Nolan, K.; Hoffmann, N.; Youssef, A.; Oelgemöller, M. Microphotochemistry—A reactor comparison study using the photosensitized addition of isopropanol to furanones as a model reaction. Photochem. Photobiol. Sci. 2011, 10, 1399–1404. [Google Scholar] [CrossRef]
- Horie, T.; Sumino, M.; Tanaka, T.; Matsushita, Y.; Ichimura, T.; Yoshida, J. Photodimerization of maleic anhydride in a microreactor without clogging. Org. Process Res. Dev. 2010, 14, 405–410. [Google Scholar] [CrossRef]
- Jähnisch, K.; Dingerdissen, U. Photochemical generation and [4+2]-cycloaddition of singlet oxygen in a falling-film microreactor. Chem. Eng. Technol. 2005, 28, 426–427. [Google Scholar] [CrossRef]
- Ehrich, H.; Linke, D.; Morgenschweis, K.; Baerns, M.; Jähnisch, K. Application of microstructured reactor technology for the photochemical chlorination of alkylaromatics. Chimia 2002, 56, 647–653. [Google Scholar] [CrossRef]
- Carofiglio, T.; Donnola, P.; Maggini, M.; Rossetto, M.; Rossi, E. Fullerene-promoted singlet-oxygen photochemical oxygenations in glass-polymer microstructured reactors. Adv. Synth. Catal. 2008, 350, 2815–2822. [Google Scholar] [CrossRef]
- Park, C.P.; Maurya, R.A.; Lee, J.H.; Kim, D.-P. Efficient photosensitized oxygenations in phase contact enhanced microreactors. Lab Chip 2011, 11, 1941–1945. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Jung, C.; Ortner, J.; Mattay, J.; Zimmermann, E. Green photochemistry: Solar photooxygenations with medium concentrated sunlight. Green Chem. 2005, 7, 35–38. [Google Scholar] [CrossRef]
- Matsubara, H.; Hino, Y.; Tokizane, M.; Ryu, I. Microflow photo-radical chlorination of cyclohexanes. Chem. Eng. J. 2011, 167, 567–571. [Google Scholar] [CrossRef]
- Freitag, A.; Dietrich, T.; Scholz, R. Energy efficient photochemistry in automated microreaction plants. In Proceedings of the 11th International Conference on Microreaction Technology (IMRET 11), Kyoto, Japan, 8-10 March 2010; pp. 24–25.
- Werner, S.; Seliger, R.; Rauter, H.; Wissmann, F. Quarzglas-Mikrophotoreaktor und Synthese von 10-Hydroxycamptothecin und 7-Alkyl-10-hydroxycamptothecin. Chem. Abstr. 2009, 150, 376721, EP 2065387A2, 2009.. [Google Scholar]
- Bou-Hamdan, F.R.; Lévesque, F.; O’Brien, A.G.; Seeberger, P.H. Continuous flow photolysis of aryl azides: Preparation of 3H-azepinones. Beilstein J. Org. Chem. 2011, 7, 1124–1129. [Google Scholar] [CrossRef]
- Maurya, R.A.; Park, C.P.; Kim, D.-P. Triple-channel microreactor for biphasic gas-liquid reactions: photosensitized oxygenations. Beilstein J. Org. Chem. 2011, 7, 1158–1163. [Google Scholar] [CrossRef]
- Gutierrez, A.C.; Jamison, T.F. Scalable and robust synthesis of CpRu(MeCN)3PF6 via continuous flow photochemistry. J. Flow Chem. 2011, 1, 24–27. [Google Scholar] [CrossRef]
- Fukuyama, T.; Kajihara, Y.; Hino, Y.; Ryu, I. Continuous microflow [2+2] photocycloadditions reactions using energy-saving compact light sources. J. Flow Chem. 2011, 1, 40–45. [Google Scholar]
- Dionigi, F.; Vesborg, P.C.K.; Pedersen, T.; Hansen, O.; Dahl, S.; Xiong, A.; Maeda, K.; Domen, K.; Chorkendorff, I. Gas phase photocatalytic water splitting with Rh2-yCryO3/GaN:ZnO in μ-reactors. Energy Environ. Sci. 2011, 4, 2937–2942. [Google Scholar]
- Ciamician, G. The photochemistry of the future. Science 1912, 36, 385–394. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oelgemöller, M.; Shvydkiv, O. Recent Advances in Microflow Photochemistry. Molecules 2011, 16, 7522-7550. https://doi.org/10.3390/molecules16097522
Oelgemöller M, Shvydkiv O. Recent Advances in Microflow Photochemistry. Molecules. 2011; 16(9):7522-7550. https://doi.org/10.3390/molecules16097522
Chicago/Turabian StyleOelgemöller, Michael, and Oksana Shvydkiv. 2011. "Recent Advances in Microflow Photochemistry" Molecules 16, no. 9: 7522-7550. https://doi.org/10.3390/molecules16097522
APA StyleOelgemöller, M., & Shvydkiv, O. (2011). Recent Advances in Microflow Photochemistry. Molecules, 16(9), 7522-7550. https://doi.org/10.3390/molecules16097522