Convergent Synthesis of the Potent P2Y Receptor Antagonist MG 50-3-1 Based on a Regioselective Ullmann Coupling Reaction


Abstract
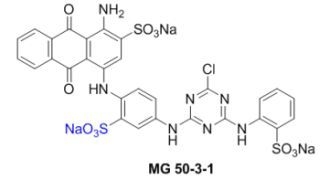
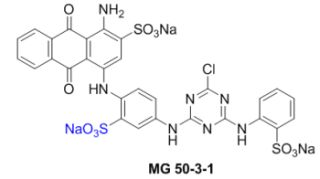
:
1. Introduction

2. Results and Discussion
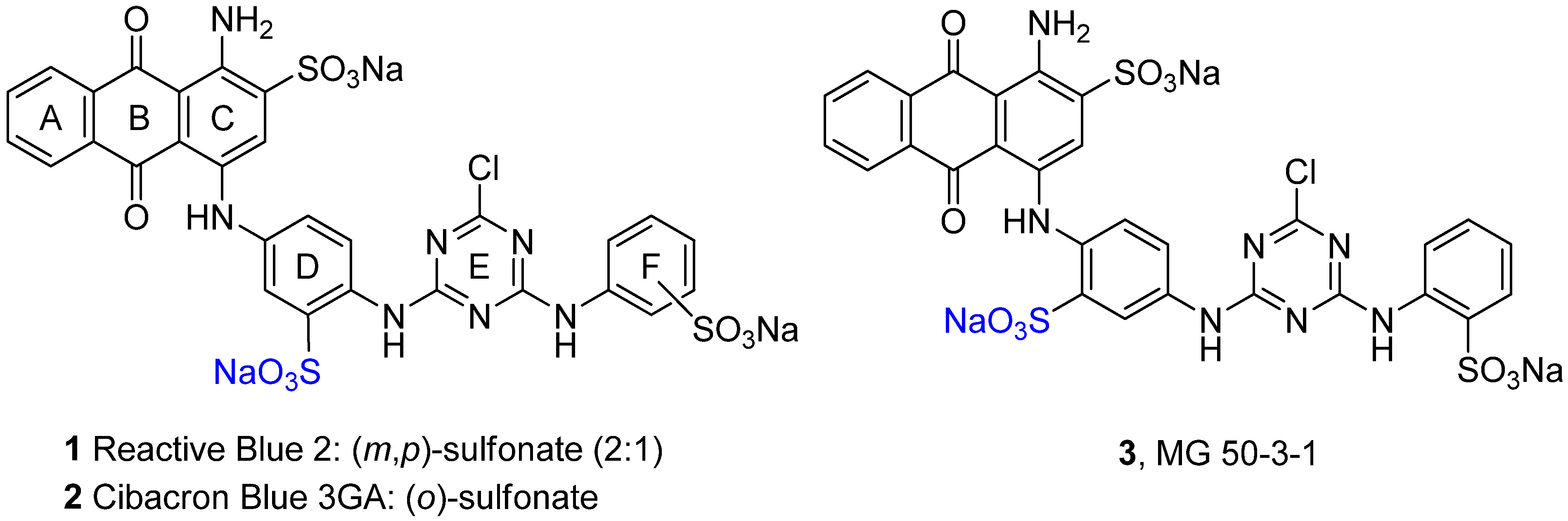
2.1. Optimization of the Ullmann Coupling Reaction of Bromaminic Acid with Aniline

{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Solvent mixture (mL) b | pH value | Cu | CuCl | CuCl2 | CuSO4 | |||||
| NaH2PO4 (0.12 M) | Na2HPO4 (0.20 M) | Time (min) | Conversion (%) c | Time (min) | Conversion (%) c | Time (min) | Conversion (%) c | Time (min) | Conversion (%) c | ||
| 1 | Water (5) | 7.0 | 20 | ca. 50 | 150 | 0 | 150 | 0 | 150 | 0 | |
| 2 | 5 | 0 | 4.8 | 25 | ca. 50 | 150 | 0 | 150 | 0 | 150 | 0 |
| 3 | 4 | 1 | 7.0 | 5 | 100 | 5 | 100 | 125 | ca. 50 | 125 | ca. 50 |
| 4 | 3 | 2 | 7.4 | 5 | 100 | 5 | 100 | 65 | 100 | 35 | 100 |
| 5 | 2 | 3 | 7.8 | 5 | 100 | 5 | 100 | 65 | 100 | 35 | 100 |
| 6 | 1 | 4 | 8.2 | 5 | 100 | 5 | 100 | 35 | 100 | 35 | 100 |
| 7 | 0 | 5 | 9.4 | 5 | 100 | 5 | 100 | 35 | 100 | 25 | 100 |
2.2. Regioselective Ullmann Coupling Reaction of Bromaminic Acid with 2,5-Diaminobenzene Sulfonate
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Solvent mixture (mL) | pH value | Time (min) | Catalyst | Conversion (%) b | ortho:meta ratio b | Yield (%) c | |
| NaH2PO4 (0.12 M) | Na2HPO4 (0.20 M) | |||||||
| 1 | 5 | 0 | 4.8 | 10 | Cu | ca. 50 | 1:1 | ca. 25 |
| 2 | 4 | 1 | 7.0 | 5 | Cu | ca. 50 | 1:1 | ca. 25 |
| 3 | 3 | 2 | 7.4 | 5 | Cu | 100 | 1:3 | > 50 |
| 4 | 2 | 3 | 7.8 | 5 | Cu | 100 | 1:3 | > 50 |
| 5 | 1 | 4 | 8.2 | 5 | Cu | 100 | 1:4 | > 75 |
| 6 | 0 | 5 | 9.4 | 5 | Cu | 100 | 1:4 | > 75 |
| 8 | 4 | 1 | 7.0 | 5 | CuCl | 100 | 1:6 | > 75 |
| 9 | 4 | 1 | 7.0 | 5 | CuSO4 | 100 | 1:6 | > 75 |
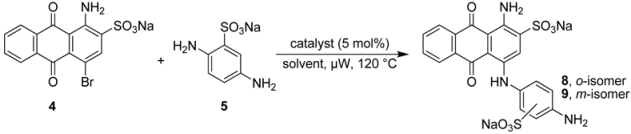 | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Time (min) | Catalyst | Conversion (%)b | ortho:meta ratio | Yield (%)c |
| 1 | water | 15 | Cu | ca. 60 | 3:1 | ca. 50 |
| 2 | water | 15 | CuCl | ca. 40 | 2:1 | ca. 25 |
| 3 | water | 20 | CuSO4 | ca. 30 | 1.5:1 | ca. 20 |
| 4 | water/Et3N | 5 | Cu | ca. 70 | 1:1 | ca. 40 |
| 5 | water/Et3N | 5 | CuCl | ca. 90 | 1:3 | > 50 |
| 6 | water/Et3N | 5 | CuSO4 | 100 | 1:3 | > 75 |
2.3. Regioselective Ullmann Coupling Reaction of Bromaminic Acid with 2,4-Diaminobenzene Sulfonate
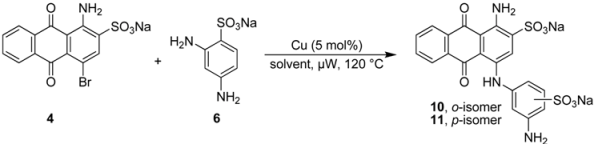 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent mixture (mL) | pH value | Time (min) | Conversion (%) b | ortho:para ratio b | Yield (%) c | |
| NaH2PO4 (0.12 M) | Na2HPO4 (0.20 M) | ||||||
| 1 | 5 | 0 | 4.8 | 10 | ca. 25 | 1.5:1 | ca. 25 |
| 2 | 4 | 1 | 7.0 | 5 | ca. 25 | 1:1 | ca. 25 |
| 3 | 3 | 2 | 7.4 | 5 | ca. 25 | 1:1.5 | ca. 25 |
| 4 | 2 | 3 | 7.8 | 5 | ca. 50 | 1:2 | >50 |
| 5 | 1 | 4 | 8.2 | 5 | 100 | 1:3 | >50 |
| 6 | 0 | 5 | 9.4 | 5 | 100 | 1:3 | >50 |
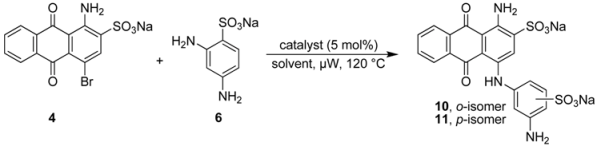 | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Time (min) | Catalyst | Conversion (%) b | ortho:para ratio | Yield (%) c |
| 1 | water | 15 | Cu | ca. 50 | >99:1 | ca. 20 |
| 2 | water | 15 | CuCl | ca. 50 | >99:1 | ca. 20 |
| 3 | water | 20 | CuSO4 | ca. 50 | >99:1 | ca. 20 |
| 4 | water/Et3N | 5 | Cu | ca. 80 | 1.5:1 | ca. 50 |
| 5 | water/Et3N | 5 | CuCl | 100 | 5:1 | >75 |
| 6 | water/Et3N | 5 | CuSO4 | 100 | 2:1 | >50 |
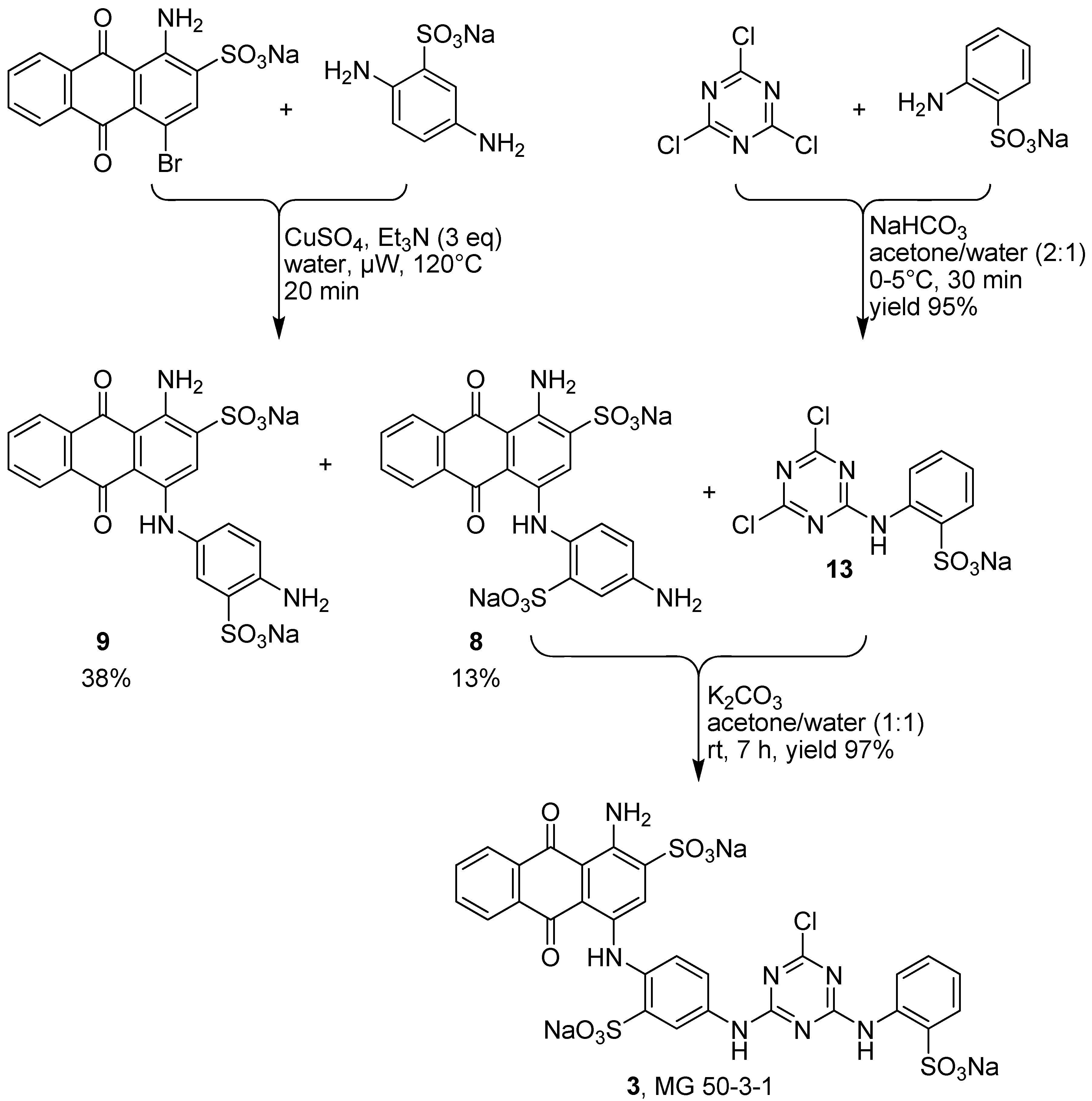
2.4. Synthesis of MG 50-3-1 (3)

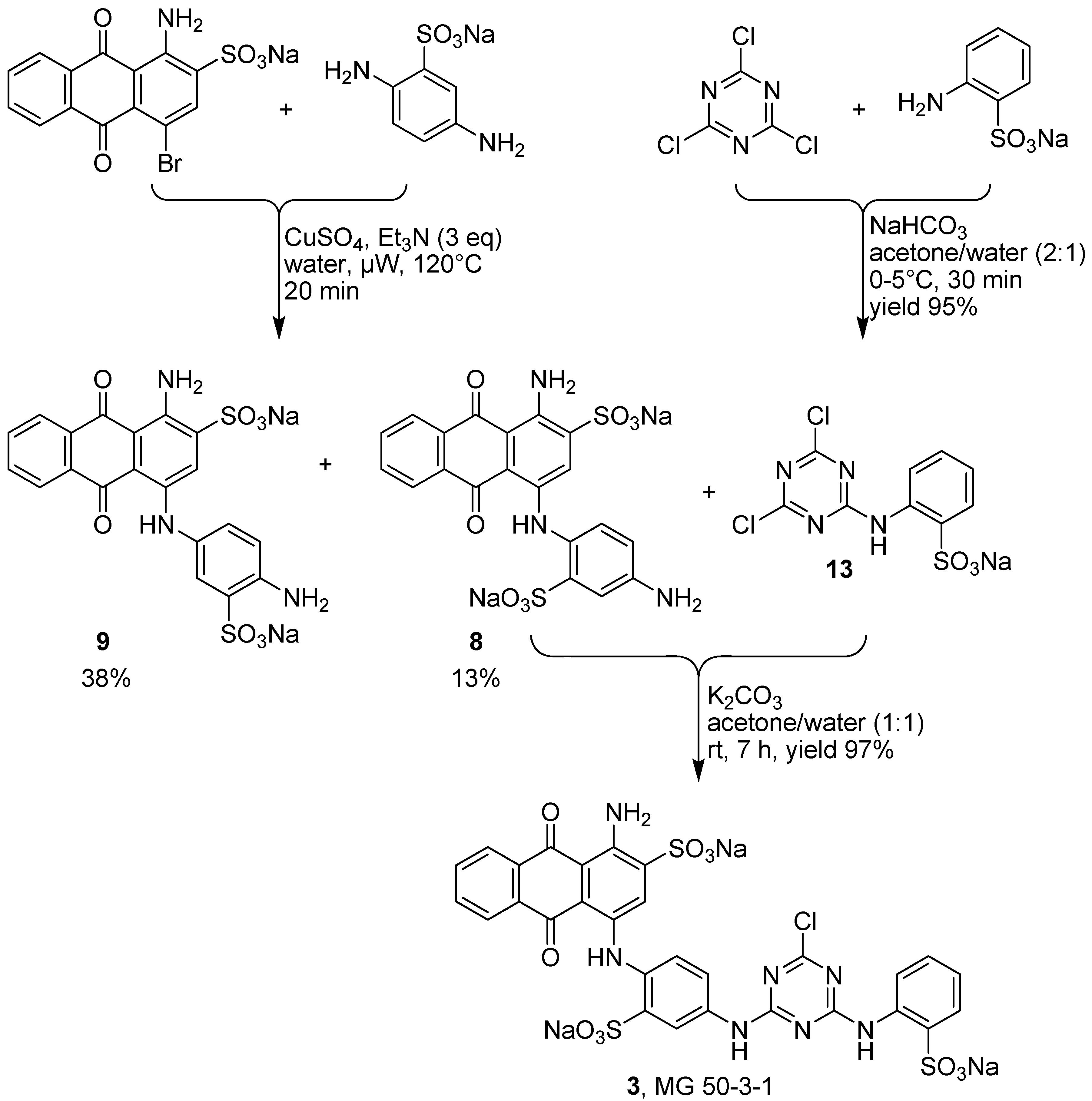
3. Experimental
3.1. General
3.2. General Procedure A: Coupling Reaction of Bromaminic Acid with Aniline
3.3. General Procedure B: Coupling Reaction of Bromaminic Acid with 2,5-Diaminobenzensulfonic Acid (5) or with 2,4-Diaminobenzensulfonic Acid (6)
3.4. Synthesis of Disodium 1-Amino-4-(4-amino-2-sulfophenylamino)-9,10-dioxo-9,10-dihydroanthracene 2-Sulfonate (8)
3.5. Synthesis of Sodium 2-(4,6-dichloro-1,3,5-triazin-2-ylamino)benzenesulfonate (13)
3.6. Synthesis of Trisodium 1-Amino-4-{4-[4-chloro-6-(2-sulfophenylamino)-1,3,5-triazin-2-ylamino]-2-sulfophenylamino}-9,10-dioxo-9,10-dihydroanthracene 2-Sulfonate (3, MG 50-3-1)
4. Conclusions
Conflict of Interest
Acknowledgments
- Sample Availability: Samples of the compounds are available from the authors.
References
- Burnstock, G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 2007, 87, 659–797. [Google Scholar] [CrossRef]
- Burnstock, G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol. Rev. 2006, 58, 58–86. [Google Scholar] [CrossRef]
- Müller, C.E. P2-pyrimidinergic receptors and their ligands. Curr. Pharm. Des. 2002, 8, 2353–2369. [Google Scholar] [CrossRef]
- Brunschweiger, A.; Müller, C.E. P2 receptors activated by uracil nucleotides—An update. Curr. Med. Chem. 2006, 13, 289–312. [Google Scholar] [CrossRef]
- Hancock, A.A. The challenge of drug discovery of a GPCR target: Analysis of preclinical pharmacology of histamine H3 antagonists/inverse agonists. Biochem. Pharmacol. 2006, 71, 1103–1113. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic signaling and vascular cell proliferation and death. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 364–373. [Google Scholar] [CrossRef]
- Gachet, C. P2 receptors, platelet function and pharmacological implications. Thromb. Haemost. 2008, 99, 466–472. [Google Scholar]
- Gachet, C. The platelet P2 receptors as molecular targets for old and new antiplatelet drugs. Pharmacol. Ther. 2005, 108, 180–192. [Google Scholar] [CrossRef]
- Gachet, C. Regulation of platelet functions by P2 receptors. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 277–300. [Google Scholar] [CrossRef]
- Cattaneo, M. P2Y12 receptor antagonists: A rapidly expanding group of antiplatelet agents. Eur. Heart J. 2006, 27, 1010–1012. [Google Scholar] [CrossRef]
- Cattaneo, M. Aspirin and clopidogrel efficacy, safety, and the issue of drug resistance Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1980–1988. [Google Scholar] [CrossRef]
- Nawarskas, J.J.; Clark, S.M. Ticagrelor: A novel reversible oral antiplatelet agent. Cardiol. Rev. 2011, 19, 95–100. [Google Scholar] [CrossRef]
- Léon, C.; Hechler, B.; Freund, M.; Eckly, A.; Vial, C.; Ohlmann, P.; Dierich, A.; LeMeur, M.; Cazenave, J.-P.; Gachet, C. Defective platelet aggregation and increased resistance to thrombosis in purinergic P2Y1 receptor-null mice. J. Clin. Invest. 1999, 104, 1731–1737. [Google Scholar] [CrossRef]
- Fabre, J.E.; Nguyen, M.; Latour, A.; Keifer, J.A.; Audoly, L.P.; Coffman, T.M.; Koller, B.H. Decreased platelet aggregation, increased bleeding time and resistance to thromboembolism in P2Y1-deficient mice. Nat. Med. 1999, 5, 1199–1202. [Google Scholar] [CrossRef]
- King, B.F.; Townsend-Nicholson, A.J. Involvement of P2Y1 and P2Y11 purinoceptors in parasympathetic inhibition of colonic smooth muscle. Pharmacol. Exp. Ther. 2008, 324, 1055–1063. [Google Scholar]
- Bean, B.P. Pharmacology and electrophysiology of ATP-activated ion channels. Trends Pharmacol. Sci. 1992, 13, 87–90. [Google Scholar] [CrossRef]
- Inoue, K.; Nakazawa, K.; Ohara-Imaizumi, M.; Obama, T.; Fujimori, K.; Takanaka, A. Antagonism by reactive blue 2 but not by brilliant blue G of extracellular ATP-evoked responses in PC12 phaeochromocytoma cells. Br. J. Pharmacol. 1991, 102, 851–854. [Google Scholar] [CrossRef]
- Nakazawa, K.; Inoue, K.; Fujimori, K.; Takanaka, A. Effects of ATP antagonists on purinoceptor-operated inward currents in rat phaeochromocytoma cells. Pflügers Arch. 1991, 418, 214–219. [Google Scholar] [CrossRef]
- Brown, J.; Brown, C.A. Evaluation of reactive blue 2 derivatives as selective antagonists for P2Y receptors. Vasc. Pharmacol. 2003, 39, 309–315. [Google Scholar] [CrossRef]
- Tuluc, F.; Bültmann, R.; Glänzel, M.; Frahm, A.W.; Starke, K. P2-receptor antagonists: IV. Blockade of P2-receptor subtypes and ecto-nucleotidases by compounds related to reactive blue 2. Naunyn Schmiedeberg’s Arch. Pharmacol. 1998, 357, 111–120. [Google Scholar] [CrossRef]
- Glänzel, M.; Bültmann, R.; Starke, K.; Frahm, A.W. Structure-activity relationships of novel P2-receptor antagonists structurally related to Reactive Blue 2. Eur. J. Med. Chem. 2005, 40, 1262–1276. [Google Scholar] [CrossRef]
- Baqi, Y.; Hausmann, R.; Rosefort, C.; Rettinger, J.; Schmalzing, G.; Müller, C.E. Discovery of potent competitive antagonists and positive modulators of the P2X2 receptor. J. Med. Chem. 2011, 54, 817–830. [Google Scholar] [CrossRef]
- Weyler, S.; Baqi, Y.; Hillmann, P.; Kaulich, M.; Hunder, A.M.; Müller, I.A.; Müller, C.E. Combinatorial synthesis of anilinoanthraquinone derivatives and evaluation as non nucleotide-derived P2Y2 receptor antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 223–227. [Google Scholar]
- Baqi, Y.; Atzler, K.; Köse, M.; Glänzel, M.; Müller, C.E. High-affinity, non nucleotide-derived competitive antagonists of platelet P2Y12 receptors. J. Med. Chem. 2009, 52, 3784–3793. [Google Scholar] [CrossRef]
- Hoffmann, K.; Baqi, Y.; Morena, M.S.; Glänzel, M.; Müller, C.E.; von Kügelgen, I. Interaction of new, very potent non-nucleotide antagonists with Arg256 of the human platelet P2Y12-receptor. J. Pharmacol. Exp. Ther. 2009, 331, 648–655. [Google Scholar] [CrossRef]
- Baqi, Y.; Weyler, S.; Iqbal, J.; Zimmermann, H.; Müller, C.E. Structure-activity relationships of anthraquinone derivatives derived from bromaminic acid as inhibitors of ectonucleoside triphosphate diphosphohydrolases (E-NTPDases). Purinerg. Signal. 2009, 5, 91–106. [Google Scholar] [CrossRef]
- Baqi, Y.; Lee, S.-Y.; Iqbal, J.; Ripphausen, P.; Lehr, A.; Scheiff, A.B.; Zimmermann, H.; Bajorath, J.; Müller, C.E. Development of potent and selective inhibitors of ecto-5’-nucleotidase based on an anthraquinone scaffold. J. Med. Chem. 2010, 53, 2076–2086. [Google Scholar]
- Baqi, Y.; Müller, C.E. Rapid and efficient microwave-assisted copper(0)-catalyzed Ullmann coupling reaction: general access to anilinoanthraquinone derivatives. Org. Lett. 2007, 9, 1271–1274. [Google Scholar] [CrossRef]
- Baqi, Y.; Müller, C.E. Synthesis of alkyl- and aryl-amino-substituted anthraquinone derivatives by microwave-assisted copper(0)-catalyzed Ullmann coupling reactions. Nat. Protoc. 2010, 5, 945–953. [Google Scholar] [CrossRef]
- Paine, A.J. Mechanisms and models for copper mediated nucleophilic aromatic substitution. 2. A single catalytic species from three different oxidation states of copper in an Ullmann synthesis of triarylamines. J. Am. Chem. Soc. 1987, 109, 1496–1502. [Google Scholar] [CrossRef]
- Lewin, A.H.; Cohen, T. The mechanism of the Ullmann reaction. Detection of an organocopper intermediate. Tetrahedron Lett. 1965, 50, 4531–4536. [Google Scholar] [CrossRef]
- Ritter, S.C. Cu(I)-Catalyzed “Click-Chemistry“ Design of a Chemical Photomultiplier Target-Guided Synthesis of Bidentate Metal-Complex Receptors. Ph.D. thesis, University of Regensburg, Germany, 2007. [Google Scholar]
- Ritter, S.C.; König, B. Signal amplification and transduction by photo-activated catalysis. Chem. Commun. 2006, 4694–4696. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Baqi, Y.; Müller, C.E. Convergent Synthesis of the Potent P2Y Receptor Antagonist MG 50-3-1 Based on a Regioselective Ullmann Coupling Reaction. Molecules 2012, 17, 2599-2615. https://doi.org/10.3390/molecules17032599
Baqi Y, Müller CE. Convergent Synthesis of the Potent P2Y Receptor Antagonist MG 50-3-1 Based on a Regioselective Ullmann Coupling Reaction. Molecules. 2012; 17(3):2599-2615. https://doi.org/10.3390/molecules17032599
Chicago/Turabian StyleBaqi, Younis, and Christa E. Müller. 2012. "Convergent Synthesis of the Potent P2Y Receptor Antagonist MG 50-3-1 Based on a Regioselective Ullmann Coupling Reaction" Molecules 17, no. 3: 2599-2615. https://doi.org/10.3390/molecules17032599