Generation of the First Structure-Based Pharmacophore Model Containing a Selective “Zinc Binding Group” Feature to Identify Potential Glyoxalase-1 Inhibitors

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
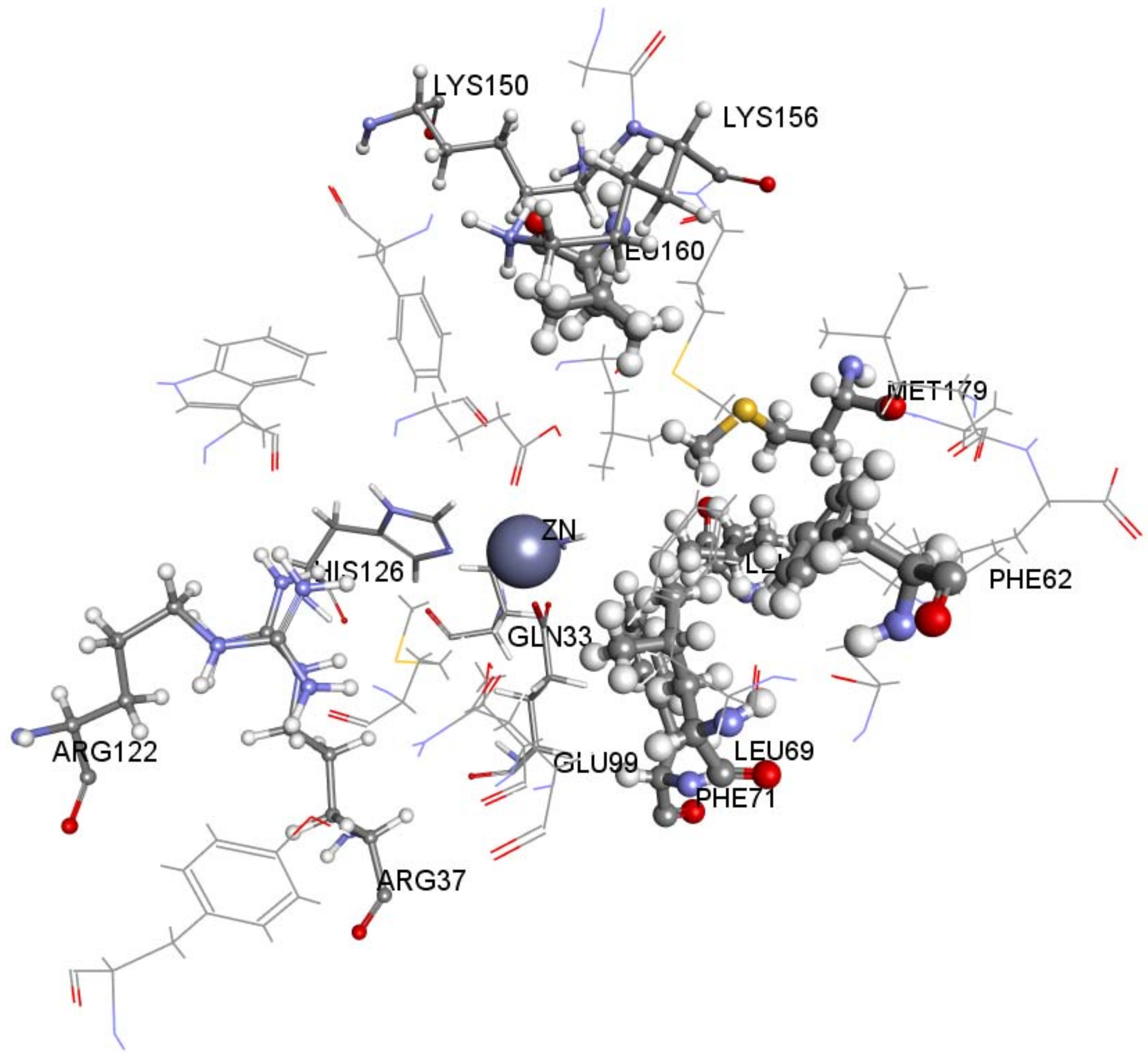
2.1. Structure-Based Pharmacophore Generation
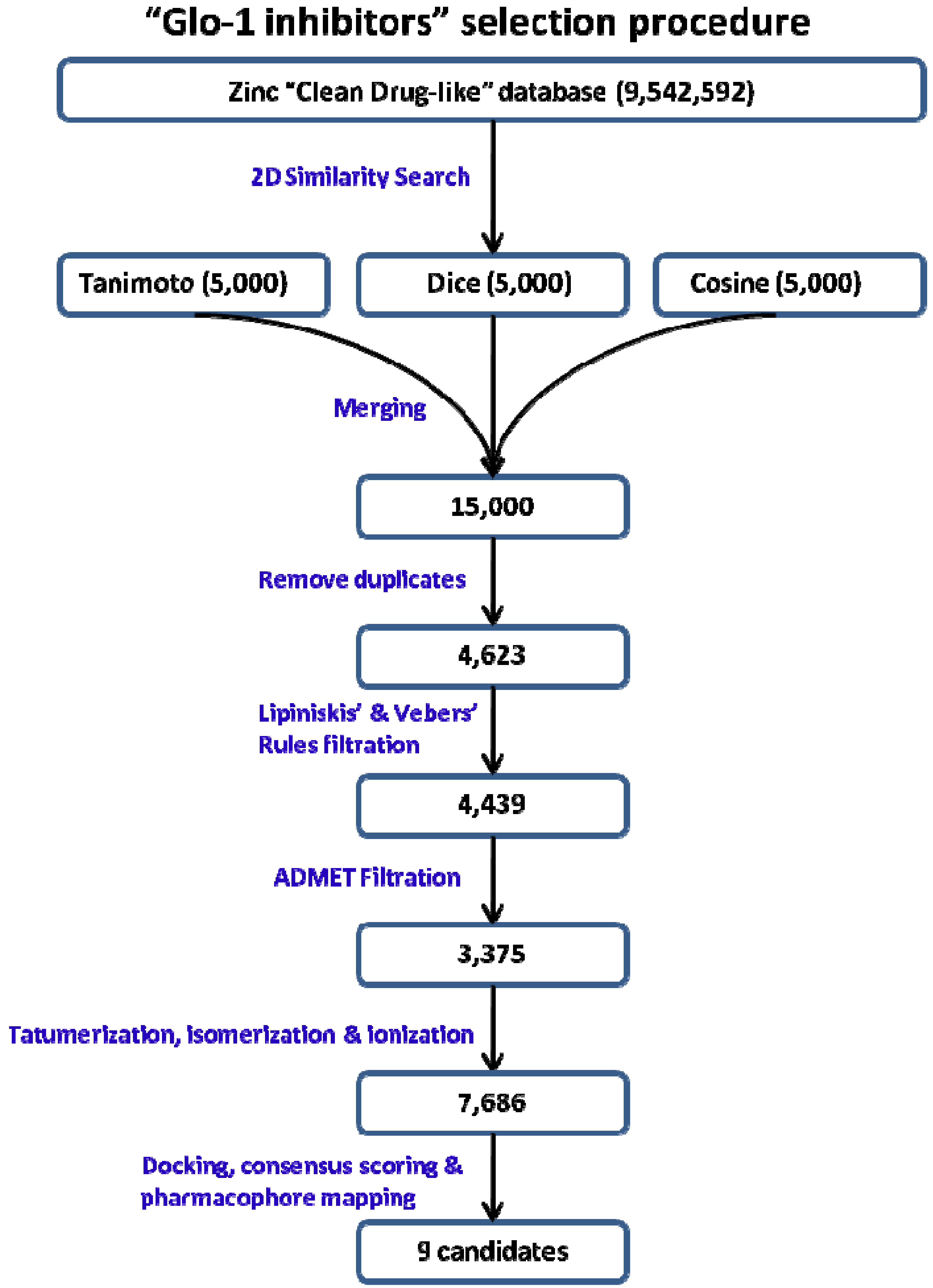
2.2. 2D Similarity Search
2.3. Mapping 2D Similarity Search Compounds over Structure-Based Pharmacophore
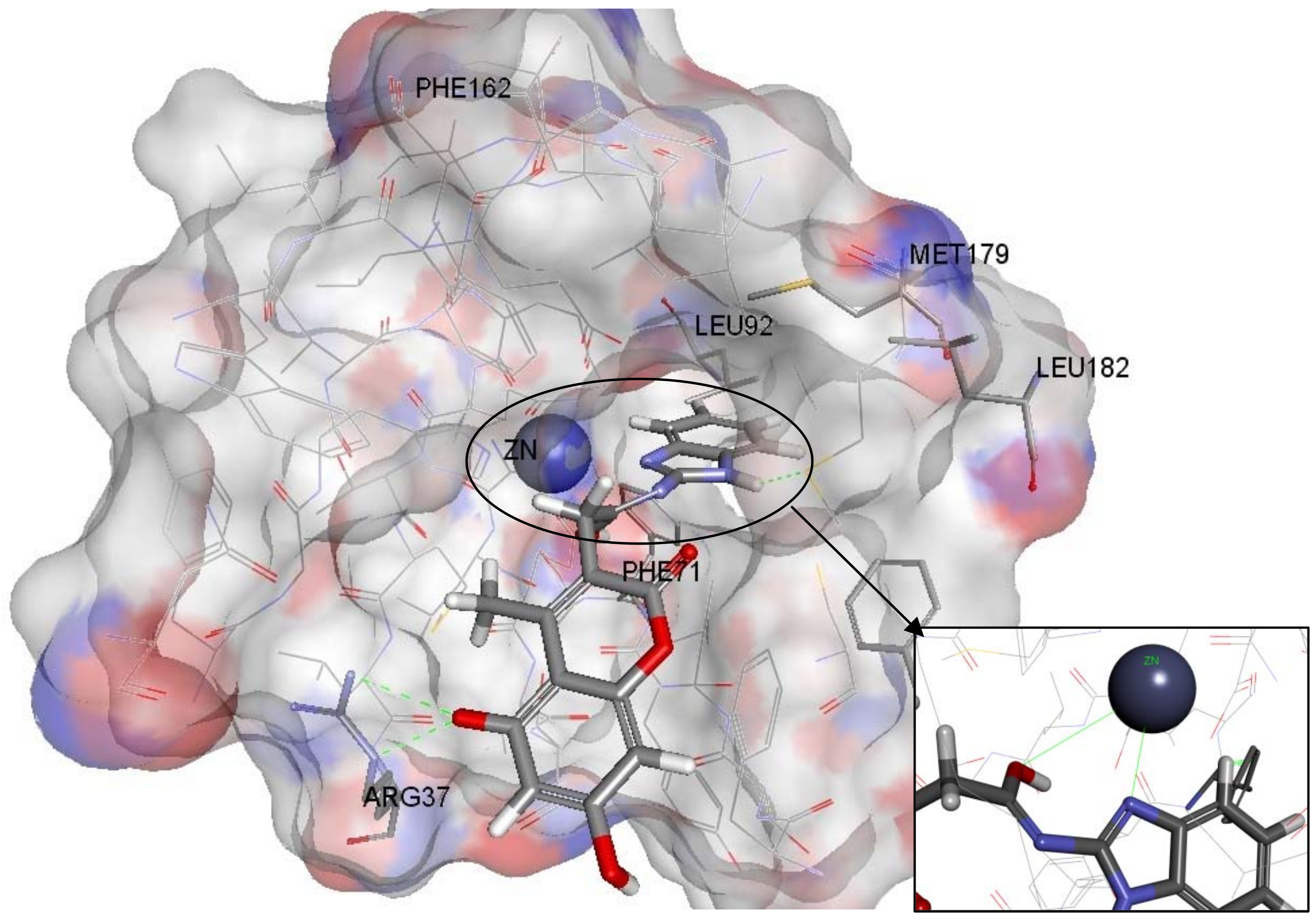
2.4. Molecular Docking
3. Experimental
3.1. Preparing Glyoxlase-1 Enzyme
3.2. Creation of Structure-Based Pharmacophore
3.3. Addition of “Zinc Binding Feature” to the Structure-Based Pharmacophore
3.4. 2D similarity Search
3.5. Mapping 2D Similarity Search Compounds over Structure-Based Pharmacophore
3.6. Molecular Docking
4. Conclusions
References
- National Cancer Institute. Available online: http://www.cancer.gov/ (accessed 2 June 2012).
- Jemal, A.; Bray, F.; Center, M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). The Global Burden of Disease: 2004 Update; WHO: Geneva, Switzerland; Available online: http://www.who.int/topics/global_burden_of_disease/en/ (accessed 2 June 2012).
- Aziz, N.M.; Rowland, J.H. Trends and advances in cancer survivorship research: Challenge and opportunity. Semin. Radiat. Oncol. 2003, 13, 248–266. [Google Scholar] [CrossRef]
- Petrelli, N.J.; Winer, E.P.; Brahmer, J.; Dubey, S.; Smith, S.; Thomas, T.; Vahdat, L.T.; Obel, J.; Vogelzang, N.; Markman, M.; et al. Clinical Cancer Advances 2009: Major Research Advances in Cancer Treatment, Prevention, and Screening: A Report from the American Society of Clinical Oncology. J. Clin. Oncol. 2009, 27, 6052–6069. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Milanesa, D.; Mallouh, C.; Choudhury, M.; Tazaki, H.; Konno, S. A possible regulatory role of glyoxalase I in cell viability of human prostate cancer. Urol. Res. 2002, 30, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P. Glyoxalase in diabetes, Obesity and related disorders. Semin. Cell. Dev. Biol. 2011, 22, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell. Dev. Biol. 2011, 22, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Rabbani, N.; Thornalley, P. Glyoxalase in ageing. Semin. Cell. Dev. Biol. 2011, 22, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mashima, T.; Kizaki, A.; Dan, S.; Hashimoto, Y.; Naito, M.; Tsuruo, T. Glyoxalase I is involved in resistance of human leukemia cells to antitumor agent-induced apoptosis. Blood 2000, 95, 3214–3218. [Google Scholar] [PubMed]
- National Toxicology Program. Final Report on Carcinogens Background Document for Formaldehyde. Rep. Carcinog. Backgr. Doc. 2010, 10-5981, i-512. [Google Scholar]
- Chaplen, F. Incidence and potential implications of the toxic metabolite methylglyoxal in cell culture: A review. Cytotechnology 1998, 26, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Suttisansanee, U.; Honek, J.F. Bacterial glyoxalase enzymes. Semin. Cell. Dev. Biol. 2011, 22, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Feierberg, I.; Luzhkov, V.; Aqvist, J. Computer Simulation of Primary Kinetic Isotope Effects in the Proposed Rate-limiting Step of the Glyoxalase I Catalyzed Reaction. J. Biol. Chem. 2000, 275, 22657–22662. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P. Protecting the genome: Defence against nucleotide glycation and emerging role of glyoxalase I overexpression in multidrug resistance in cancer chemotherapy. Biochem. Soc. Trans. 2003, 31, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P. Glyoxalase I—Structure, Function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.; Olin, B.; Ridderström, M.; Mannervik, B.; Jones, T. Crystal structure of human glyoxalase I-evidence for gene duplication and 3D domain swapping. EMBO J. 1997, 16, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.; Ridderström, M.; Olin, B.; Kavarana, M.; Creighton, D.; Mannervik, B. Reaction Mechanism of Glyoxalase I Explored by an X-ray Crystallographic Analysis of the Human Enzyme in Complex with a Transition State Analogue. Biochemistry 1999, 38, 13480–13490. [Google Scholar] [CrossRef] [PubMed]
- Vince, R.; Daluge, S.; Wadd, W. Inhibition of glyoxalase I by S-substituted glutathiones. J. Med. Chem. 1971, 14, 402–404. [Google Scholar] [CrossRef] [PubMed]
- More, S.; Vince, R. A metabolically stable tight-binding transition-state inhibitor of glyoxalase-I. Bioorg. Med. Chem. Lett. 2006, 16, 6039–6042. [Google Scholar] [CrossRef] [PubMed]
- More, S.; Vince, R. Design, synthesis, and binding studies of bidentate Zn-chelating peptidic inhibitors of glyoxalase-I. Bioorg. Med. Chem. Lett. 2007, 17, 3793–3797. [Google Scholar] [CrossRef] [PubMed]
- More, S.; Vince, R. Inhibition of Glyoxalase I: The First Low-Nanomolar Tight-Binding Inhibitors. J. Med. Chem. 2009, 52, 4650–4656. [Google Scholar] [CrossRef] [PubMed]
- Takayoshi, N. Rational Design of Non-Hydroxamate Histone Deacetylase Inhibitors. Mini Rev. Med. Chem. 2006, 6, 515–526. [Google Scholar]
- Maingot, L.; Leroux, F.; Landry, V.; Dumont, J.; Nagase, H.; Villoutreix, B.; Sperandio, O.; Deprez-Poulain, R.; Deprez, B. New non-hydroxamic ADAMTS-5 inhibitors based on the 1,2,4-triazole-3-thiol scaffold. Bioorg. Med. Chem. Lett. 2010, 20, 6213–6216. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, R.; Saeki, K.; Tao, A.; Yoshimori, A.; Uchiro, H.; Fujiwara, M.; Tanuma, S. Delphinidin, a dietary anthocyanidin in berry fruits, inhibits human glyoxalase I. Bioorg. Med. Chem. 2010, 18, 7029–7033. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, R.; Takahashi, S.; Saeki, K.; Sunaga, S.; Yoshimori, A.; Tanuma, S. Structure activity relationship of human GLO I inhibitory natural flavonoids and their growth inhibitory effects. Bioorg. Med. Chem. 2008, 16, 3969–3975. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, R.; Tao, A.; Saeki, K.; Shionozaki, N.; Tanaka, R.; Uchiro, H.; Takahashi, S.; Yoshimori, A.; Tanuma, S. Discovery of a new type inhibitor of human glyoxalase I by myricetin-based 4-point pharmacophore. Bioorg. Med. Chem. Lett. 2011, 21, 4337–4342. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yuan, M.; Luo, M.; Bu, X.; Luo, H.B.; Hu, X. Binding of curcumin with glyoxalase I: Molecular docking, Molecular dynamics simulations, and kinetics analysis. Biophys. Chem. 2010, 147, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Luo, M.; Song, Y.; Xu, Q.; Wang, X.; Cao, Y.; Bu, X.; Ren, Y.; Hu, X. Identification of curcumin derivatives as human glyoxalase I inhibitors: A combination of biological evaluation, Molecular docking, 3D-QSAR and molecular dynamics simulation studies. Bioorg. Med. Chem. 2011, 19, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Glen, R. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef] [PubMed]
- Mark, A.; Johnson, G. Concepts and Applications of Molecular Similarity, 1st ed.; Wiley-Interscience: New York, NY, USA, 1990. [Google Scholar]
- Eckert, H.; Bajorath, J. Molecular similarity analysis in virtual screening: Foundations, Limitations and novel approaches. Drug Discov. Today 2007, 12, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.; Raushel, F.; Shoichet, B. Virtual Screening against Metalloenzymes for Inhibitors and Substrates. Biochemistry 2005, 44, 12316–12328. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F.; Vulpetti, A. Tautomer Preference in PDB Complexes and its Impact on Structure-Based Drug Discovery. J. Chem. Inf. Model. 2010, 50, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y. Let’s not forget tautomers. J. Comput.-Aided. Mol. Des. 2009, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Mulder, G.; Meerman, J. Sulfation and glucuronidation as competing pathways in the metabolism of hydroxamic acids: the role of N,O-sulfonation in chemical carcinogenesis of aromatic amines. Environ. Health Perspect. 1983, 49, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, S.; Mucha, A.; Cuniasse, P.; Georgiadis, D.; Lucet-Levannier, K.; Beau, F.; Kannan, R.; Murphy, G.; Knäuper, V.; Rio, M.C.; et al. Phosphinic Pseudo-Tripeptides as Potent Inhibitors of Matrix Metalloproteinases: A Structure-Activity Study. J. Med. Chem. 1999, 42, 2610–2620. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, M.; Floyd, C.D.; Brown, P.; Gearing, A.J. Design and Therapeutic Application of Matrix Metalloproteinase Inhibitors. Chem. Rev. 1999, 99, 2735–2776. [Google Scholar] [CrossRef] [PubMed]
- Douglass, R.; Heckman, G. Drug-related taste disturbance. Can. Fam. Physician 2010, 56, 1142–1147. [Google Scholar] [PubMed]
- Irwin, J.; Shoichet, B. ZINC: A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2004, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, in press. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins: Struct. Funct. Bioinf. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Diller, D.; Merz, K. High throughput docking for library design and library prioritization. Proteins: Struct. Funct. Bioinf. 2001, 43, 113–124. [Google Scholar] [CrossRef]
- Diller, D.; Li, R. Kinases, Homology Models, and High Throughput Docking. J. Med. Chem. 2003, 46, 4638–4647. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.N.; Head, M.S.; Kulkarni, A.; LaLonde, J.M. Validation Studies of the Site-Directed Docking Program LibDock. J. Chem. Inf. Model. 2007, 47, 2159–2171. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacophore Summary | |||
|---|---|---|---|
| Pharmacophore | Number of Features | Feature Set | Selectivity Score |
| Pharmacophore_01 | 6 | DDDHNN a | 8.5416 |
| Pharmacophore_02 | 6 | ADDHNN | 8.0771 |
| Pharmacophore_03 | 6 | ADDHNN | 8.0771 |
| Pharmacophore_04 | 6 | DDHHNN | 7.9040 |
| Pharmacophore_05 | 6 | DDDHNN | 7.9040 |
| Pharmacophore_06 | 6 | ADDHNN | 7.7474 |
| Pharmacophore_07 | 6 | ADDHNN | 7.6186 |
| Pharmacophore_08 | 6 | ADDHNN | 7.6186 |
| Pharmacophore_09 | 6 | ADDHNN | 7.5951 |
| Pharmacophore_10 | 6 | ADDHNN | 7.5951 |
| Name | Structure | Consensus score | ZB group |
|---|---|---|---|
| Zinc02120846 | 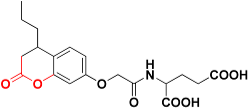 | 492.4 | 2H-pyran-2-one |
| Zinc05528245 |  | 486.9 | 4-oxo-4H-chromen-5-olate |
| Zinc13100715 |  | 485.8 | Sulfonamide |
| Zinc01747330 | 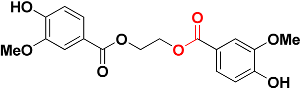 | 482.6 | Ester |
| Zinc06403375 | 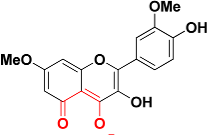 | 467.2 | 5-oxo-5H-chromen-4-olate |
| Zinc08791976 | 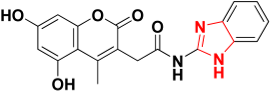 | 465.9 | Imidazole ring |
| Zinc05732763 | 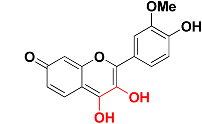 | 463.9 | 3,4-dihydroxy-7H-chromen-7-one |
| Zinc00044208 |  | 463.3 | Carboxylic acid |
| Zinc01863036 | 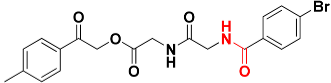 | 463.1 | Amide |
© 2012 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Balas, Q.; Hassan, M.; Al-Oudat, B.; Alzoubi, H.; Mhaidat, N.; Almaaytah, A. Generation of the First Structure-Based Pharmacophore Model Containing a Selective “Zinc Binding Group” Feature to Identify Potential Glyoxalase-1 Inhibitors. Molecules 2012, 17, 13740-13758. https://doi.org/10.3390/molecules171213740
Al-Balas Q, Hassan M, Al-Oudat B, Alzoubi H, Mhaidat N, Almaaytah A. Generation of the First Structure-Based Pharmacophore Model Containing a Selective “Zinc Binding Group” Feature to Identify Potential Glyoxalase-1 Inhibitors. Molecules. 2012; 17(12):13740-13758. https://doi.org/10.3390/molecules171213740
Chicago/Turabian StyleAl-Balas, Qosay, Mohammad Hassan, Buthina Al-Oudat, Hassan Alzoubi, Nizar Mhaidat, and Ammar Almaaytah. 2012. "Generation of the First Structure-Based Pharmacophore Model Containing a Selective “Zinc Binding Group” Feature to Identify Potential Glyoxalase-1 Inhibitors" Molecules 17, no. 12: 13740-13758. https://doi.org/10.3390/molecules171213740