Two New Iridoid Glycosides from the Root Barks of Sambucus williamsii Hance

Abstract
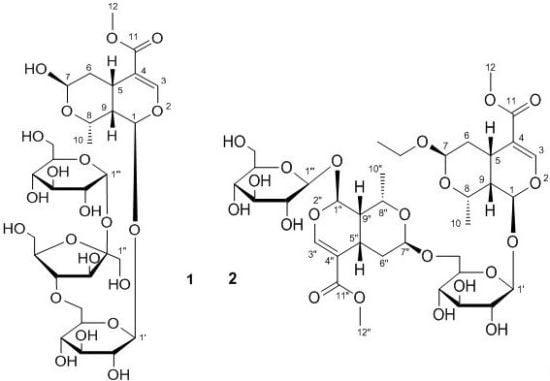
:
1. Introduction

2. Results and Discussion


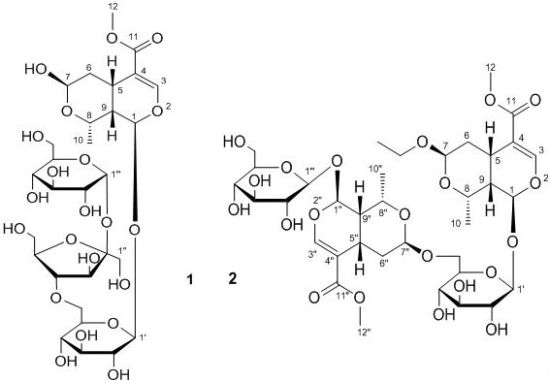{kind=link}
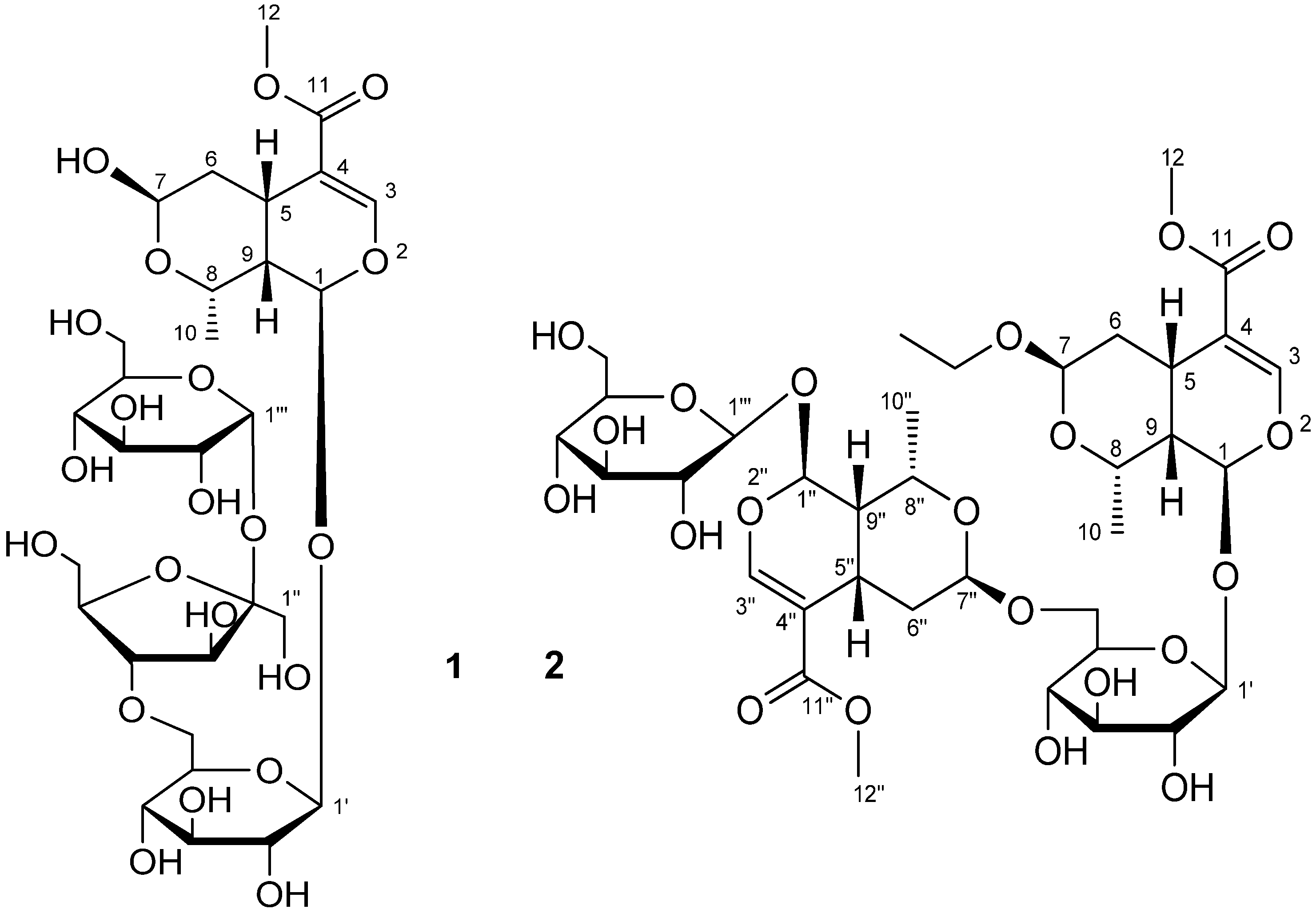{kind=link}
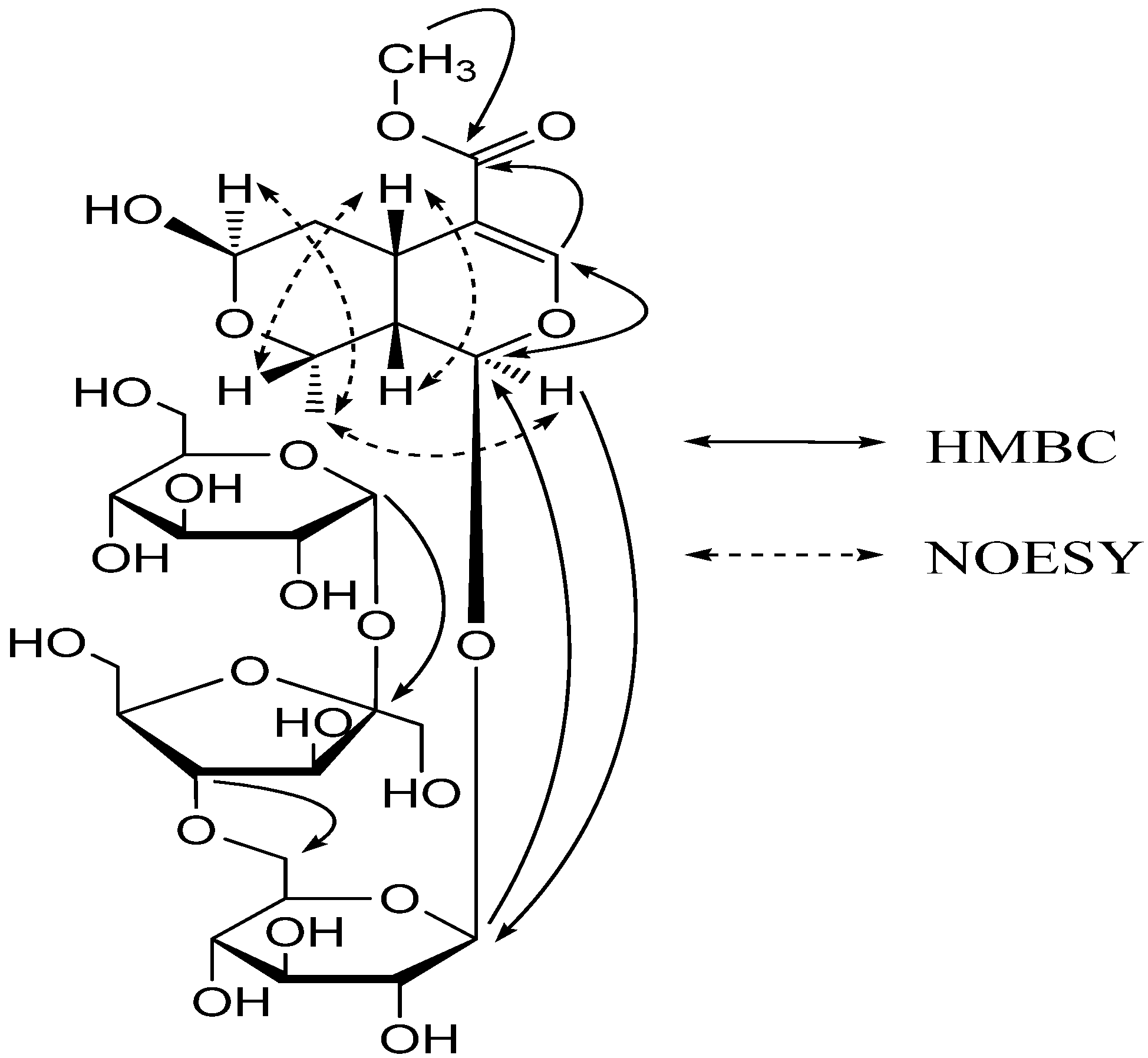{kind=link}
| No. | 1 | No. | 2 | ||
|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | ||
| 1 | 95.7 | 5.89 (1H, d, 9.2) | 1 | 96.0 | 5.84 (1H, d, 9.5) |
| 3 | 154.5 | 7.50 (1H, s) | 3 | 154.5 | 7.50 (1H, s) |
| 4 | 111.7 | - | 4 | 111.7 | - |
| 5 | 28.0 | 3.06 (1H, dt, 4.4, 12.8) | 5 | 27.8 | 3.06 (1H, m) |
| 6 | 33.7 | 1.51 (1H, dt, 4.8, 13.6) | 6 | 34.0 | 1.39 (1H, dt, 3.7, 13.6) |
| 1.97 (1H, dd, 3.6, 13.6) | 1.85 (1H, dd, 4.7, 13.6) | ||||
| 7 | 92.7 | 5.00 (1H, br.d, 3.2) | 7 | 97.8 | 4.76 (1H, br.d, 4.6) |
| 8 | 66.5 | 4.36 (1H, dq, 2.0, 6.8) | 8 | 66.0 | 4.26 (1H, dq, 2.1, 6.9) |
| 9 | 40.3 | 1.83 (1H, m) | 9 | 40.4 | 1.79 (1H, m) |
| 10 | 19.7 | 1.33 (3H, d, 6.8) | 10 | 19.7 | 1.29 (3H, d, 6.9) |
| 11 | 168.7 | - | 11 | 168.6 | - |
| 12 | 51.8 | 3.68 (3H, s) | 12 | 51.8 | 3.68 (3H, s) |
| 1′ | 100.1 | 4.78 (1H, d, 7.6) | -OCH2CH3 | 63.6 | 3.41 (1H, o) |
| 2′ | 75.0 | 3.21 (1H, m) | 3.64 (1H, o) | ||
| 3′ | 78.0 | 3.36 (1H, m) | -OCH2CH3 | 15.4 | 1.19 (3H, t, 7.1) |
| 4′ | 71.6 | 3.26 (1H, m) | 1′ | 100.4 | 4.78 (1H, d, 7.9) |
| 5′ | 78.6 | 3.06 (1H, dt, 4.4, 12.8) | 2′ | 75.4 | 3.20 (1H, m) |
| 6′ | 69.4 | 3.84 (2H, o) | 3′ | 78.0 | 3.37 (1H, m) |
| 1′′ | 63.8 | 3.62 (2H, s) | 4′ | 71.8 | 3.34 (1H, m) |
| 2′′ | 105.6 | - | 5′ | 76.8 | 3.47 (1H, m) |
| 3′′ | 78.7 | 4.10 (1H, d, 8.4) | 6′ | 68.0 | 3.62 (1H, o) |
| 4′′ | 76.3 | 3.99 (1H, t, 8.2) | 3.96 (1H, dd, 1.8, 12.0) | ||
| 5′′ | 82.1 | 3.90 (1H, o) | 1′′ | 95.7 | 5.87 (1H, d, 9.7) |
| 6′′ | 62.3 | 3.80 (1H, o) | 3′′ | 154.5 | 7.50 (1H, s) |
| 3.72 (1H, o) | 4′′ | 111.8 | - | ||
| 1′′′ | 93.4 | 5.43 (1H, d, 3.8) | 5′′ | 28.0 | 3.06 (1H, m) |
| 2′′′ | 73.3 | 3.41 (1H, m) | 6′′ | 34.0 | 1.49 (1H, dt, 3.7, 13.6) |
| 3′′′ | 74.8 | 3.68 (1H, m) | 1.97 (1H, dd, 4.6, 13.7) | ||
| 4′′′ | 71.4 | 3.34 (1H, m) | 7′′ | 99.3 | 4.94 (1H, d, 3.2) |
| 5′′′ | 74.2 | 3.84 (1H, m) | 8′′ | 66.4 | 4.30 (1H, dq, 2.1, 7.0) |
| 6′′′ | 62.8 | 3.67 (1H, dd, 4.8, 11.8) | 9′′ | 40.3 | 1.79 (1H, m) |
| 3.86 (1H, dd, 2.0, 11.8) | 10′′ | 19.8 | 1.34 (3H, d, 6.9) | ||
| 11′′ | 168.7 | - | |||
| 12′′ | 51.8 | 3.66 (3H, s) | |||
| 1′′′ | 100.1 | 4.78 (1H, d, 7.9) | |||
| 2′′′ | 75.0 | 3.20 (1H, m) | |||
| 3′′′ | 78.0 | 3.37 (1H, m) | |||
| 4′′′ | 71.6 | 3.28 (1H, m) | |||
| 5′′′ | 78.5 | 3.28 (1H, m) | |||
| 6′′′ | 62.8 | 3.63 (1H, o) | |||
| 3.86 (1H, dd, 2.0, 11.6) | |||||
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
4. Conclusions
Acknowledgments
- Sample Availability: Samples of williamsosides C and D are available from the authors.
References and Notes
- Hance, H.F. Sambucus williamsii Hance. Ann. Sci. Nat. Bot. 1866, 5. Available online: http://www.tropicos.org/Name/6000520/ (accessed on 22 December 2011). Series 4. Missouri Botanical Garden.
- Liu, W.; Wu, C.F.; Guo, Y.Y.; Yu, Q.H. Anti-inflammatory activity of aqueous extract of the root of Sambucus williamsii. Fitoterapia 1991, 62, 83–85. [Google Scholar]
- Han, H.; Yan, X.Y.; Kuang, H.X.; Dong, P.L. Advances in research of Sambucus williamsii hance. Inf. Tradit. Chin. Med. 2008, 25, 14–16. [Google Scholar]
- Ouyang, F.; Liu, Y.; Li, R.; Li, L.; Wang, N.L.; Yao, X.S. Five lignans and an iridoid from Sambucus williamsii. Chin. J. Nat. Med. 2011, 9, 26–29. [Google Scholar]
- Wang, Z.Y.; Han, H.; Yang, B.Y.; Xia, Y.G.; Kuang, H.X. Two new iridoid glycosides from the root barks of Sambucus williamsii Hance. Molecules 2011, 16, 3869–3874. [Google Scholar] [CrossRef]
- Furuya, T.; Ushiyama, M.; Asada, Y.; Yoshikawa, T.; Orihara, Y. Biotransformation of phenylacetic acid and 2-phenyl-propionic acid in suspension culture of Coffea Arabica. Phytochemistry 1988, 27, 803–807. [Google Scholar] [CrossRef]
- Furuya, T.; Ushiyama, M.; Asada, Y.; Yoshikawa, T. Glycosylation of 2-phenylpropionic acid and its ethyl ester in suspension cultures of Nicotiana tabacum, Dioscoreophyllum cumminsii and Aconitum japonicum. Phytochemistry 1987, 26, 2983–2989. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Gan, M.L.; Lin, S.; Liu, M.T.; Song, W.X.; Zi, J.C.; Wang, S.J.; Li, S.; Yang, Y.C.; Shi, J.G. Glycosides from the bark of Adina polycephala. J. Nat. Prod. 2008, 71, 905–909. [Google Scholar] [CrossRef]
- de Bruyn, A.; Alvarez, A.P.; Sandra, P.; de Leenheer, L. Isolation and identification of O-beta-D-fructofuranosyl-(2→1)-O-beta-D-fructofuranosyl-(2→1)-D-fructose, a product of the enzymic hydrolysis of the inulin from Cichorium intybus. Carbohydr. Res. 1992, 235, 303–308. [Google Scholar] [CrossRef]
- Angyal, S.J.; Bethell, G.S. Conformational analysis in carbohydrate chemistry. III. The 13C-NMR spectra of the hexuloses. Aust. J. Chem. 1976, 29, 1249–1265. [Google Scholar] [CrossRef]
- Ren, F.Z.; Chen, S.H.; Zhang, X.X.; Li, L.H.; Gao, Y.Q.; Wang, N. Structure analysis of an oligosaccharide isolated from Bixo orellana by NMR spectroscopy. Chin. J. Magn. Reson. 2011, 28, 160–167. [Google Scholar]
- Chen, Y.W.; Xue, Z. Studies on the immunoactive constituents of zhi yu rou. J. Chin. Jpn. Friendsh. Hosp. 1992, 6, 231–234. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kuang, H.-X.; Han, H.; Yang, B.-Y.; Yang, L.; Jiang, H.; Wang, Q.-H. Two New Iridoid Glycosides from the Root Barks of Sambucus williamsii Hance. Molecules 2012, 17, 1830-1836. https://doi.org/10.3390/molecules17021830
Kuang H-X, Han H, Yang B-Y, Yang L, Jiang H, Wang Q-H. Two New Iridoid Glycosides from the Root Barks of Sambucus williamsii Hance. Molecules. 2012; 17(2):1830-1836. https://doi.org/10.3390/molecules17021830
Chicago/Turabian StyleKuang, Hai-Xue, Hua Han, Bing-You Yang, Liu Yang, Hai Jiang, and Qiu-Hong Wang. 2012. "Two New Iridoid Glycosides from the Root Barks of Sambucus williamsii Hance" Molecules 17, no. 2: 1830-1836. https://doi.org/10.3390/molecules17021830