Synthesis of Amino Core Compounds of Galactosyl Phytosyl Ceramide Analogs for Developing iNKT-Cell Inducers

Abstract
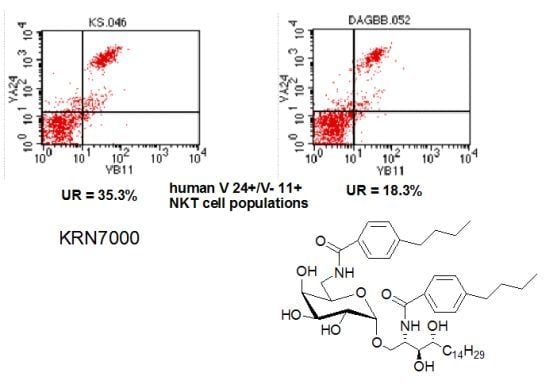
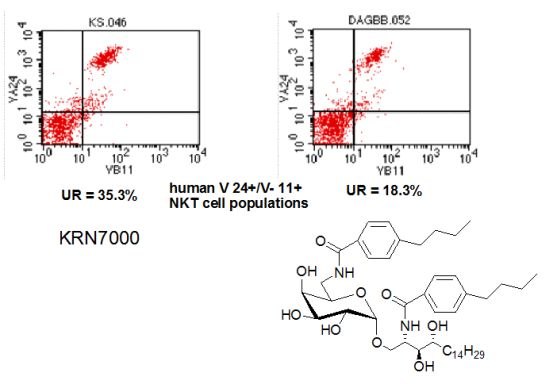
:
1. Introduction

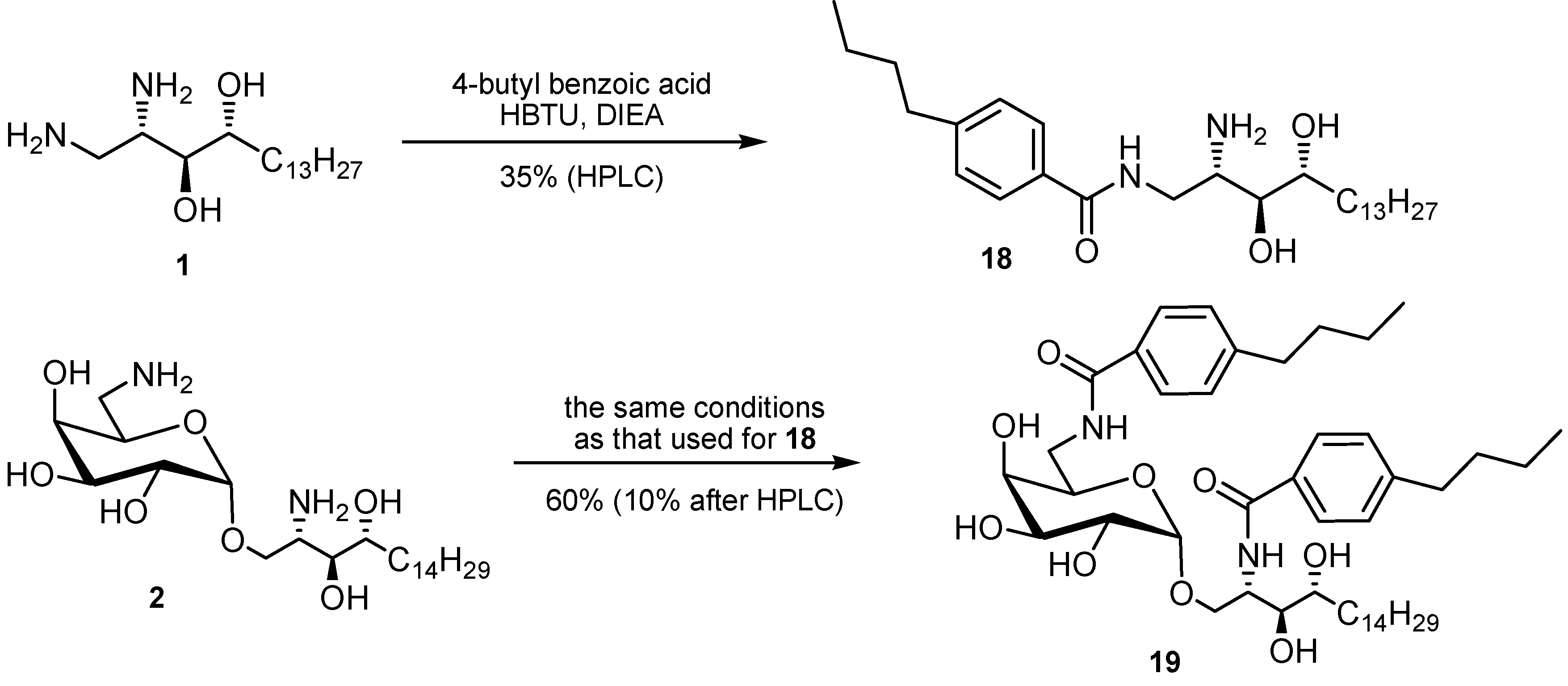
2. Results and Discussion





{kind=link}
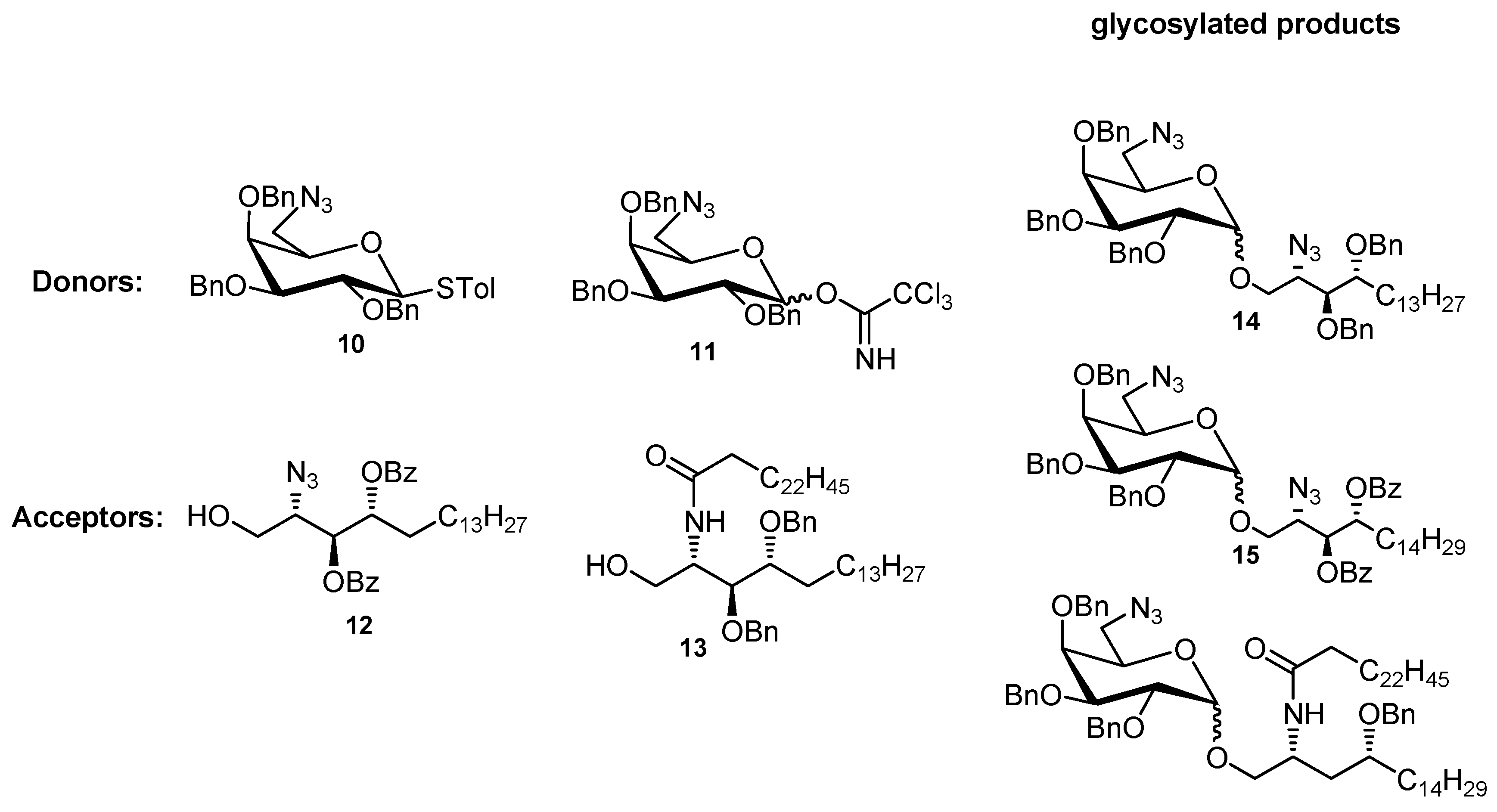{kind=link}
{kind=link}
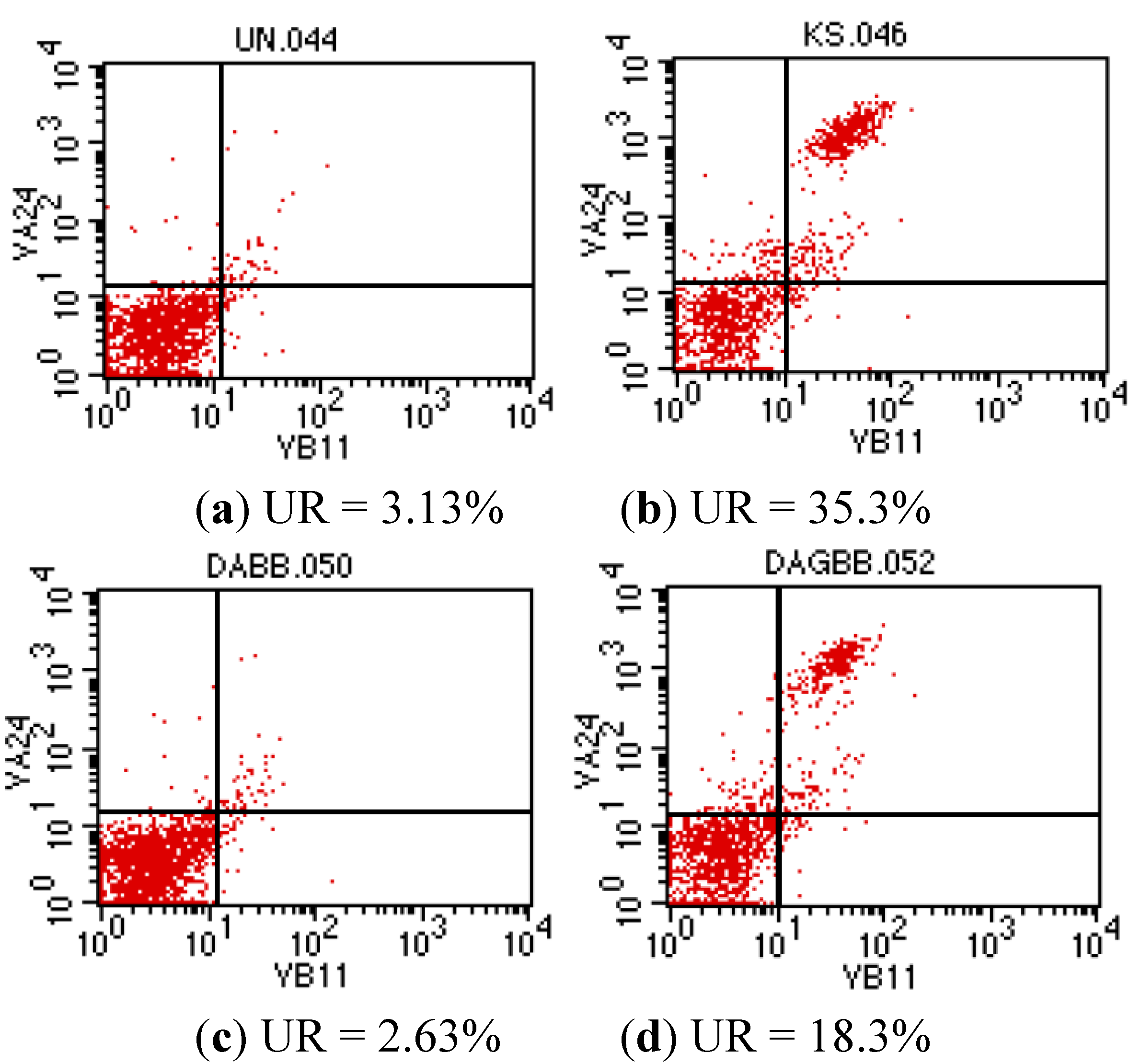{kind=link}
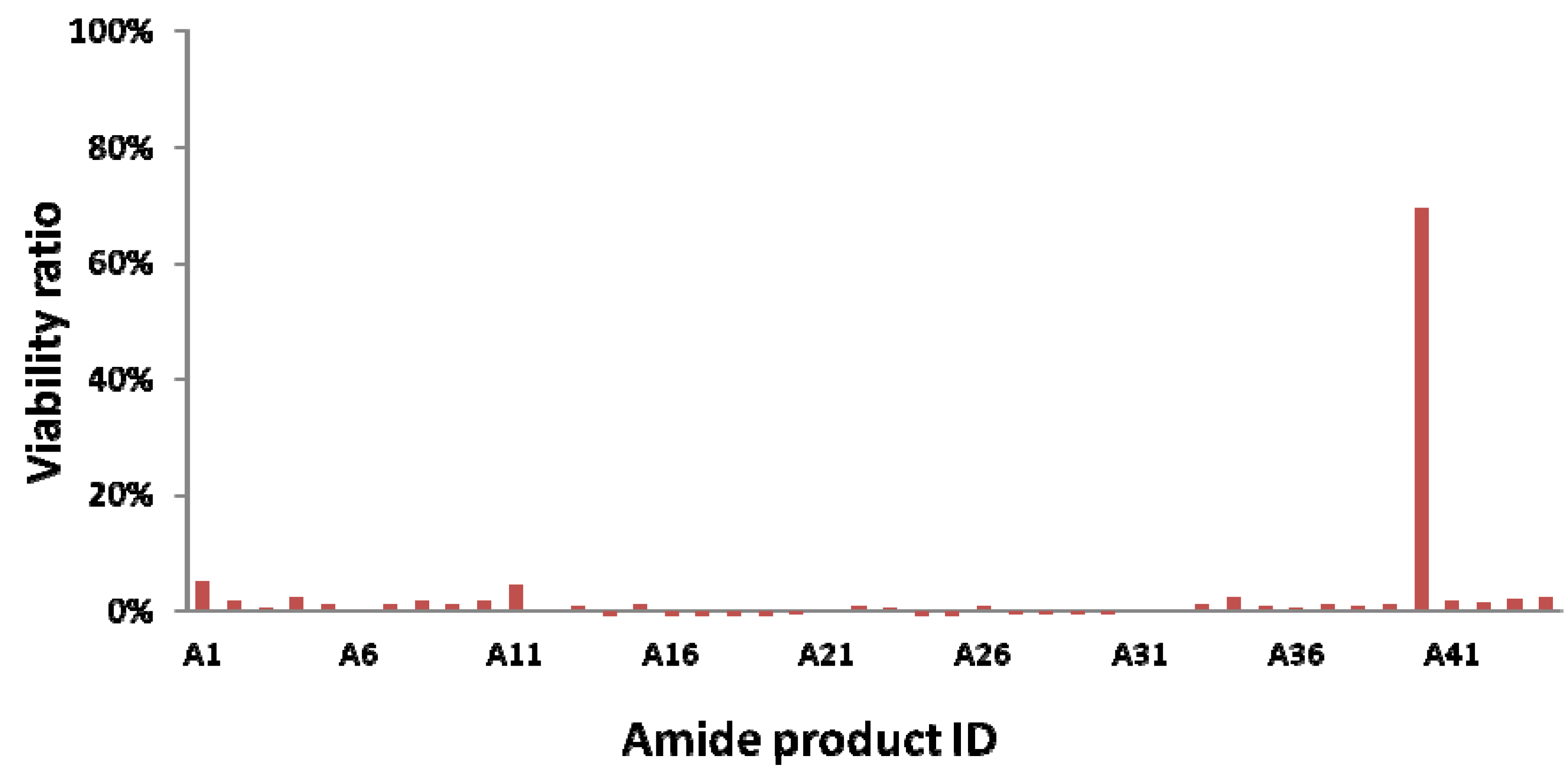{kind=link}
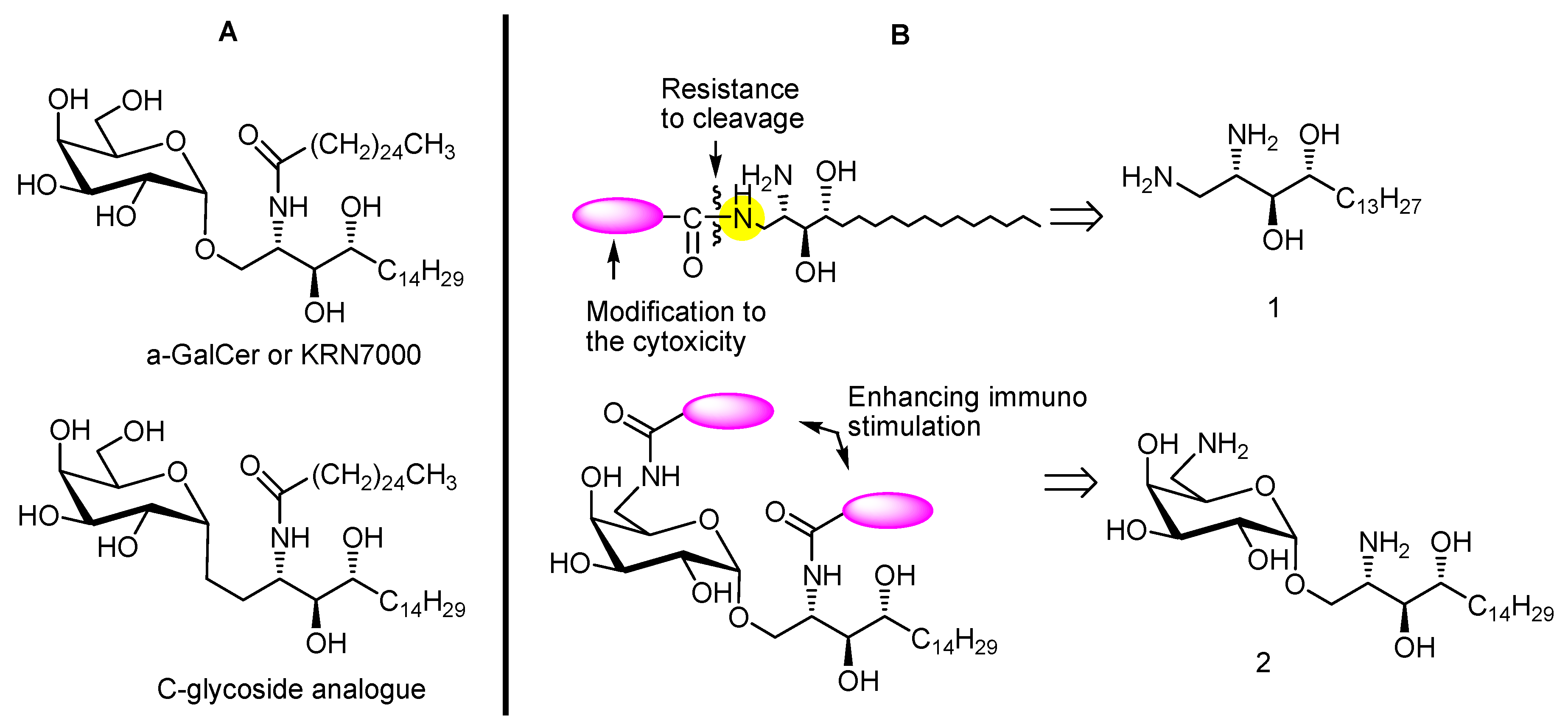{kind=link}
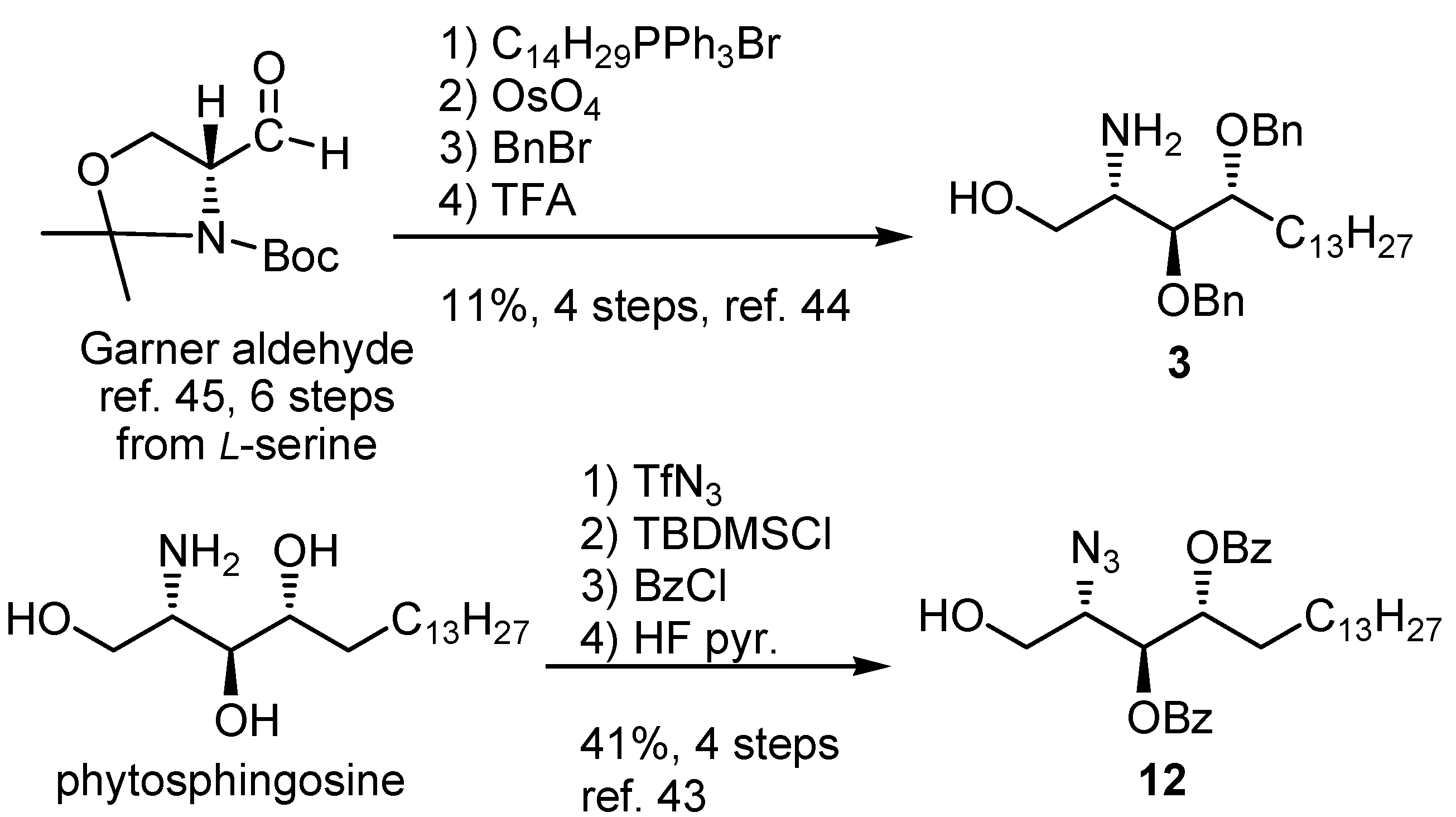{kind=link}
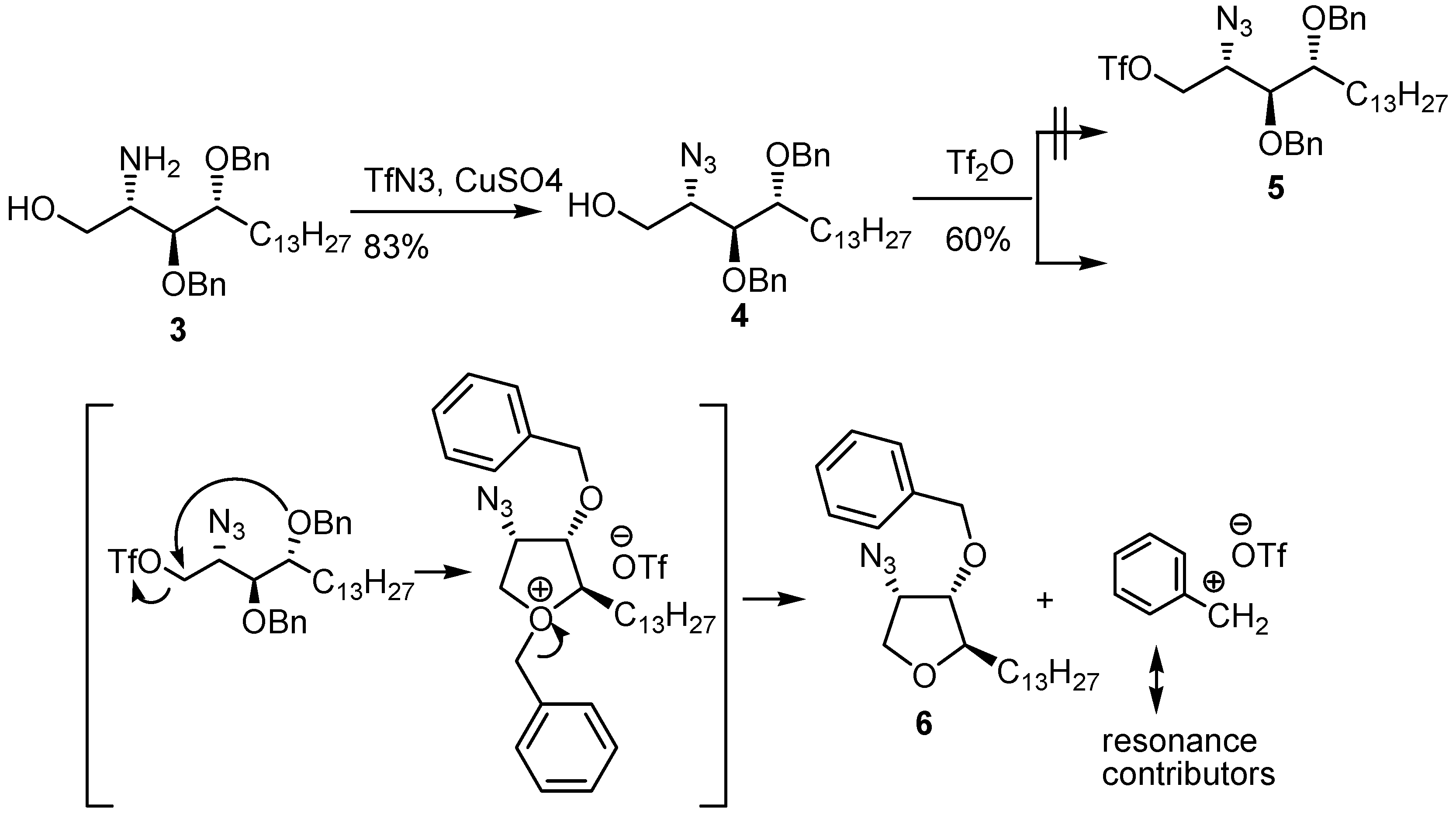{kind=link}
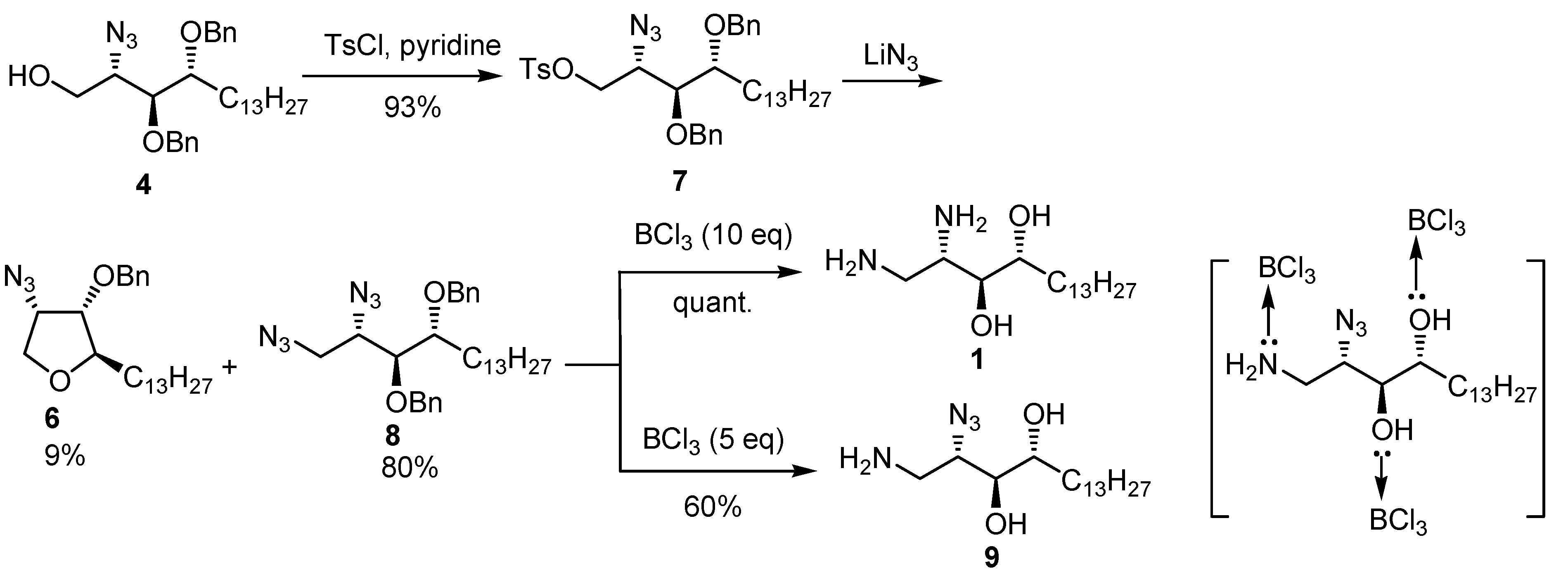{kind=link}
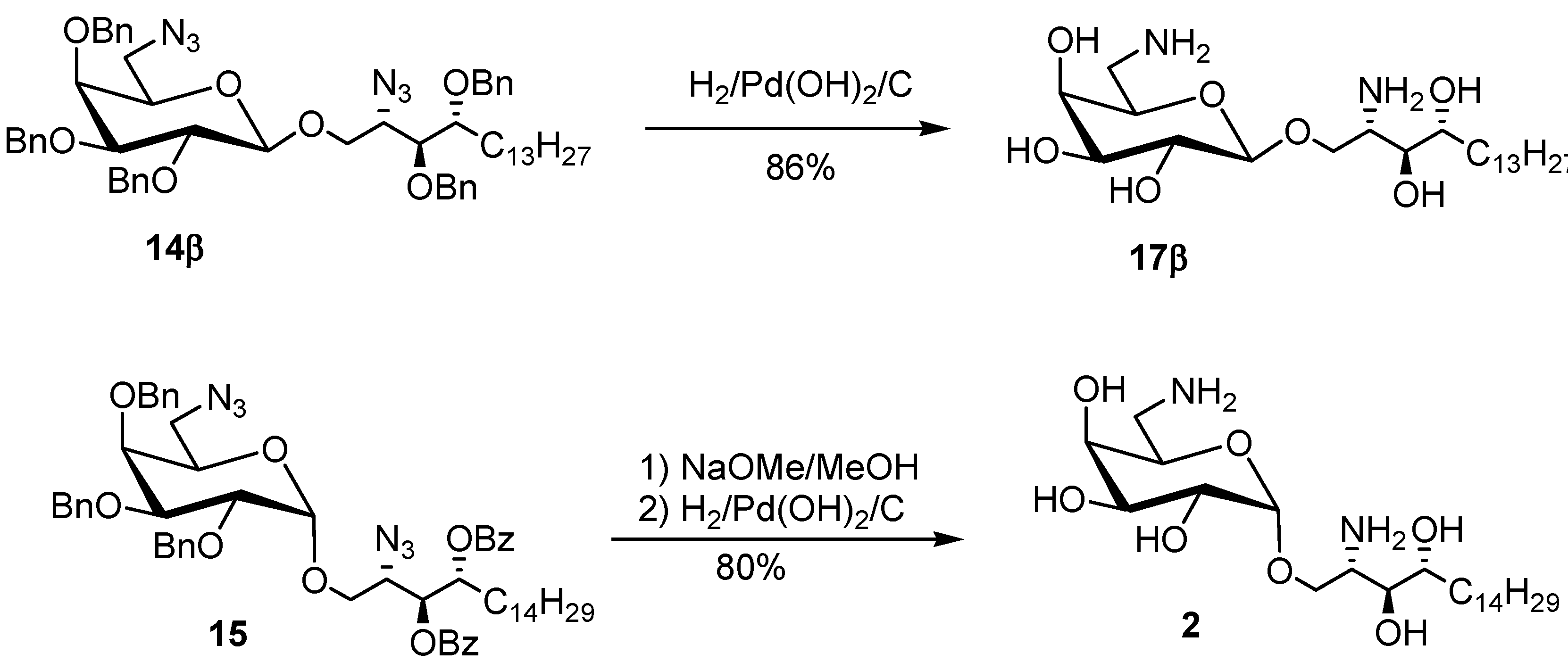{kind=link}
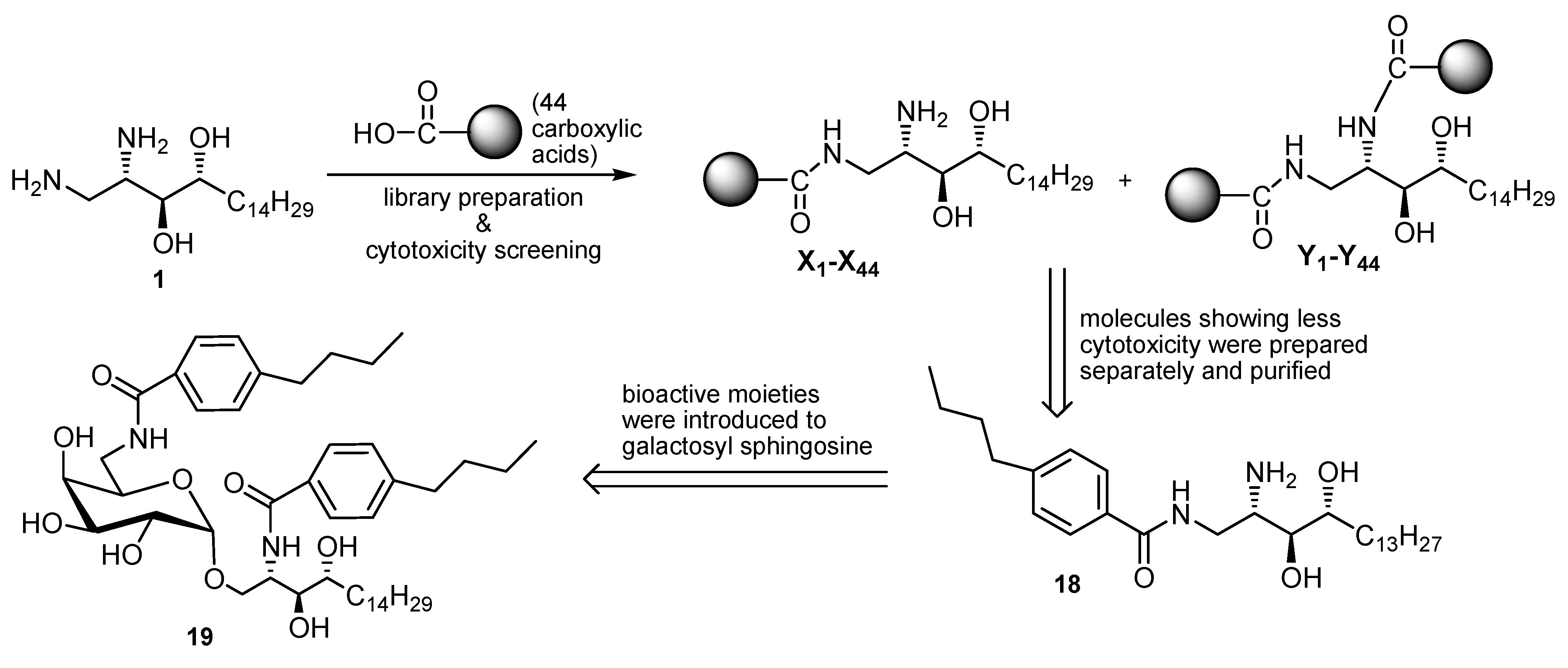{kind=link}
{kind=link}
| Entry | Donor | Acceptor | Time | Product | Yield | α/β |
|---|---|---|---|---|---|---|
| 1 † | 10 | 4 | 30 min | 14 | 95% | 51/44 |
| 2 ‡ | 10 | 12 | 1 h | 15 | 65% | 2/1 |
| 3 § | 10 | 13 | 1 h | 16 | <2% | N.A. |
| 4 Ұ | 11 | 12 | 1 h | 15 | N.F. | N.A. |
| 5 Ұ | 11 | 13 | 1 h | 16 | N.O. | N.A. |





3. Experimental
3.1. General
3.2. Synthesis of the Compounds
3.3. Preparation of Cell Lines and MTT Assay
3.3.1. Cell Culture
3.3.2. MTT Assay of Amide-Bond Formation Products
3.3.2.1. Cell Plating
3.3.2.2. Construction of Amide Bonding Libraries and Their Cytotoxicity Screening

3.4. Invariant Nature Killer Cell Quantification
4. Conclusions
Supplementary Materials
Acknowledgments
Conflict of Interest
References and Notes
- Morita, M.; Motoki, K.; Akimoto, K.; Natori, T.; Sakai, T.; Sawa, E.; Yamaji, K.; Koezuka, Y.; Kobayashi, E.; Fukushima, H. Structure-activity relationship of alpha-galactosylceramides against B16-bearing mice. J. Med. Chem. 1995, 38, 2176–2187. [Google Scholar] [CrossRef]
- Natori, T.; Morita, M.; Akimoto, K.; Koezuka, Y. Agelasphins, novel antitumor and immunostimulatory cerebrosides from the marine sponge Agelas mauritianus. Tetrahedron 1994, 50, 2771–2784. [Google Scholar] [CrossRef]
- Singh, R.; Sharma, M.; Joshi, P.; Rawat, D.S. Clinical status of anti-cancer agents derived from marine sources. Anti-Cancer Agents Med. Chem. 2008, 8, 603–610. [Google Scholar]
- Padron, J.M. Sphingolipids in anticancer therapy. Curr. Med. Chem. 2006, 13, 755–770. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Kronenberg, M. Going both ways: Immune regulation via CD1d-dependent NKT cells. J. Clin. Invest. 2004, 114, 1379–1388. [Google Scholar]
- Kawano, T.; Cui, J.Q.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Motoki, K.; Ueno, H.; Nakagawa, R.; Sato, H.; Kondo, E.; et al. CD1d-restricted and TCR-mediated activation of V(alpha)14 NKT cells by glycosylceramides. Science 1997, 278, 1626–1629. [Google Scholar]
- Zajonc, D.M.; Cantu, C.; Mattner, J.; Zhou, D.P.; Savage, P.B.; Bendelac, A.; Wilson, I.A.; Teyton, L. Structure and function of a potent agonist for the semi-invariant natural killer T cell receptor. Nat. Immunol. 2005, 6, 810–818. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Gervay-Hague, J. Chemical Glycobiology I: Glycoconjugates and Carbohydrate-Protein Interactions; American Chemical Society: Washington, DC, USA, 2008; pp. 153–166, ACS Symposium Series 990. [Google Scholar]
- Wu, D.; Fujio, M.; Wong, C.-H. Glycolipids as immunostimulating agents. Bioorg. Med. Chem. 2008, 16, 1073–1083. [Google Scholar] [CrossRef]
- Kinjo, Y.; Wu, D.; Kim, G.S.; Xing, G.W.; Poles, M.A.; Ho, D.D.; Tsuji, M.; Kawahara, K.; Wong, C.H.; Kronenberg, M. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature 2005, 4341, 520–525. [Google Scholar]
- Wu, D.; Xing, G.W.; Poles, M.A.; Horowitz, A.; Kinjo, Y.; Sullivan, B.; Bodmer-Narkevitch, V.; Plettenburg, O.; Kronenberg, M.; Tsuji, M.; et al. Bacterial glycolipids and analogs as antigens for CD1d-restricted NKT cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1351–1356. [Google Scholar]
- Fujio, M.; Wu, D.G.; Garcia-Navarro, R.; Ho, D.D.; Tsuji, M.; Wong, C.H. Structure-based discovery of glycolipids for CD1d-mediated NKT cell activation: Tuning the adjuvant versus immunosuppression activity. J. Am. Chem. Soc. 2006, 128, 9022–9023. [Google Scholar]
- Gonzalez-Aseguinolaza, G.; Van Kaer, L.; Bergmann, C.C.; Wilson, J.M.; Schmieg, J.; Kronenberg, M.; Nakayama, K.; Taniguchi, M.; Koezuka, Y.; Tsuji, M. Natural killer T cell ligand alpha-galactosylceramide enhances protective immunity induced by malaria vaccines. J. Exp. Med. 2002, 195, 617–624. [Google Scholar] [CrossRef]
- Savage, P.B.; Teyton, L.; Bendelac, A. Glycolipids for natural killer T cells. Chem. Soc. Rev. 2006, 35, 771–779. [Google Scholar]
- Giaccone, G.; Punt, C.J.A.; Ando, Y.; Ruijter, R.; Nishi, N.; Peters, M.; von Blomberg, B.M.E.; Scheper, R.J.; van der Vliet, H.J.J.; van den Eertwegh, A.J.M.; et al. A phase I study of the natural killer T-cell ligand alpha-Galactosylceramide (KRN7000) in patients with solid tumors. Clin. Cancer Res. 2002, 8, 3702–3709. [Google Scholar]
- Subleski, J.J.; Hall, V.L.; Wolfe, T.B.; Scarzello, A.J.; Weiss, J.M.; Chan, T.; Hodge, D.L.; Back, T.C.; Ortaldo, J.R.; Wiltrout, R.H. TCR-Dependent and -independent activation underlie liver-specific regulation of NKT cells. J. Immunol. 2011, 186, 838–847. [Google Scholar] [CrossRef]
- Lu, X.; Song, L.; Metelitsa, L.S.; Bittman, R. Synthesis and evaluation of an alpha-C-Galactosylceramide analogue that induces Th1-biased responses in human natural killer T cells. ChemBioChem 2006, 7, 1750–1756. [Google Scholar] [CrossRef]
- Chen, W.L.; Xia, C.F.; Cai, L.; Wang, P.G. Efficient synthesis of galactosylceramide analogues for iNKT cell stimulation. Bioorg. Med.Chem. Lett. 2010, 20, 3859–3862. [Google Scholar] [CrossRef]
- Miyamoto, K.; Miyake, S.; Yamamura, T. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing T(H)2 bias of natural killer T cells. Nature 2001, 413, 531–534. [Google Scholar] [CrossRef]
- Li, Q.; Ndonye, R.M.; Illarionov, P.A.; Yu, K.O.A.; Jerud, E.S.; Diaz, K.; Bricard, G.; Porcelli, S.A.; Besra, G.S.; Chang, Y.T.; et al. Rapid identification of immunostimulatory alpha-galactosylceramides using synthetic combinatorial libraries. J. Comb. Chem. 2007, 9, 1084–1093. [Google Scholar] [CrossRef]
- Oki, S.; Tomi, C.; Yamamura, T.; Miyake, S. Preferential T(h)2 polarization by OCH is supported by incompetent NKT cell induction of CD40L and following production of inflammatory cytokines by bystander cells in vivo. Int. Immunol. 2005, 17, 1619–1629. [Google Scholar]
- Yu, K.O.A.; Im, J.S.; Molano, A.; Cutronc, Y.; Illarinov, P.A.; Forestier, C.; Fujiwara, N.; Arias, I.; Miyake, S.; Yamamura, T.; et al. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceram ides. Proc. Natl. Acad. Sci. USA 2005, 102, 3383–3388. [Google Scholar]
- Im, J.S.; Arora, P.; Bricard, G.; Molano, A.; Venkataswamy, M.M.; Baine, I.; Jerud, E.S.; Goldberg, M.F.; Baena, A.; Yu, K.O.A.; et al. Kinetics and cellular site of glycolipid loading control the outcome of natural killer T cell activation. Immunity 2009, 30, 888–898. [Google Scholar] [CrossRef]
- Yang, G.L.; Schmieg, J.; Tsuji, M.; Franck, R.W. The C-glycoside analogue of the immunostimulant alpha-galactosylceramide (KRN7000): Synthesis and striking enhancement of activity. Angew. Chem. Int. Ed. 2004, 43, 3818–3822. [Google Scholar] [CrossRef]
- Goff, R.D.; Gao, Y.; Mattner, J.; Zhou, D.P.; Yin, N.; Cantu, C.; Teyton, L.; Bendelac, A.; Savage, P.B. Effects of lipid chain lengths in alpha-galactosylceramides on cytokine release by natural killer T cells. J. Am. Chem. Soc. 2004, 126, 13602–13603. [Google Scholar]
- Trappeniers, M.; Van Beneden, K.; Decruy, T.; Hillaert, U.; Linclau, B.; Elewaut, D.; Van Calenbergh, S. 6'-Derivatised alpha-GalCer analogues capable of inducing strong CD1d-mediated Th1-biased NKT cell responses in mice. J. Am. Chem. Soc. 2008, 130, 16468–16469. [Google Scholar]
- Koch, M.; Stronge, V.S.; Shepherd, D.; Gadola, S.D.; Mathew, B.; Ritter, G.; Fersht, A.R.; Besra, G.S.; Schmidt, R.R.; Jones, E.Y.; et al. The crystal structure of human CD1d with and without alpha-galactosylceramide. Nat. Immunol. 2005, 6, 819–826. [Google Scholar]
- Borg, N.A.; Wun, K.S.; Kjer-Nielsen, L.; Wilce, M.C.J.; Pellicci, D.G.; Koh, R.; Besra, G.S.; Bharadwaj, M.; Godfrey, D.I.; McCluskey, J.; et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature 2007, 448, 44–49. [Google Scholar] [CrossRef]
- Liu, Y.; Goff, R.D.; Zhou, D.P.; Mattner, J.; Sullivan, B.A.; Khurana, A.; Cantu, C.; Ravkov, E.V.; Lbegbu, C.C.; Altman, J.D.; et al. A modified alpha-galactosyl ceramide for staining and stimulating natural killer T cells. J. Immunol. Methods 2006, 312, 34–39. [Google Scholar] [CrossRef]
- Xia, C.F.; Zhang, W.P.; Zhang, Y.L.; Woodward, R.L.; Wang, J.H.; Wang, P.G. Facile synthesis of biotin-labelled alpha-galactosylceramide as antigen for invariant natural killer T cells. Tetrahedron 2009, 65, 6390–6395. [Google Scholar] [CrossRef]
- Liu, Y.; Goff, R.D.; Zhou, D.P.; Mattner, J.; Sullivan, B.A.; Khurana, A.; Cantu, C.; Ravkov, E.V.; Lbegbu, C.C.; Altman, J.D.; et al. Synthesis and NKT cell stimulating properties of fluorophore- and biotin-appended 6''-amino-6''-deoxy-galactosylceramides. Org. Lett. 2002, 4, 1267–1270. [Google Scholar]
- Prigozy, T.I.; Naidenko, O.; Qasba, P.; Elewaut, D.; Brossay, L.; Khurana, A.; Natori, T.; Koezuka, Y.; Kulkarni, A.; Kronenberg, M. Glycolipid antigen processing for presentation by CD1d molecules. Science 2001, 291, 664–667. [Google Scholar]
- Jervis, P.J.; Cox, L.R.; Besra, G.S. Synthesis of a versatile building block for the preparation of 6-N-derivatized alpha-galactosyl ceramides: Rapid access to biologically active glycolipids. J. Org. Chem. 2011, 76, 320–323. [Google Scholar] [CrossRef]
- Schombs, M.; Park, F.E.; Du, W.J.; Kulkarni, S.S.; Gervay-Hague, J. One-pot syntheses of immunostimulatory glyeolipids. J. Org. Chem. 2010, 75, 4891–4898. [Google Scholar]
- Du, W.J.; Gervay-Hague, J. Efficient synthesis of alpha-galactosyl ceramide analogues using glycosyl iodide donors. Org. Lett. 2005, 7, 2063–2065. [Google Scholar]
- Du, W.; Kulkarni, S.S.; Gervay-Hague, J. Efficient, one-pot syntheses of biologically active alpha-linked glycolipids. Chem. Commun. 2007, 23, 2336–2338. [Google Scholar]
- Xing, G.W.; Wu, D.; Poles, M.A.; Horowitz, A.; Tsuji, M.; Ho, D.D.; Wong, C.H. Synthesis and human NKT cell stimulating properties of 3-O-sulfo-alpha/beta-galactosylceramides. Bioorg. Med. Chem. 2005, 13, 2907–2916. [Google Scholar]
- Schmidt, R.R.; Zimmermann, P. Glycosylimidates. 23. synthesis of glycosphingolipids and psychosines. Angew. Chem. Int. Ed. Engl. 1986, 25, 725–726. [Google Scholar] [CrossRef]
- Chiang, L.W.; Pei, K.; Chen, S.W.; Huang, H.L.; Lin, K.J.; Yen, T.C.; Yu, C.S. Combining a solution-phase derived library with in-situ cellular bioassay: Prompt screening of amide-forming minilibraries using mtt assay. Chem. Pharm. Bull. 2009, 57, 714–718. [Google Scholar]
- Su, Y.H.; Chiang, L.W.; Jeng, K.C.; Huang, H.L.; Chen, J.T.; Lin, W.J.; Huang, C.W.; Yu, C.S. Solution-phase parallel synthesis and screening of anti-tumor activities from fenbufen and ethacrynic acid libraries. Bioorg. Med. Chem. Lett. 2011, 21, 1320–1324. [Google Scholar]
- Zhang, L.; Sun, F.; Li, Y.X.; Sun, X.; Liu, X.M.; Huang, Y.S.; Zhang, L.H.; Ye, X.S.; Xiao, J. Rapid synthesis of iminosugar derivatives for cell-based in situ screening: Discovery of "Hit" compounds with anticancer activity. ChemMedChem 2007, 2, 1594–1597. [Google Scholar]
- Lin, K.-I.; Yang, C.-H.; Huang, C.-W.; Jian, J.-Y.; Huang, Y.-C.; Yu, C.-S. Synthesis and structure-activity relationships of fenbufen amide analogs. Molecules 2010, 15, 8796–8803. [Google Scholar] [CrossRef]
- Su, W.-C. Preparation of 6-amino galactosyl sphingosine analogs and its elaboration to amide libraries. Master thesis, National Tsing-Hua University, Hsinchu, Taiwan. 2010. [Google Scholar]
- Chiang, L.-W.; Pan, S.-D.; Lo, J.-M.; Yu, C.-S. Triflic acid-promoted formylation of ceramide in dimethylformamide. Chin. J. Chem. 2009, 27, 2296–2299. [Google Scholar] [CrossRef]
- Garner, P.; Park, J.M. The synthesis and configurational stability of differentially protected beta-hydroxy-alpha-amino aldehydes. J. Org. Chem. 1987, 52, 2361–2364. [Google Scholar] [CrossRef]
- Alper, P.-B.; Hung, S.-C.; Wong, C.-H. Metal catalyzed diazo transfer for the synthesis of azides from amines. Tetrahedron Lett. 1996, 37, 6029–6032. [Google Scholar]
- Kuroda, I.; Musman, M.; Ohtani, I.; Ichiba, T.; Tanaka, J.; Gravalos, D.G.; Higa, T. Pachastrissamine, a cytotoxic anhydrophytosphingosine from a marine sponge, Pachastrissa sp. J. Nat. Prod. 2002, 65, 1505–1506. [Google Scholar]
- Noyce, D.S.; Virglio, J.A. Synthesis and solvolysis of 1-phenylethyl disubstituted phosphinates. J. Org. Chem. 1972, 37, 2643–2647. [Google Scholar]
- Yu, C.-S.; Wang, R.-T.; Chiang, L.-W.; Lee, M.-H. Synthesis of 4',4'-C-diaminomethyl nucleoside derivative as a building block for constructing libraries via amide bond formation. Tetrahedron Lett. 2007, 48, 2979–2982. [Google Scholar]
- Jagdhane, R.C.; Shashidhar, M.S. Orthogonally protected cyclohexanehexols by a "One Reaction—One Product" approach: Efficient access to cyclitols and their analogs. Eur.J. Org. Chem. 2010, 2945–2953. [Google Scholar]
- Mitra, A.; DePue, L.J.; Struss, J.E.; Patel, B.P.; Parkin, S.; Atwood, D.A. Mononuclear Schiff base boron halides: Synthesis, characterization, and dealkylation of trimethyl phosphate. Inorg. Chem. 2006, 45, 9213–9224. [Google Scholar]
- Keizer, T.S.; DePue, L.J.; Parkin, S.; Atwood, D.A. Boron halide chelate compounds and their activity towards the demethylation of trimethylphosphate. Can. J. Chem. 2002, 80, 1463–1468. [Google Scholar] [CrossRef]
- Patel, A.; Lindhorst, T.K. Synthesis of "mixed type" oligosaccharide mimetics based on a carbohydrate scaffold. Eur. J. Org. Chem. 2002, 1, 79–86. [Google Scholar]
- Plettenburg, O.; Bodmer-Narkevitch, V.; Wong, C.-H. Synthesis of alpha-galactosyl ceramide, a potent immunostimulatory agent. J. Org. Chem. 2002, 67, 4559–4564. [Google Scholar] [CrossRef]
- Hansen, H.C.; Magnusson, G. Synthesis of selected aminodeoxy analogues of galabiose and globotriose. Carbohydr. Res. 1999, 322, 166–180. [Google Scholar] [CrossRef]
- Greenberg, W.A.; Priestley, E.S.; Sears, P.S.; Alper, P.B.; Rosenbohm, C.; Hendrix, M.; Hung, S.C.; Wong, C.H. Design and synthesis of new aminoglycoside antibiotics containing neamine as an optimal core structure: Correlation of antibiotic activity with in vitro inhibition of translation. J. Am. Chem. Soc. 1999, 121, 6527–6541. [Google Scholar]
- Mydock, L.K.; Demchenko, A.V. Superarming the S-benzoxazolyl glycosyl donors by simple 2-O-benzoyl-3,4,6-tri-O-benzyl protection. Org. Lett. 2008, 10, 2103–2106. [Google Scholar] [CrossRef]
- Morales-Serna, J.A.; Boutureira, O.; Diaz, Y.; Matheu, M.I.; Castillon, S. Recent advances in the glycosylation of sphingosines and ceramides. Carbohydr. Res. 2007, 342, 1595–1612. [Google Scholar]
- Xia, C.F.; Yao, Q.J.; Schumann, J.; Rossy, E.; Chen, W.L.; Zhu, L.Z.; Zhang, W.P.; De Libero, G.; Wang, P.G. Synthesis and biological evaluation of alpha-galactosylceramide (KRN7000) and isoglobotrihexosylceramide (iGb3). Bioorg. Med. Chem. Lett. 2006, 16, 2195–2199. [Google Scholar]
- Polt, R.; Szabo, L.; Treiberg, J.; Li, Y.; Hruby, V.J. Generalmethods for alpha-o-ser/thr or beta-o-ser/thr glycosides and glycopeptides solidphase synthesis of o-glycosyl cyclic enkephalin analogs. J. Am. Chem. Soc. 1992, 114, 10249–10258. [Google Scholar] [CrossRef]
- Fan, G.T.; Pan, Y.S.; Lu, K.C.; Cheng, Y.P.; Lin, W.C.; Lin, S.; Lin, C.H.; Wong, C.H.; Fang, J.M.; Lin, C.C. Synthesis of alpha-galactosyl ceramide and the related glycolipids for evaluation of their activities on mouse splenocytes. Tetrahedron 2005, 61, 1855–1862. [Google Scholar]
- Sample Availability: Contact the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, Y.-C.; Chiang, L.-W.; Chang, K.-S.; Su, W.-C.; Lin, Y.-H.; Jeng, K.-C.; Lin, K.-I.; Liao, K.-Y.; Huang, H.-L.; Yu, C.-S. Synthesis of Amino Core Compounds of Galactosyl Phytosyl Ceramide Analogs for Developing iNKT-Cell Inducers. Molecules 2012, 17, 3058-3081. https://doi.org/10.3390/molecules17033058
Huang Y-C, Chiang L-W, Chang K-S, Su W-C, Lin Y-H, Jeng K-C, Lin K-I, Liao K-Y, Huang H-L, Yu C-S. Synthesis of Amino Core Compounds of Galactosyl Phytosyl Ceramide Analogs for Developing iNKT-Cell Inducers. Molecules. 2012; 17(3):3058-3081. https://doi.org/10.3390/molecules17033058
Chicago/Turabian StyleHuang, Yin-Cheng, Li-Wu Chiang, Kai-Shiang Chang, Wen-Chin Su, Yi-Hsian Lin, Kee-Ching Jeng, Kun-I Lin, Kuo-Yen Liao, Ho-Lein Huang, and Chung-Shan Yu. 2012. "Synthesis of Amino Core Compounds of Galactosyl Phytosyl Ceramide Analogs for Developing iNKT-Cell Inducers" Molecules 17, no. 3: 3058-3081. https://doi.org/10.3390/molecules17033058