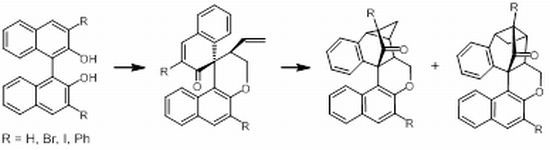
Tandem RCM–Claisen Rearrangement–[2+2] Cycloaddition of O,O'-(But-2-en-1,4-diyl)-bridged Binaphthols

Abstract
:
1. Introduction
2. Results and Discussion
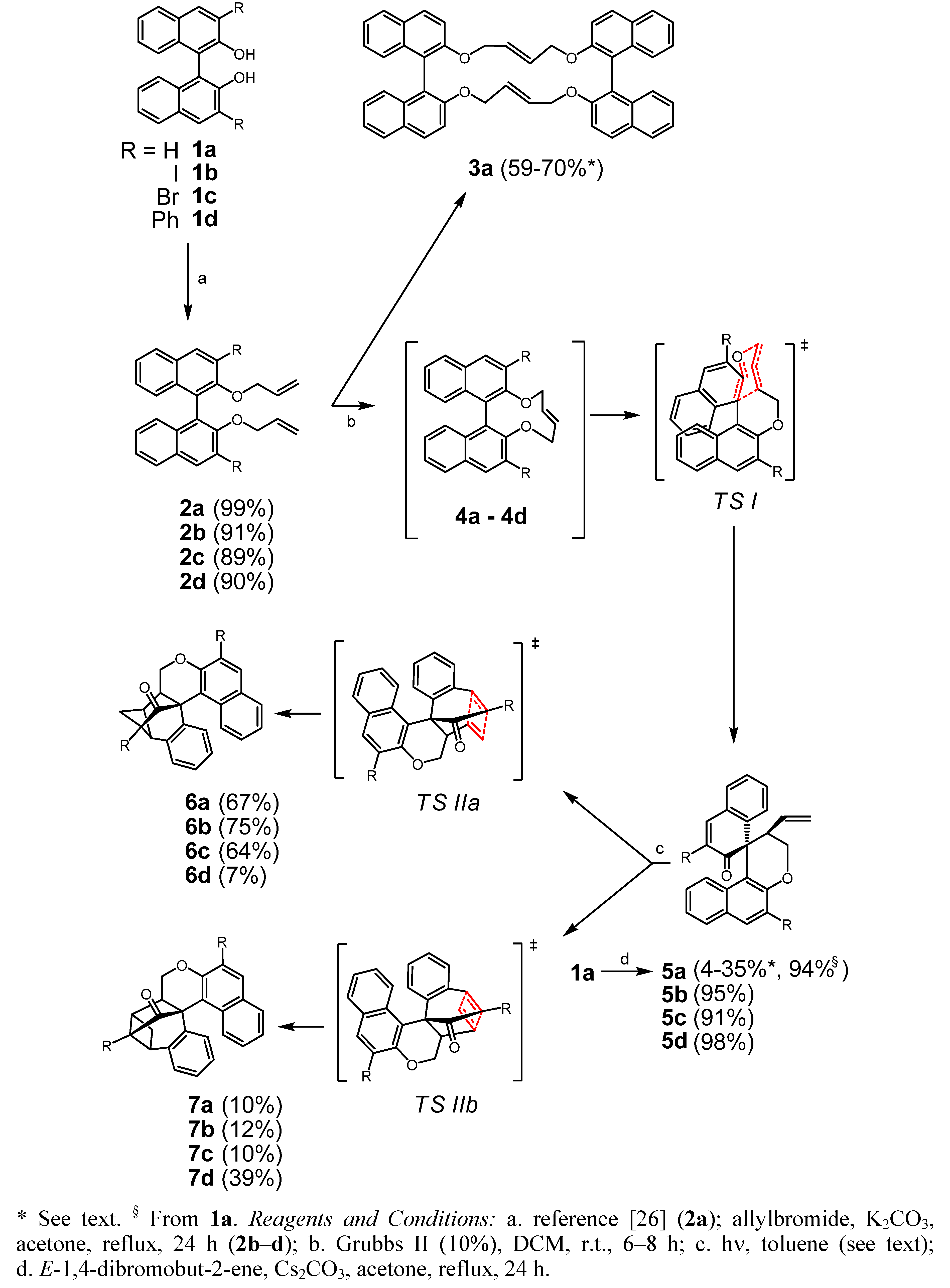
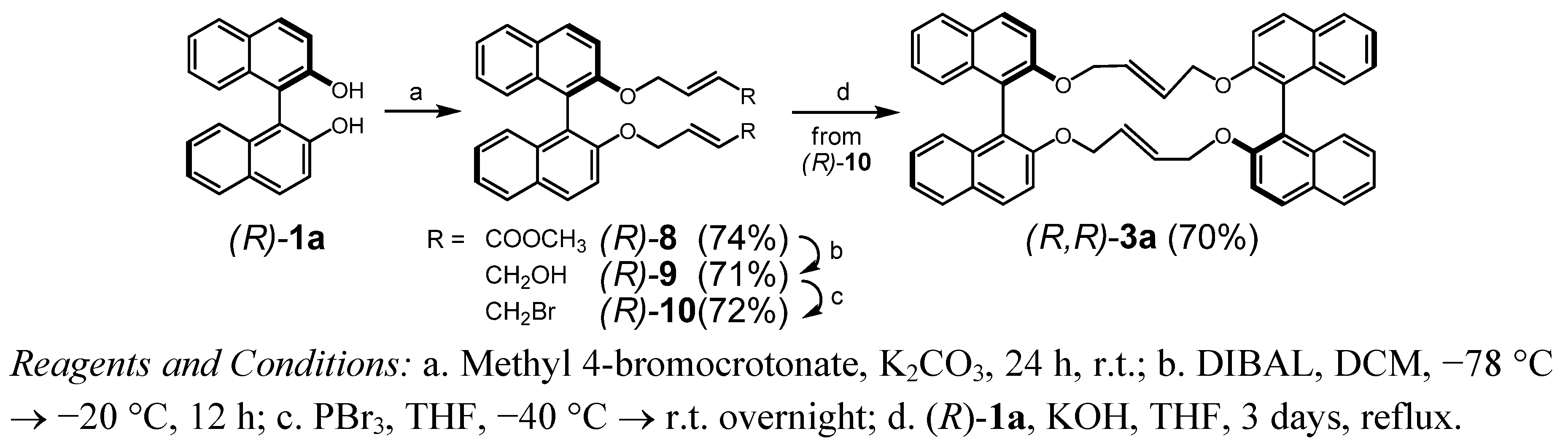
2.1. Synthesis and Rearrangements





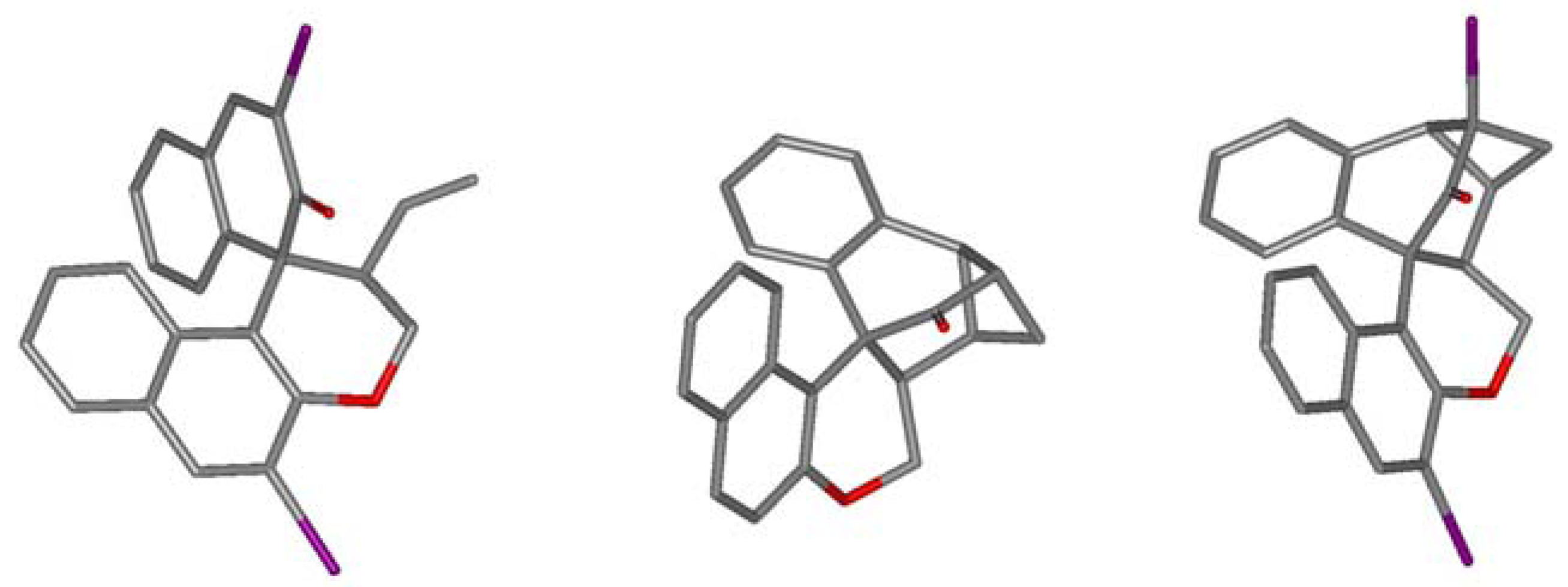
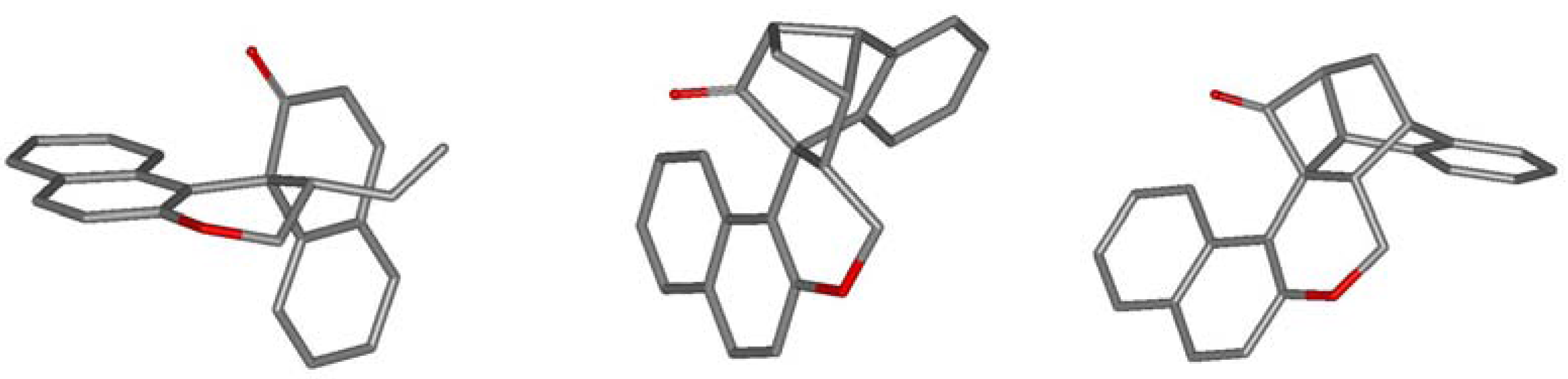
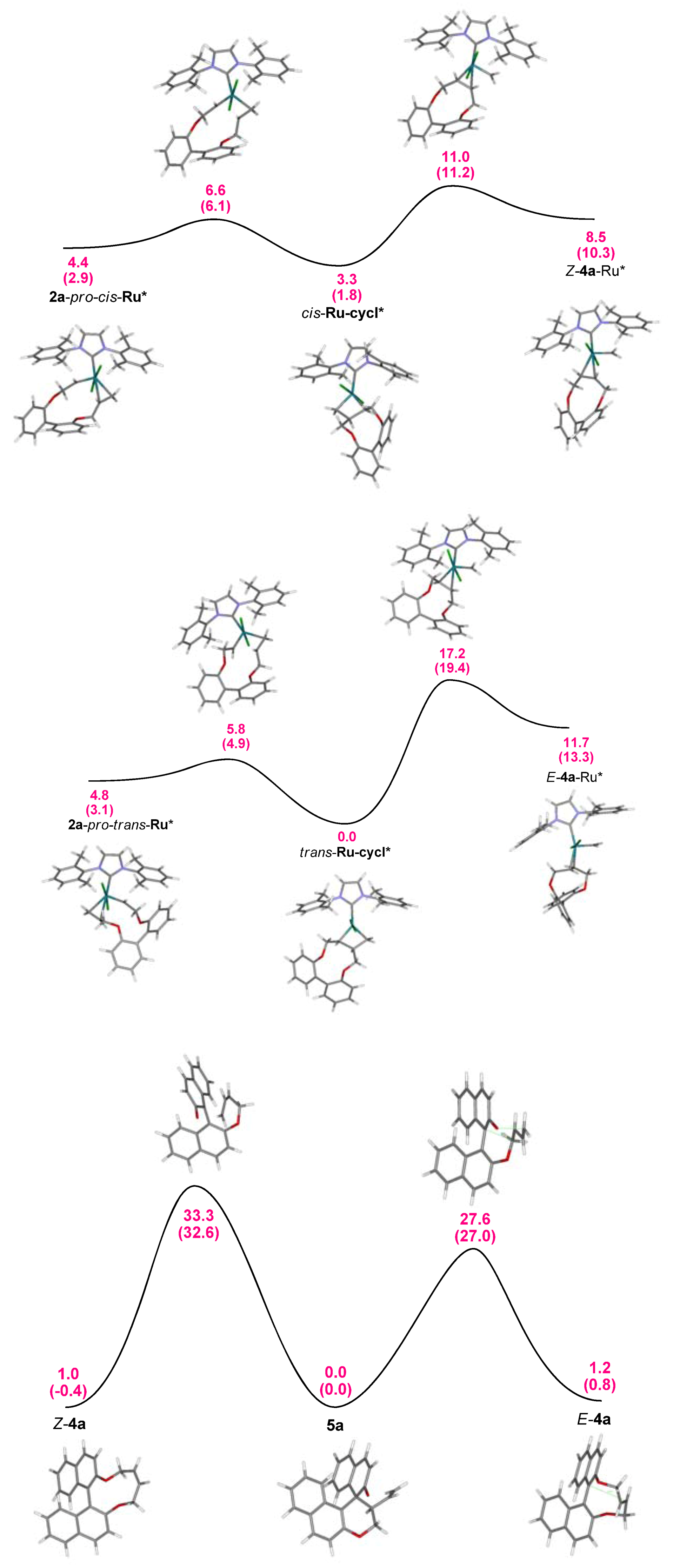
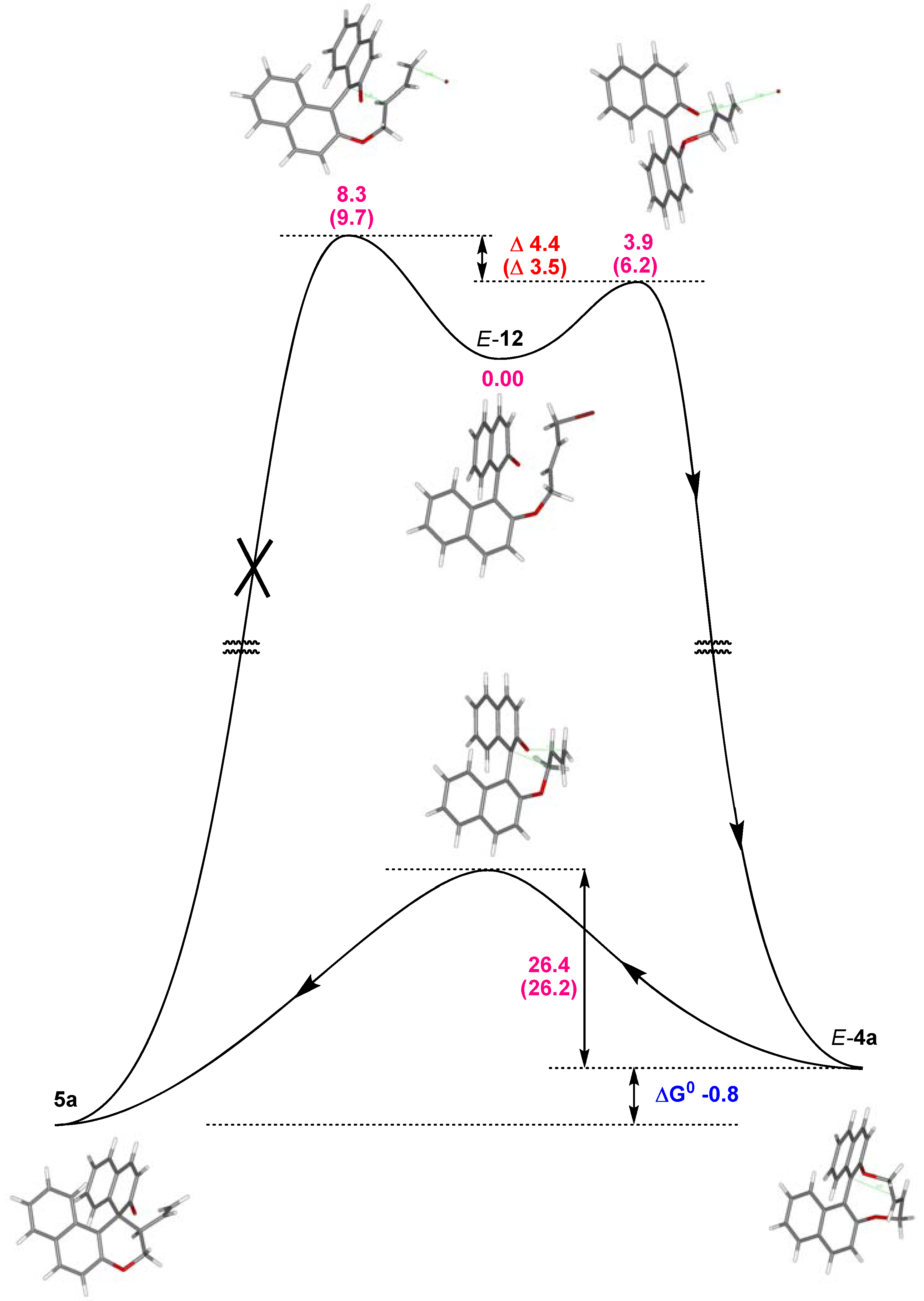
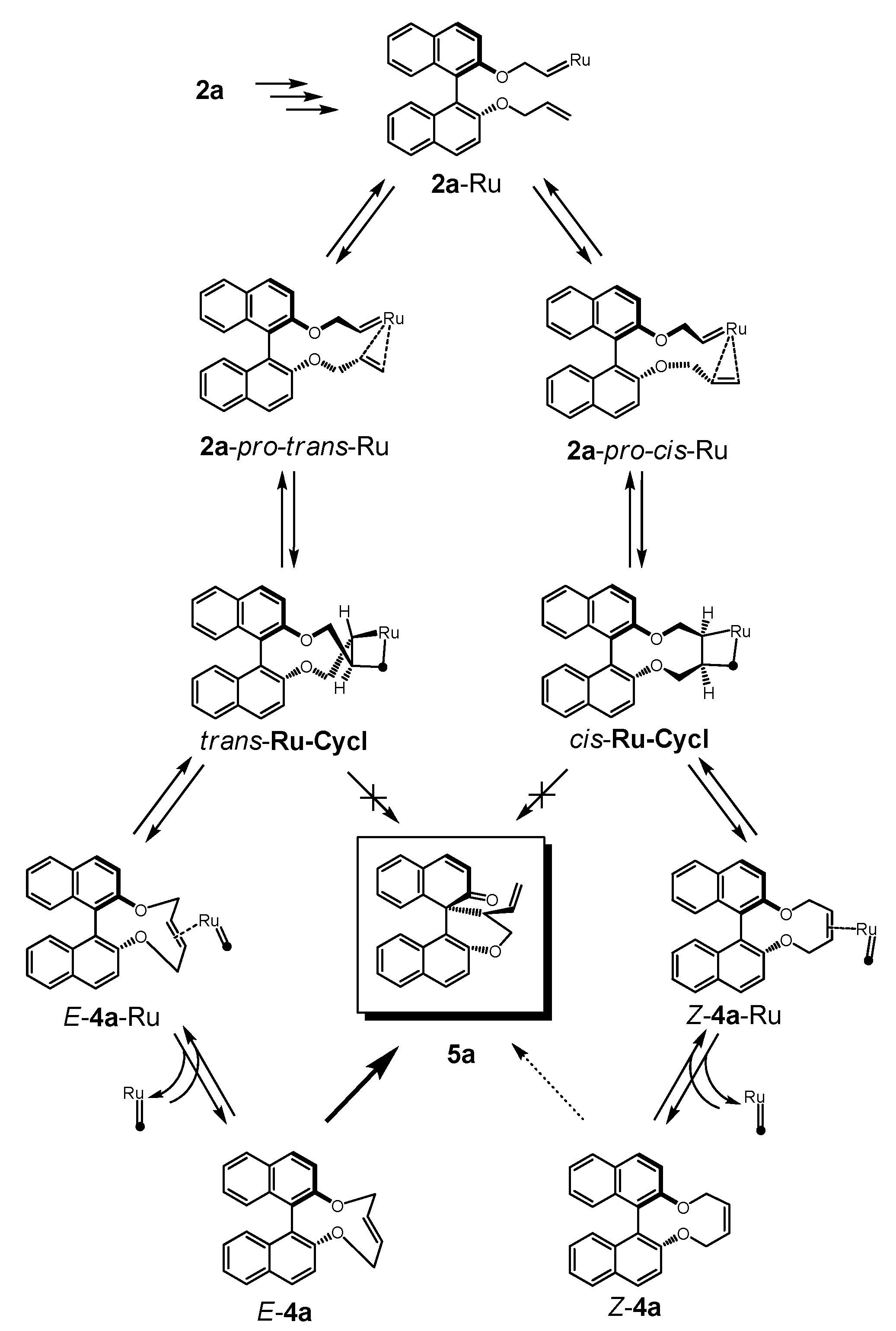
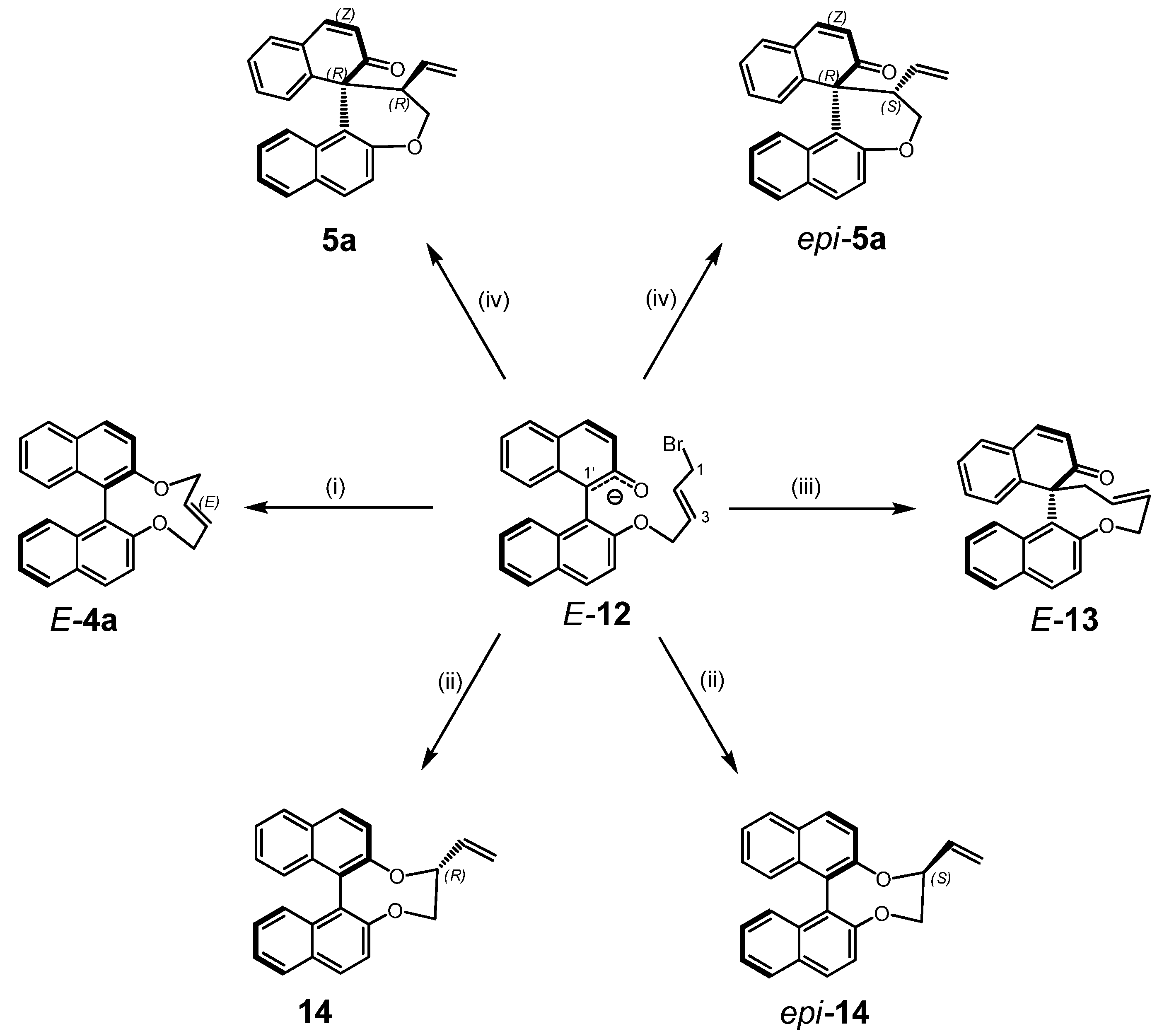
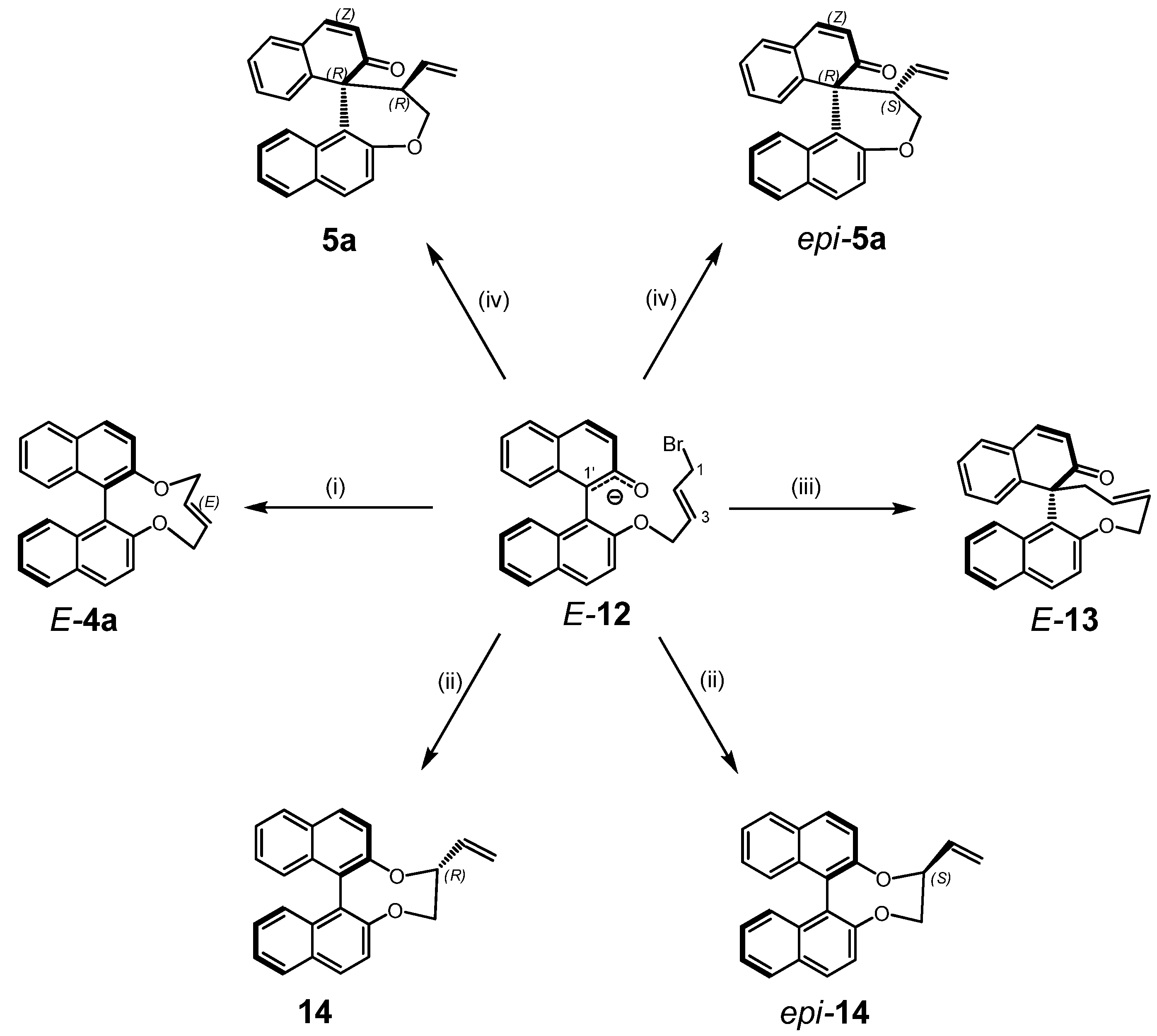
2.2. Calculations, Evidence for Existence of 4a, Its Geometry and Rearrangement to 5a

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔG‡ b | |||
|---|---|---|---|
| B3LYP | MO6 | MP2 | |
| 2a-pro-trans-Ru* → trans-Ru-Cycl* | 1.0 (1.8) | ||
| 2a-pro-cis-Ru* → cis-Ru-Cycl* | 2.2 (3.2) | ||
| trans-Ru-Cycl* → E-4a-Ru* | 17.2 (19.4) | ||
| cis-Ru-Cycl* → Z-4a-Ru* | 7.7 (9.4) | ||
| E-4a → 5a | 26.4 (26.2) | 27.3 | 21.0 |
| Z-4a → 5a | 32.3 (33.0) | 32.2 | 26.9 |
| E-12 → E-4a | 3.9 (6.2) | 8.3 | 6.0 |
| E-12 → 5a | 8.3 (9.7) | 10.6 | 8.2 |
| E-12 → epi-5a | 8.6 (9.8) | 10.4 | 7.8 |
| E-12 → E-13 | 39.1 | 40.0 | 35.2 |
| E-12 → 14 | 7.3 | 13.5 | 13.6 |
| E-12 → epi-14 | 7.1 | 14.4 | 16.2 |
| E-4a → epi-5a | 39.1 | 41.4 | 39.2 |



3. Experimental
3.1. General
3.2. Syntheses and Rearrangements
3.2.1. Typical Procedure: 2,2'-Bis(allyloxy)-3,3'-diiodo-1,1'-binaphthyl (2b)
3.2.2. (15E,33E)-14,17,32,35-Tetrahydrotetranaphtho[2,1-b:1',2'-d:2'',1''-l:1''',2''' n][1,6,11,16]tetraoxa-cycloicosine (3a) and 2-Vinyl-2,3-dihydro-2'H-spiro[benzo[f]chromene-1,1'-naphthalen]-2'-one (5a)
3.2.3. One Step Preparation of 5a from 1a
3.2.4. Photoisomerisation of 5a
3.2.5. Photoisomerisation of epi-5a
3.2.6. 3',5-Diiodo-2-vinyl-2,3-dihydro-2'H-spiro[benzo[f]chromene-1,1'-naphthalen]-2'-one (5b)
3.2.7. Photoisomerisation of 5b (Typical Procedure)
3.2.8. Synthesis of 6b/7b from 2b (One Pot Procedure)
3.3. Crystallographic Structure Determination
| Compound | epi-5a | 5b | 6a | epi-6a | 6b | 11 |
|---|---|---|---|---|---|---|
| formula | C24H18O2 | C24H16I2O2 | C24H18O2 | C24H18O2 | C73.88H52.25I6O7.13 | C24H18O2 |
| Fw | 338.38 | 590.17 | 338.38 | 338.38 | 1815.31 | 338.38 |
| space group | P21212 | P-1 | P212121 | P21 | C2/c | P212121 |
| a [Å] | 40.275(2) | 8.9274(6) | 9.3461(5) | 11.9996(7) | 28.1310(16) | 7.9726(2) |
| b [Å] | 14.7521(9) | 14.7039(14) | 10.0950(6) | 9.7117(6) | 11.8338(8) | 11.6491(4) |
| c [Å] | 8.8731(5) | 15.9680(16) | 18.2172(11) | 15.2652(9) | 37.359(2) | 17.7858(6) |
| α [°] | 90.792(4) | |||||
| β [°] | 105.289(3) | 108.528(3) | 95.219(6) | |||
| γ [°] | 90.064(3) | |||||
| V [Å3] | 5271.9(5) | 2021.7(3) | 1718.77(17) | 1686.75(17) | 12385.1(13) | 1651.84(9) |
| Z | 12 | 4 | 4 | 4 | 8 | 4 |
| λ [Å] | 0.71073 | 0.70713 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| ρcalcd [g cm−3] | 1.279 | 1.939 | 1.308 | 1.333 | 1.947 | 1.361 |
| T [K] | 100(2) | 100(2) | 100(2) | 150(2) | 100(2) | 150(2) |
| μ [mm−1] | 0.080 | 3.128 | 0.082 | 0.084 | 3.068 | 0.085 |
| R1a | 0.0565 | 0.0628 | 0.0294 | 0.0431 | 0.0483 | 0.0384 |
| wR2b | 0.1402 | 0.1592 | 0.0781 | 0.1011 | 0.0960 | 0.1032 |
| GOFc | 1.054 | 1.191 | 1.034 | 1.045 | 1.083 | 1.049 |
3.4. Calculations
4. Conclusions
Supplementary Materials
Supplementary File 1- Sample Availability: Samples of the compounds 3a and 5a are available from the authors.
References and Notes
- Kataoka, T.; Watanabe, S. Tandem reactions initiated by the conjugate addition of chalcogen compounds utilization and synthesis of heterocycles. Heterocycles 2011, 83, 447–489. [Google Scholar] [CrossRef]
- Li, J.; Lee, D. Enyne-Metathesis-Based Tandem Processes. Eur. J. Org. Chem. 2011, 4269–4287. [Google Scholar]
- Porta, M.; Blechert, S. Cascade metathesis in natural product synthesis. In Metathesis in Natural Product Synthesis; Cossy, J., Arseniyadis, S., Meyer, C., Eds.; John Wiley & Sons: New York, NY, USA, 2010; pp. 313–341. [Google Scholar]
- Fustero, S.; Sanchez-Rosello, M.; del Pozo, C. Asymmetric tandem reactions: New synthetic strategies. Pure Appl. Chem. 2010, 82, 669–677. [Google Scholar] [CrossRef]
- Zhou, J. Recent advances in multicatalyst promoted asymmetric tandem reactions. Chem. Asian J. 2010, 5, 422–434. [Google Scholar] [CrossRef]
- Arns, S.; Barriault, L. Cascading pericyclic reactions: Building complex carbon frameworks for natural product synthesis. Chem. Commun. 2007, 2211–2221. [Google Scholar]
- Mori, M. Synthesis of natural products and related compounds using enyne metathesis. Adv. Synth. Catal. 2007, 349, 121–135. [Google Scholar] [CrossRef]
- Eilbracht, P.; Schmidt, A.M. New synthetic applications of tandem reactions under hydroformylation conditions. In Transition Metals for Organic Synthesis, 2nd; Beller, M., Bolm, C., Eds.; John Wiley & Sons: New York, NY, USA, 2004; Volume 1, pp. 57–85. [Google Scholar]
- Padwa, A. Tandem methodology for heterocyclic synthesis. Pure Appl. Chem. 2004, 76, 1933–1952. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Montagnon, T.; Snyder, S.A. Tandem reactions, cascade sequences, and biomimetic strategies in total synthesis. Chem. Commun. 2003, 551–564. [Google Scholar]
- Schmalz, H.-G.; Geis, O. Tandem and cascade processes terminated by carbonylative esterification, amidation, and related reactions. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; John Wiley & Sons: New York, NY, USA, 2002; Volume 2, pp. 2377–2397. [Google Scholar]
- The Claisen Rearrangement; Hiersemann, M.; Nubbemeyer, U. (Eds.) Willey-VCH: Weinheim, Germany, 2007.
- Martin Castro, A.M. Claisen Rearrangement over the past nine decades. Chem. Rev. 2004, 104, 2939–3002. [Google Scholar] [CrossRef]
- Nakazaki, A.; Kobayashi, S. Stereocontrolled synthesis of functionalized spirocyclic compounds based on Claisen rearrangement and its application to the synthesis of spirocyclic sesquiterpenes and pyrrolidinoindoline alkaloids. Synlett 2012, 23, 1427–1445. [Google Scholar] [CrossRef]
- Kotha, S.; Krishna, N.G.; Halder, S.; Misra, S. A synergistic approach to polycyclics via a strategic utilization of Claisen rearrangement and olefin metathesis. Org. Biomol. Chem. 2011, 9, 5597–5624. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Bhattacharyya, T.; Chattopadhyay, B.; Sinha, B. Recent advances in the aza-Claisen rearrangement. Synthesis 2009, 2117–2142. [Google Scholar]
- Hiratani, K.; Albrecht, M. The tandem Claisen rearrangement in the construction of building blocks for supramolecular chemistry. Chem. Soc. Rev. 2008, 37, 2413–2421. [Google Scholar] [CrossRef]
- Majumdar, K.C. New variation of the aromatic ortho-Claisen rearrangement: synthesis of fused thiophenes and pyrroles. Synlett 2008, 2400–2411. [Google Scholar]
- Tymoshenko, D.O. Microwave-assisted Claisen and aza-Claisen rearrangements. Mini-Rev. Org. Chem. 2008, 5, 85–95. [Google Scholar]
- Majumdar, K.C.; Alam, S.; Chattopadhyay, B. Catalysis of the Claisen rearrangement. Tetrahedron 2007, 64, 597–643. [Google Scholar]
- Fanning, K.N.; Jamieson, A.G.; Sutherland, A. Palladium(II)-catalyzed rearrangement reactions. Curr. Org. Chem. 2006, 10, 1007–1020. [Google Scholar] [CrossRef]
- Ramesha, A.R.; Vishnumurthy, K.; Guru, R.; Tayur, N.; Chandrasekaran, S. Interesting reaction of 2,2'-binaphthol with 1,2-dibromoethane: Synthesis of a novel spirodienone. Indian J. Chem. Section B 1999, 38B, 1015–1017. [Google Scholar]
- Kyba, E.P.; Gokel, G.W.; De Jong, F.; Koga, K.; Sousa, L.R.; Siegel, M.G.; Kaplan, L.; Sogah, G.D.Y.; Cram, D.J. Host-guest complexation 7. The binaphthyl structural unit in host compounds. J. Org. Chem. 1977, 42, 4173–4184. [Google Scholar] [CrossRef]
- Piedra, E.; Francos, J.; Nebra, N.; Suárez, F.J.; Díez, J.; Cadierno, V. Access to unusual polycyclic spiro-enones from 2,2′-bis(allyloxy)-1,1′-binaphthyls using Grubbs' catalysts: An unprecedented one-pot RCM/Claisen sequence. Chem. Commun. 2011, 47, 7866–7868. [Google Scholar]
- Angelovski, G.; Eilbracht, P. Synthesis of hydroquinone-, biphenol-, and binaphthol-containing aza macroheterocycles via regioselective hydroformylation and reductive amination. Tetrahedron 2003, 59, 8265–8274. [Google Scholar] [CrossRef]
- Rasmussen, B.S.; Elezcano, U.; Skrydstrup, T. Synthesis and binding properties of chiral macrocyclic barbiturate receptors: application to nitrile oxide cyclizations. J. Chem. Soc. Perkin 1 2002, 1723–1733. [Google Scholar]
- Garas, A.; Bremner, J.B.; Coates, J.; Deadman, J.; Keller, P.A.; Pyne, S.G.; Rhodes, D.I. Binaphthyl scaffolded peptoids via ring-closing metathesis reactions and their anti-bacterial activities. Bioorg. Med. Chem. Lett. 2009, 19, 3010–3013. [Google Scholar] [CrossRef]
- Widhalm, M. University of Vienna: Wien, Austria, 2010; Unpublished work.
- Nakamura, Y.; Hollenstein, R.; Zsindely, J.; Schmid, H.; Oberhaensli, W.E. Intramolecular cycloadditions in the binaphthyl series. Helv. Chim. Acta 1975, 58, 1949–1977. [Google Scholar] [CrossRef]
- Schlosser, M.; Bailly, F. Embedding an allylmetal dimer in a chiral cavity: The unprecedented stereoselectivity of a twofold Wittig [1,2]-Rearrangement. J. Am. Chem. Soc. 2006, 128, 16042–16043. [Google Scholar] [CrossRef]
- Waespe, H.-R.; Heimgartner, H.; Schmid, H.; Hansen, H.-J.; Paul, H.; Fischer, H. Photoreactions. Part 55. The photochemistry of allyl aryl ethers. Helv. Chim. Acta 1978, 61, 401–429. [Google Scholar] [CrossRef]
- Lustenberger, P.; Martinborough, E.; Denti, T.M.; Diederich, F. Geometrical optimization of 1,1'-binaphthalene receptors for enantioselective molecular recognition of excitatory amino acid derivatives. J. Chem. Soc. Perkin Trans. 2 1998, 747–762. [Google Scholar]
- Cookson, R.C.; Crundwell, E.; Hill, R.R.; Hudec, J. Photochemical cyclization of Diels-Alder adducts. J. Chem. Soc. 1964, 3062–3075. [Google Scholar]
- Ogino, T.; Awano, K.; Ogihara, T.; Isogai, K. Intramolecular olefin metathesis of tricyclic bridged dienones. Regiospecific and reversible cycloreversion of trishomocubanones to cis,syn,cis-tricyclopentanoids. Tetrahedron Lett. 1983, 24, 2781–2782. [Google Scholar]
- Brooke, G.M.; Matthews, R.S.; Robson, N.S. Photochemically-allowed [3,5] sigmatropic rearrangements. Chem. Commun. 1980, 194–195. [Google Scholar]
- Geraghty, N.W.A.; Monaghan, M.J.; Hanley, N.M. Photochemistry of 1-allyl-2(1H)-naphthalenones: a revised mechanism for the formation of benzotricyclo[3.3.1.0.2.7]nonen-8-ones. Tetrahedron Lett. 1987, 28, 4729–4732. [Google Scholar]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O'Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Trnka, T.M.; Grubbs, R.H. The Development of L2X2Ru=CHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res. 2001, 34, 18–29. [Google Scholar] [CrossRef]
- Adlhart, C.; Chen, P. Mechanism and Activity of Ruthenium Olefin Metathesis Catalysts: The Role of Ligands and Substrates from a Theoretical Perspective. J. Am. Chem. Soc. 2004, 126, 3496–3510. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Benchmark Energetic Data in a Model System for Grubbs II Metathesis Catalysis and Their Use for the Development, Assessment, and Validation of Electronic Structure Methods. J. Chem. Theory Comput. 2009, 5, 324–333. [Google Scholar] [CrossRef]
- Vyboishchikov, S.F.; Bühl, M.; Thiel, W. Mechanism of Olefin Metathesis with Catalysis by Ruthenium Carbene Complexes: Density Functional Studies on Model Systems. Chem. Eur. J. 2002, 8, 3962–3975. [Google Scholar] [CrossRef]
- Since more than 85% of conformers were found within a narrow energy range of ~1 kcal the Curtin-Hammet Principle for Competing Reactions can be applied.
- Moreover, calculations showed very similar activation energies for direct transformations E-12→5a and E-12→epi-5a. If such reaction path would be operative similar amounts of products 5a and epi-5a should be isolated which disagrees with experimental results.
- Garcia-Exposito, E.; Bearpark, M.J.; Ortuno, R.M.; Robb, M.A.; Branchadell, V. Theoretical Study of the Photochemical [2+2]-Cycloadditions of Cyclic and Acyclic α,β-Unsaturated Carbonyl Compounds to Ethylene. J. Org. Chem. 2002, 67, 6070–6077. [Google Scholar]
- Bradley, S.A.; Bresnan, B.J.; Draper, S.M.; Geraghty, N.W.A.; Jeffares, M.; McCabe, T.; McMurry, T.B.H.; O'Brien, J.E. Photochemical [2+2] cycloaddition reactions of 6-alkenyl-3-phenylcyclohex-2-en-1-ones: using biradical conformation control to account for exceptions to the “rule of five”. Org. Biomol. Chem. 2011, 9, 2959–2968. [Google Scholar] [CrossRef]
- Cruciani, G.; Margaretha, P. Regiochemical trends in intramolecular [2+2] photocycloadditions of 6-(prop-2-enyl)cyclohex-2-enones and 5-(prop-2-enyl)cyclopent-2-enones. Helv.Chim. Acta 1990, 73, 288–297. [Google Scholar] [CrossRef]
- Wu, T.R.; Shen, L.; Chong, J.M. Asymmetric Allylboration of Aldehydes and Ketones Using 3,3'-Disubstituted Binaphthol-Modified Boronates. Org. Lett. 2004, 6, 2701–2704. [Google Scholar] [CrossRef]
- Zimmer, R.; Schefzig, L.; Peritz, A.; Dekaris, V.; Reissig, H.-U. Functionalized BINOL Derivatives as Ligands for Enantioselectively Catalyzed Aldol Additions: Highly Enantioselective Synthesis of Chiral β-Hydroxy Thioesters. Synthesis 2004, 1439–1445. [Google Scholar]
- Ooi, T.; Kameda, M.; Maruoka, K. Design of N-Spiro C2-Symmetric Chiral Quaternary Ammonium Bromides as Novel Chiral Phase-Transfer Catalysts: Synthesis and Application to Practical Asymmetric Synthesis of a-Amino Acids. J. Am. Chem. Soc. 2003, 125, 5139–5151. [Google Scholar] [CrossRef]
- Saelinger, D.; Brueckner, R. The First Asymmetric Halogen/Metal-Exchange Reaction: Desymmetrization of Alcohols with Enantiotopic Bromoarene Substituents. Chem. Eur. J. 2009, 15, 6688–6703. [Google Scholar] [CrossRef]
- Wipf, P.; Jung, J.-K. Formal Total Synthesis of (+)-Diepoxin α. J. Org. Chem. 2000, 65, 6319–6337. [Google Scholar] [CrossRef]
- SAINT-Plus (version 7.06a) and APEX2. Bruker-Nonius AXS Inc.: Madison, WI, USA, 2004.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A46, 112–122. [Google Scholar]
- Johnson, C.K. Report ORNL-5138. Oak Ridge National Laboratory: 840 Oak Ridge, TN, USA, 1976. [Google Scholar]
- Becke, A.D. Densityfunctional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H.; Schwerdtfeger, P.; Pitzer, R.M. Accuracy of energy-adjusted, quasirelativistic, ab initio pseudopotentials: all-electron and pseudopotential benchmark calculations for mercury, mercury hydride (HgH), and their cations. Mol. Phys. 1993, 78, 1211–1224. [Google Scholar] [CrossRef]
- Kuechle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P. The accuracy of the pseudopotential approximation. II. A comparison of various core sizes for indium pseudopotentials in calculations for spectroscopic constants of InH, InF, and InCl. J. Chem. Phys. 1996, 105, 1052–1059. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Gaussian basis set for molecular wavefunctions containing third-row atoms. Chem. Phys. 1970, 52, 1033–1036. [Google Scholar]
- Hay, P.J. Gaussian basis sets for molecular calculations. The representation of 3d orbitals in transition-metal atoms. J. Chem. Phys. 1977, 66, 4377–4384. [Google Scholar]
- Raghavachari, K.; Trucks, G.W. Highly correlated systems. Ionization energies of first row transition metals from scandium to zinc. J. Chem. Phys. 1989, 91, 2457–2460. [Google Scholar] [CrossRef]
- Curtiss, L.A.; McGrath, M.P.; Blaudeau, J.-P.; Davis, N.E.; Binning, R.C., Jr.; Radom, L. Extension of Gaussian-2 theory to molecules containing third-row atoms Ga-Kr. J. Comput. Chem. 1995, 103, 6104–6113. [Google Scholar]
- McGrath, M.P.; Radom, L. Extension of Gaussian-1(G1) theory of the bromine-containing molecules. J. Chem. Phys. 1991, 94, 511–516. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent . Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abraham, M.; Reischl, W.; Kirchner, K.A.; Roller, A.; Veiros, L.F.; Widhalm, M. Tandem RCM–Claisen Rearrangement–[2+2] Cycloaddition of O,O'-(But-2-en-1,4-diyl)-bridged Binaphthols. Molecules 2012, 17, 14531-14554. https://doi.org/10.3390/molecules171214531
Abraham M, Reischl W, Kirchner KA, Roller A, Veiros LF, Widhalm M. Tandem RCM–Claisen Rearrangement–[2+2] Cycloaddition of O,O'-(But-2-en-1,4-diyl)-bridged Binaphthols. Molecules. 2012; 17(12):14531-14554. https://doi.org/10.3390/molecules171214531
Chicago/Turabian StyleAbraham, Michael, Wolfgang Reischl, Karl A. Kirchner, Alexander Roller, Luis F. Veiros, and Michael Widhalm. 2012. "Tandem RCM–Claisen Rearrangement–[2+2] Cycloaddition of O,O'-(But-2-en-1,4-diyl)-bridged Binaphthols" Molecules 17, no. 12: 14531-14554. https://doi.org/10.3390/molecules171214531
APA StyleAbraham, M., Reischl, W., Kirchner, K. A., Roller, A., Veiros, L. F., & Widhalm, M. (2012). Tandem RCM–Claisen Rearrangement–[2+2] Cycloaddition of O,O'-(But-2-en-1,4-diyl)-bridged Binaphthols. Molecules, 17(12), 14531-14554. https://doi.org/10.3390/molecules171214531