Acylated mono-, bis- and tris- Cinchona-Based Amines Containing Ferrocene or Organic Residues: Synthesis, Structure and in Vitro Antitumor Activity on Selected Human Cancer Cell Lines

Abstract
:1. Introduction
2. Results and Discussion
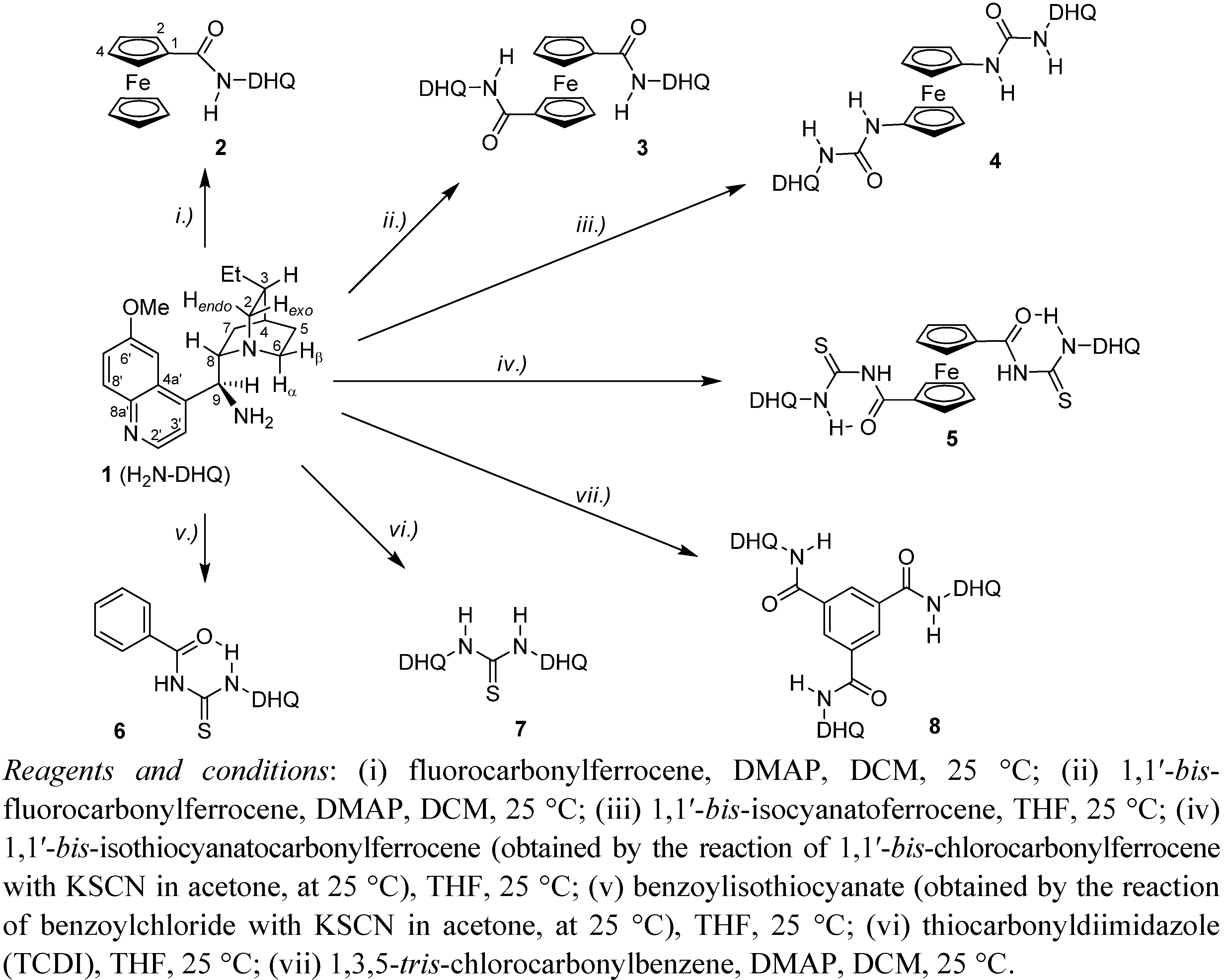
2.1. Synthesis of the Model Compounds

2.2. Theoretical Calculations
2.3. Structure Determination
2.4. In Vitro Activity of the Compounds on Human Tumor Cell Cultures

{kind=link}
| Cell line | ||||
| HepG2 | SH-SY5Y | HL-60 | MCF-7 | |
| Compd. | Cytotoxicity (IC50a in µM) | |||
| 1 | >100 | >100 | >100 | > 100 |
| 2 | 33.10 ± 3.04 | 29.80 ± 4.24 | 37.70 ± 3.67 | 25.32 ± 4.60 |
| 3 | 0.72 ± 0.01 | 0.78 ± 0.02 | 1.70 ± 0.05 | 0.75 ± 0.02 |
| 4 | 4.24 ± 1.12 | 0.82 ± 0.54 | 0.86 ± 0.02 | 21.70 ± 3.23 |
| 5 | >100 | >100 | 6.70 ± 0.02 | >100 |
| 6 | 17.60 ± 0.25 | 21.20 ± 3.24 | 32.20 ± 4.67 | >100 |
| 7 | 3.34 ± 1.02 | 0.84 ± 0.02 | 1.80 ± 0.56 | 5.34 ± 1.78 |
| 8 | 8.90 ± 0.23 | 1.50 ± 0.02 | 2.30 ± 0.05 | >100 |
| Cytostatic effect (IC50 in µM) | ||||
| 1 | >100 | >100 | >100 | > 100 |
| 2 | 65.00 ± 6.70 | 80.70 ± 5.78 | 41.90 ± 1.45 | 56.00 ± 4.56 |
| 3 | 0.40 ± 0.17 | 0.99 ± 0.10 | 0.76 ± 0.01 | 1.00 ± 0.34 |
| 4 | 3.40 ± 0.12 | 1.30 ± 0.54 | 0.94 ± 0.02 | 5.10 ± 0.67 |
| 5 | >100 | >100 | 6.50 ± 3.56 | 21.80 ± 3.18 |
| 6 | 65.60 ± 3.40 | 82.90 ± 6.78 | >100 | 82.90 ± 7.98 |
| 7 | 4.60 ± 0.02 | 4.20 ± 2.30 | 10.20 ± 1.65 | 3.89 ± 1.18 |
| 8 | 19.60 ± 2.12 | 17.20 ± 3.45 | 4.50 ± 0.01 | 2.36 ± 0.01 |
3. Experimental
3.1. General
3.2.Synthesis of the Novel Quinine Derivatives
3.2.1. N-{(S)-[(2S,4R,8R)-8-Ethylquinuclidin-2-yl](6-methoxyquinolin-4-yl)methyl)}ferrocene-carboxamide (2)
3.2.2. N-{(S)-[(2S,4R,8R)-8-Ethylquinuclidin-2-yl](6-methoxyquinolin-4-yl)methyl)}ferrocene-1,1'-bis-carboxamide (3)
3.2.3. 1,1'-(Ferrocene-1,1'-diyl)-bis-{3-[(S)-((2S,4R,8R)-8-ethylquinuclidin-2-yl](6-methoxyquinolin-4-yl)methyl)}urea (4)
3.2.4. 1,1'-(Ferrocene-1,1'-dicarbonyl-diyl)-bis-{3-[(S)-((2S,4R,8R)-8-ethylquinuclidin-2-yl](6-methoxyquinolin-4-yl)methyl)}thiourea (5)
3.2.5. 1-Benzoyl-3-[(S)-((2S,4R,8R)-8-ethylquinuclidin-2-yl](6-methoxyquinolin-4-yl)methyl)thiourea (6)
3.2.6. 1,3-Bis-{(S)-[(2S,4R 8R)-8-ethylquinuclidin-2-yl](6-methoxyquinolin-4-yl)methyl)}thiourea (7)
3.2.7. N-{(S)-[(2S,4R,8R)-8-Ethylquinuclidin-2-yl](6-methoxyquinolin-4-yl)methyl)}benzene-1,3,5-tris-carboxamide (8)
3.3. In Vitro Cytostatic and Cytotoxic Activity of the Compounds
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds 2–8 are available from the authors.
References and Notes
- Ding, Y.; Bao, Y.; An, L. Progress in antitumor agents, vinblastine analogues. Zhongguo Yiyao Gongye Zazhi 2005, 36, 424–428. [Google Scholar]
- Gao, H. Research status of antitumor drug camptothecin and its derivatives. Hebei Yiyao 2008, 30, 1786–1788. [Google Scholar]
- Prudhomme, M. Staurosporines and structurally related indolocarbazoles as antitumor agents. Anticancer Agents Nat. Prod. 2005, 499–517. [Google Scholar]
- Ohashi, M.; Oki, T. Ellipticine and related anticancer agents. Expert Opin. Ther. Pat. 1996, 6, 1285–1294. [Google Scholar] [CrossRef]
- Kaur, K.; Jain, M.; Reddy, R.P.; Jain, R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010, 45, 3245–3264. [Google Scholar] [CrossRef]
- Wolf, R.; Baroni, A.; Greco, R.; Donnarumma, G.; Ruocco, E.; Tufano, M.A.; Ruocco, V. Quinine sulfate and bacterial invasion. Ann. Clin. Microbiol. Antimicrob. 2002, 1–5. [Google Scholar]
- Kelsey, F.E.; Brunschwig, A. Concentration of quinine in gastrointestinal cancers; preliminary report. Cancer Res. 1947, 7, 355–356. [Google Scholar]
- Kim, J.; Lee, K.; Jung, W.; Lee, O.; Kim, T.; Kim, H.; Lee, J.; Passaro, D.J. Validity of serum pepsinogen levels and quininium resin test combined for gastric cancer screening. Cancer Detect. Prev. 2005, 29, 570–575. [Google Scholar] [CrossRef]
- Lehnert, M.; Dalton, W.S.; Roe, D.; Emerson, S.; Salmon, S.E. Synergistic inhibition by verapamil and quinine of P-glycoprotein-mediated multidrug resistance in a human myeloma cell line model. Blood 1991, 77, 348–354. [Google Scholar]
- Taylor, C.W.; Dalton, W.S.; Mosley, K.; Dorr, R.T.; Salmon, S.E. Combination chemotherapy with cyclophosphamide, vincristine, adriamycin, and dexamethasone (CVAD) plus oral quinine and verapamil in patients with advanced breast cancer. Breast Cancer Res. Treat. 1997, 42, 7–14. [Google Scholar] [CrossRef]
- Genne, P.; Dimanche-Boitrel, M.T.; Mauvernay, R.Y.; Gutierrez, G.; Duchamp, O.; Petit, J.M.; Martin, F.; Chauffert, B. Cinchonine, a potent efflux inhibitor to circumvent anthracycline resistance in vivo. Cancer Res. 1992, 52, 2797–2801. [Google Scholar]
- Baraniak, D.; Kacprzak, K.; Celewicz, L. Synthesis of 3′-azido-3′-deoxythymidine (AZT) cinchona alkaloid conjugates via click chemistry: Toward novel fluorescent markers and cytostatic agents. Bioorg. Med. Chem. Lett. 2011, 21, 723–726. [Google Scholar] [CrossRef]
- Sohue, N. Quinine derivatives and the transplantable tumor. III. The effect of quinine derivatives upon the growth rate of Fujinawa's rat sarcoma in the tissue culture. Folia Pharm. Jpn. 1941, 31, 1–7. [Google Scholar]
- Sakai, S.; Minoda, K.; Saito, G.; Akagi, S.; Ueno, A.; Fukuoka, F. The anticancer action of quinoline derivatives. Gann 1955, 46, 605–616. [Google Scholar]
- Fiorina, V.J.; Dubois, R.J.; Brynes, S. Ferrocenyl polyamines as agents for the chemoimmunotherapy of cancer. J. Med. Chem. 1978, 21, 393–395. [Google Scholar] [CrossRef]
- Koepf-Maier, P.; Koepf, H.; Neuse, E.W. Ferrocenium salts—The first antitumor iron compounds. Angew. Chem. Int. Ed. 1984, 96, 446–447. [Google Scholar] [CrossRef]
- Neuse, E.W.; Kanzawa, F. Evaluation of the activity of some water-soluble ferrocene and ferricenium compounds against carcinoma of the lung by the human tumor clonogenic assay. Appl. Org.-Met. Chem. 1990, 4, 19–26. [Google Scholar]
- Snegur, L.V.; Nekrasov, S.; Gumenyuk, V.V.; Zhilina, Z.V.; Morozova, N.B.; Skviridova, I.K.; Rodina, I.A.; Sergeeva, N.S.; Shchitkov, K.G.; et al. Ferrocenylalkylazoles, a new class of low-toxicity compounds with antitumor activity. Rossiiskii Khim. Zhurn. 1998, 42, 178–183. [Google Scholar]
- Osella, D.; Ferrali, M.; Zanello, P.; Laschi, F.; Fontani, M.; Nervi, C.; Cavigiolio, G. On the mechanism of the antitumor activity of ferrocenium derivative. Inorg. Chim. Acta 2000, 306, 42–48. [Google Scholar] [CrossRef]
- Gormen, M.; Pigeon, P.; Top, S.; Vessieres, A.; Plamont, M.A.; Hillard, E.A.; Jaouen, G. Facile synthesis and strong antiproliferative activity of disubstituted diphenylmethylidenyl-[3]ferrocenophanes on breast and prostate cancer cell lines. Med. Chem. Commun. 2010, 1, 149–151. [Google Scholar] [CrossRef]
- Monserrat, J.P.; Chabot, G.G.; Hamon, L.; Quentin, L.; Scherman, D.; Jaouen, G.; Hillard, E.A. Synthesis of cytotoxic ferrocenyl flavones via a ferricenium-mediated 1,6-oxidative cyclization. Chem. Commun. 2010, 46, 5145–5147. [Google Scholar] [CrossRef]
- Hillard, E.A.; Vessieres, A.; Thouin, L.; Jaouen, G.; Amatore, C. Ferrocene-mediated proton-coupled electron transfer in a series of ferrocifen -type breast-cancer drug candidates. Angew. Chem. Int. Ed. 2006, 45, 285–290. [Google Scholar]
- Li, H.; Lv, P.; Yan, T.; Zhu, H. Urea derivatives as anticancer agents. Anticancer Agents Med. Chem. 2009, 9, 471–480. [Google Scholar]
- Jordan, A.M.; Khan, T.H.; Malkin, H.; Osborn, H.M.I. Synthesis and analysis of urea and carbamate prodrugs as candidates for melanocyte-directed enzyme prodrug therapy (MDEPT). Bioorg. Med. Chem. 2002, 10, 2625–2633. [Google Scholar] [CrossRef]
- Ma, Z.; Saluta, G.; Kucera, G.L.; Bierbach, U. Effect of linkage geometry on biological activity in thiourea and guanidine-substituted acridines and platinum-acridines. Bioorg. Med. Chem. Lett. 2008, 18, 3799–3801. [Google Scholar] [CrossRef]
- Cesarini, S.; Spallarossa, A.; Ranise, A.; Schenone, S.; Rosano, C.; La Colla, P.; Sanna, G.; Busonera, B.; Loddo, R. N-Acylated and N,N'-diacylated imidazolidine-2-thione derivatives and N,N'-diacylated tetrahydropyrimidine-2(1H)-thione analogues: Synthesis and antiproliferative activity. Eur. J. Med. Chem. 2009, 44, 1106–1118. [Google Scholar] [CrossRef]
- Rao, X.; Wu, Y.; Song, Z.; Shang, S.; Wang, Z. Synthesis and antitumor activities of unsymmetrically disubstituted acylthioureas fused with hydrophenanthrene structure. Med. Chem. Res. 2011, 20, 333–338. [Google Scholar] [CrossRef]
- Suda, Y.; Egami, K.; Fujita, H. Preparation of acylthiourea compounds as c-Met kinase inhibitors. PCT Int. Appl. 2009, 79. [Google Scholar]
- Ruat, M.; Faure, H.; Traiffort, E.; Schoenfelder, A.; Mann, A.; Taddei, M.; Solinas, A.; Manetti, F. Preparation of aromatic N- acylthiourea and N-acylurea as inhibitors of the Hedgehog protein signalling pathway for the treatment of cancer, neurodegenerative diseases and diabetes. Fr. Demande 2009, 57. [Google Scholar]
- Garcia-Martin, F.; Cruz, L.J.; Rodriguez-Mias, R.A.; Giralt, E.; Albericio, F. Design and synthesis of FAJANU: A de Novo C2 symmetric cyclopeptide family. J. Med. Chem. 2008, 51, 3194–3202. [Google Scholar] [CrossRef]
- Manna, C.M.; Tshuva, E.Y. arkedly different cytotoxicity of the two enantiomers of C2-symmetrical Ti(IV) phenolato complexes; mechanistic implications. Dalton T. 2010, 39, 1182–1184. [Google Scholar] [CrossRef]
- Rabouin, D.; Perron, V.; N'Zemba, B.C.; Gaudreault, R.; Berube, G. A facile synthesis of C2-symmetric 17b-estradiol dimers. Bioorg. Med. Chem. Lett. 2003, 13, 557–560. [Google Scholar] [CrossRef]
- Raynes, K.; Galatis, D.; Cowman, A.F.; Tilley, L.; Deady, L.W. Synthesis and activity of some antimalarial bisquinolines. J. Med. Chem. 1995, 38, 204–206. [Google Scholar] [CrossRef]
- Ayad, F.; Tilley, L.; Deady, L.W. Synthesis, antimalarial activity and inhibition of haem detoxification of novel bisquinolines. Bioorg. Med. Chem. Lett. 2001, 11, 2075–2077. [Google Scholar]
- Cowman, A.F.; Deady, L.W.; Deharo, E.; Desneves, J.; Tilley, L. Synthesis and activity of some antimalarial bisquinolinemethanols. Aust. J. Chem. 1997, 50, 1091–1096. [Google Scholar] [CrossRef]
- Brunner, H.; Buegler, J. Enantioselective catalysis. 106. 9-Amino-(9-deoxy)cinchona alkaloids and their derivatives. B. Soc. Chim. Belg. 1997, 106, 77–84. [Google Scholar]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. GAUSSIAN 03, Rev. A. 1; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Galow, T.H.; Rodrigo, J.; Cleary, K.; Cooke, G.; Rotello, V.M. Fluorocarbonylferrocene. A versatile intermediate for ferrocene esters and amides. J. Org. Chem. 1999, 64, 3745–3746. [Google Scholar] [CrossRef]
- Van Leusen, D.; Hessen, B. 1,1'-Diisocyanoferrocene and a convenient synthesis of ferrocenylamine. Organometallics 2001, 20, 224–226. [Google Scholar]
- Yuan, Y.; Ye, S.; Zhang, L.; Wang, B.; Xu, Y.; Wang, J.; Wang, H. Studies on intramolecular hydrogen bonding of 1,1'-bis[N-formyl-N'-p-chlorophenylthiourea]ferrocene. Inorg. Chim. Acta 1997, 256, 313–318. [Google Scholar] [CrossRef]
- Slater, T.F.; Sawyer, B.; Sträuli, U. Studies on succinate-tetrazolium reductase systems: III. Points of coupling of four different tetrazolium salts III. Points of coupling of four different tetrazolium salts. Biochim. Biophys. Acta 1963, 77, 383–393. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Liu, Y.B.; Peterson, D.A.; Kimura, H.; Schubert, D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J. Neurochem. 1997, 69, 581–593. [Google Scholar]
- Altman, F.P. Tetrazolium salts and formazans. Prog. Histochem. Cytochem. 1976, 9, 1–56. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Datki, Z.; Juhász, A.; Gálfi, M.; Soós, K.; Papp, R.; Zádori, D.; Penke, B. Method for measuring neurotoxicity of aggregating polypeptides with the MTT assay on differentiated neuroblastoma cells. Brain Res. Bull. 2003, 62, 223–229. [Google Scholar]
- Datki, Z.; Papp, R.; Zádori, D.; Soós, K.; Fülöp, L.; Juhász, A.; Laskay, G.; Hetényi, C.; Mihalik, E.; Zarándi, M.; Penke, B. In vitro model of neurotoxicity of Aβ 1-42 and neuroprotection by a pentapeptide: irreversible events during the first hour. Neurobiol. Dis. 2004, 17, 507–515. [Google Scholar]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple Neurotransmitter Synthesis by Human Neuroblastoma Cell Lines and Clones. Cancer Res. 1978, 38, 3751–3757. [Google Scholar]
- Biedler, J.L.; Helson, L.; Spengler, B.A. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973, 33, 2643–2652. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Károlyi, B.I.; Bősze, S.; Orbán, E.; Sohár, P.; Drahos, L.; Gál, E.; Csámpai, A. Acylated mono-, bis- and tris- Cinchona-Based Amines Containing Ferrocene or Organic Residues: Synthesis, Structure and in Vitro Antitumor Activity on Selected Human Cancer Cell Lines. Molecules 2012, 17, 2316-2329. https://doi.org/10.3390/molecules17032316
Károlyi BI, Bősze S, Orbán E, Sohár P, Drahos L, Gál E, Csámpai A. Acylated mono-, bis- and tris- Cinchona-Based Amines Containing Ferrocene or Organic Residues: Synthesis, Structure and in Vitro Antitumor Activity on Selected Human Cancer Cell Lines. Molecules. 2012; 17(3):2316-2329. https://doi.org/10.3390/molecules17032316
Chicago/Turabian StyleKárolyi, Benedek Imre, Szilvia Bősze, Erika Orbán, Pál Sohár, László Drahos, Emese Gál, and Antal Csámpai. 2012. "Acylated mono-, bis- and tris- Cinchona-Based Amines Containing Ferrocene or Organic Residues: Synthesis, Structure and in Vitro Antitumor Activity on Selected Human Cancer Cell Lines" Molecules 17, no. 3: 2316-2329. https://doi.org/10.3390/molecules17032316