A QSAR Study of Environmental Estrogens Based on a Novel Variable Selection Method

Abstract
:
Abbreviations:
| QSAR | quantitative structure-activity relationship |
| LOO | leave-one-out cross validation |
| VSMVI | variable selection method based on variable interaction |
| LMO | leave-multiple-out |
| CV | cross validation |
| LOOCV | leave-one-out cross validation |
| LMOCV | leave-multiple-out cross validation |
| MCCV | Monte Carlo cross validation |
| OECD | Organization for Economic Co-operation and Development |
| EDCs | endocrine disrupting chemicals |
| ER | estrogen receptor |
| MLR | multiple-linear regression |
| ASR | all-subsets regression |
| PLS | partial least squares |
| VSMP | variable selection and modeling method based on the prediction |
| EA | evolutionary algorithms |
| UFS | unsupervised forward selection |
| LASSO | least absolute shrinkage and selection operator |
| GA | genetic algorithms |
| kNN | k-nearest neighbor |
| RMSE | root-mean-square errors |
| PSO | particle swarm optimization |
| RMSEV | root-mean-square errors of leave-one-out cross validation |
| RMSEP | root-mean-square error of the test set |
| logRBA | middle logarithm of relative binding affinities |
| STD | standard deviation |
| RBA | relative binding affinities |
| VCCLAB | Virtual Computational Chemistry Laboratory |
| EDKB | endocrine disruptor knowledge base |
| CoMFA | comparative molecular field analysis |
| CoMSIA | comparative molecular similarity indices analysis |
| GMDH | group method of data handling |
| NCTR | National Center for Toxicological Research |
| CODESSA | comprehensive descriptors for structural and statistical analysis |
| HQSAR | hologram quantitative structure-activity relationship |
1. Introduction
2. Results and Discussion
2.1. Construction and Validation of Model
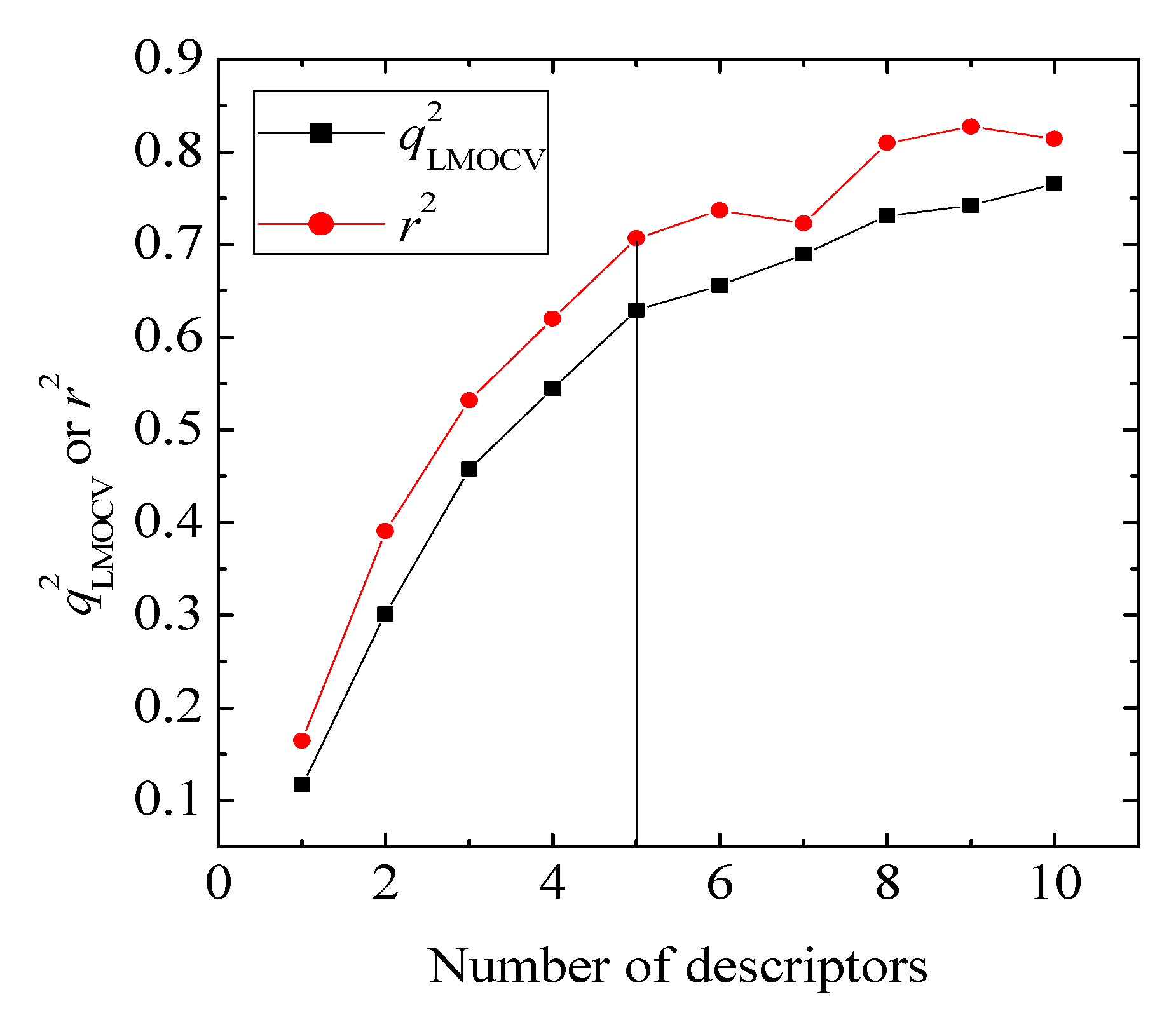
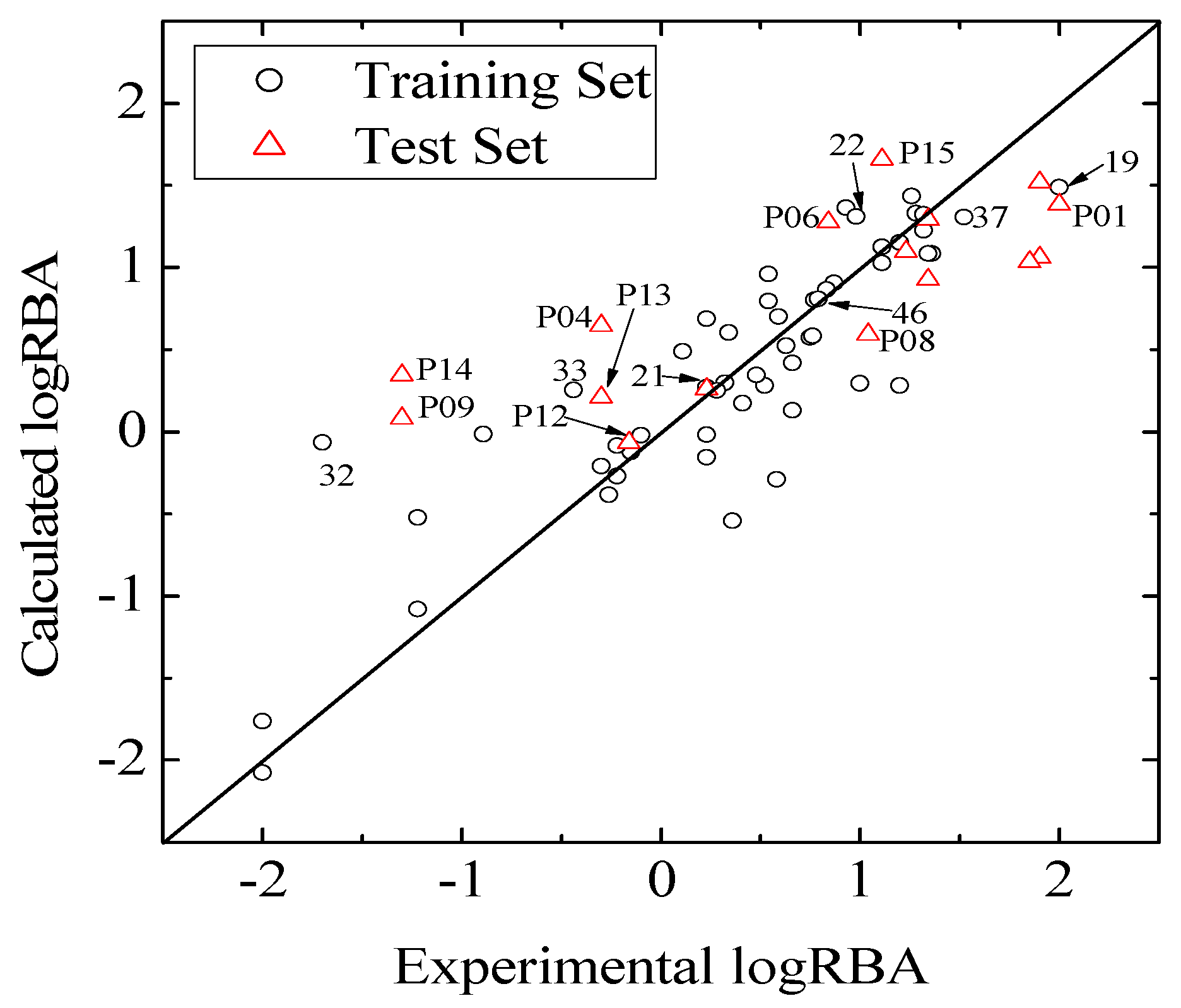
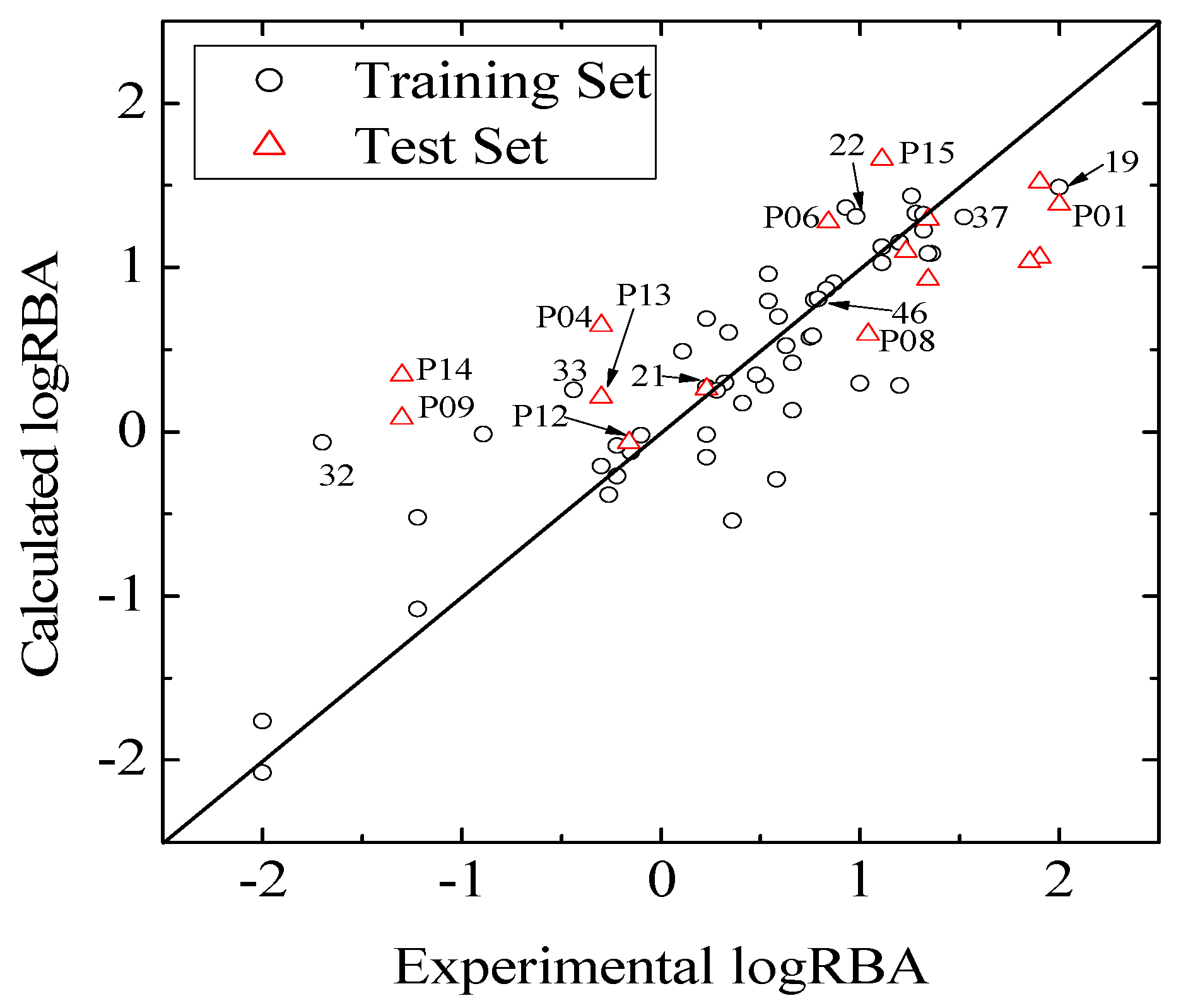
 values against the number of descriptors (Figure 1), which provide guidance in deciding the number of descriptors for constructing models, suggest that the optimal models include five descriptors, because the increase in with the five descriptor is less than 5%. The names, descriptions, and types of the descriptors are listed in Table 1. As shown in Table S3 (supporting information), there is non-correlation between any two variances (correlation coefficient r < 0.8). The experimental and predicted logRBA values for the 53 compounds were summarized in Table S1 (supporting information) and the plots of the experimental logRBA values versus calculated values of the training and test sets were described in Figure 2. The best multiple linear regression model developed using the optimal subset was presented as Equation (1) and Table 2:
values against the number of descriptors (Figure 1), which provide guidance in deciding the number of descriptors for constructing models, suggest that the optimal models include five descriptors, because the increase in with the five descriptor is less than 5%. The names, descriptions, and types of the descriptors are listed in Table 1. As shown in Table S3 (supporting information), there is non-correlation between any two variances (correlation coefficient r < 0.8). The experimental and predicted logRBA values for the 53 compounds were summarized in Table S1 (supporting information) and the plots of the experimental logRBA values versus calculated values of the training and test sets were described in Figure 2. The best multiple linear regression model developed using the optimal subset was presented as Equation (1) and Table 2:
 value of QSAR model [Equation (1)] is 0.0967 and the cr2p value 0.7040. Thus, it can be inferred that the QSAR model developed in the present study is not only the outcome of chance.) vs. number of descriptors.
value of QSAR model [Equation (1)] is 0.0967 and the cr2p value 0.7040. Thus, it can be inferred that the QSAR model developed in the present study is not only the outcome of chance.) vs. number of descriptors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Descriptor Type |
|---|---|---|
| Mor28u | signal 28 / unweighted | 3D MoRSE descriptors |
| E1u | 1st component accessibility directional WHIM index / unweighted | WHIM descriptors |
| E3u | 3rd component accessibility directional WHIM index / unweighted | WHIM descriptors |
| HATS0m | leverage weighted autocorrelation of lag 0 / weighted by mass | GETAWAY descriptors |
| R2m+ | R maximal autocorrelation of lag 2 / weighted by mass | GETAWAY descriptors |
| n | m | r2 or 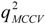 | RMSE | F | Rp | |
|---|---|---|---|---|---|---|
| Estimation | 53 | 5 | 0.7540 | 0.4275 | 28.8090 | 0.7043 |
| MCCV | 53 | 5 | 0.6375 | 0.5166 | ||
| LOOCV | 53 | 5 | 0.6909 | 0.4792 | ||
| External test | 16 | 5 | 0.5308 | 0.7098 |
 and root-mean-square error of the test set (RMSEP) are 0.5308 and 0.7098, respectively, which demonstrate that the derived model exhibits quite good predictive ability. The experimental and predicted LogRBA of 16 compounds in the test set are shown in Table S1 and Figure 2.
and root-mean-square error of the test set (RMSEP) are 0.5308 and 0.7098, respectively, which demonstrate that the derived model exhibits quite good predictive ability. The experimental and predicted LogRBA of 16 compounds in the test set are shown in Table S1 and Figure 2. 2.2. Application Domain and Outlier Analysis of Models




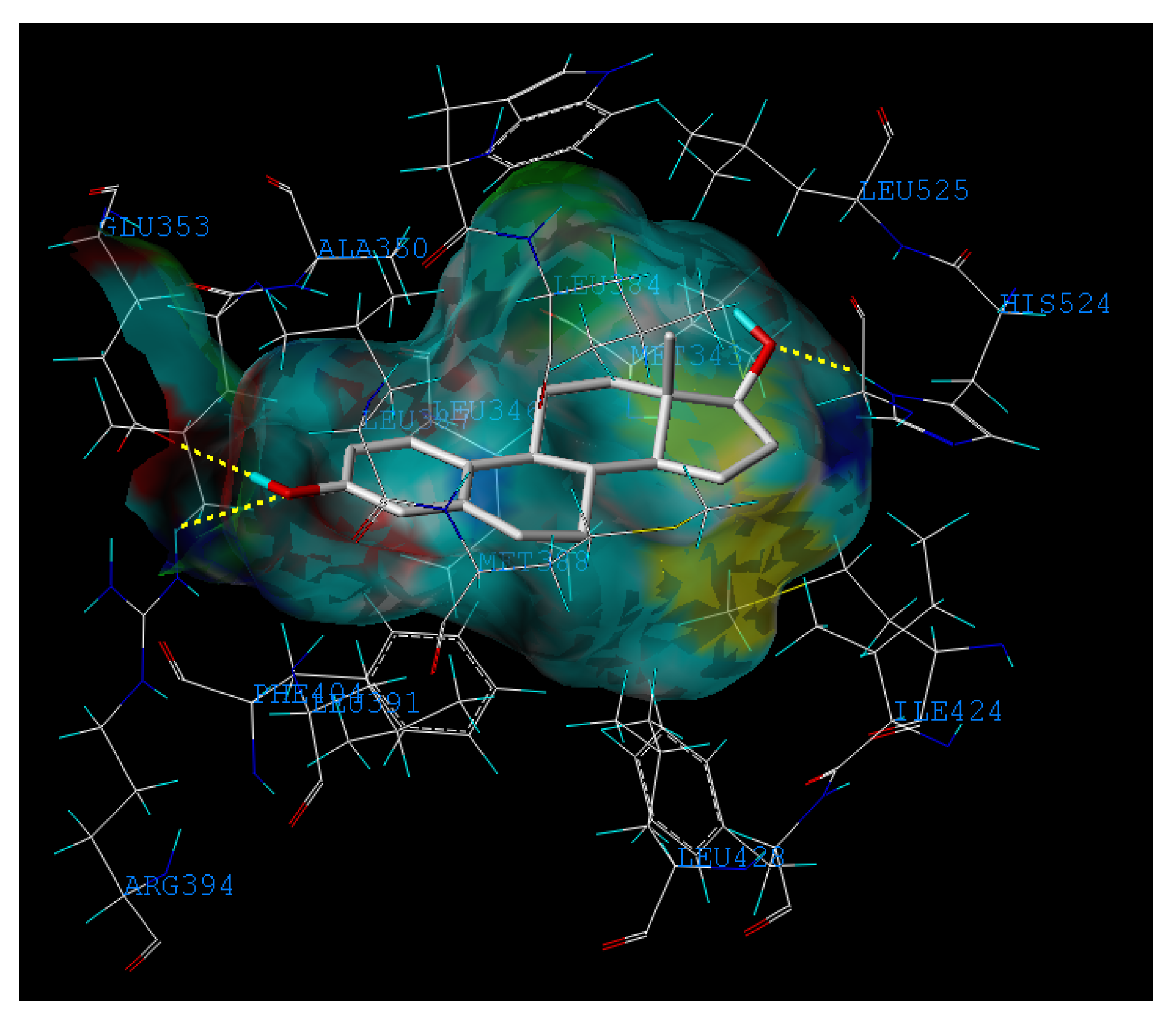
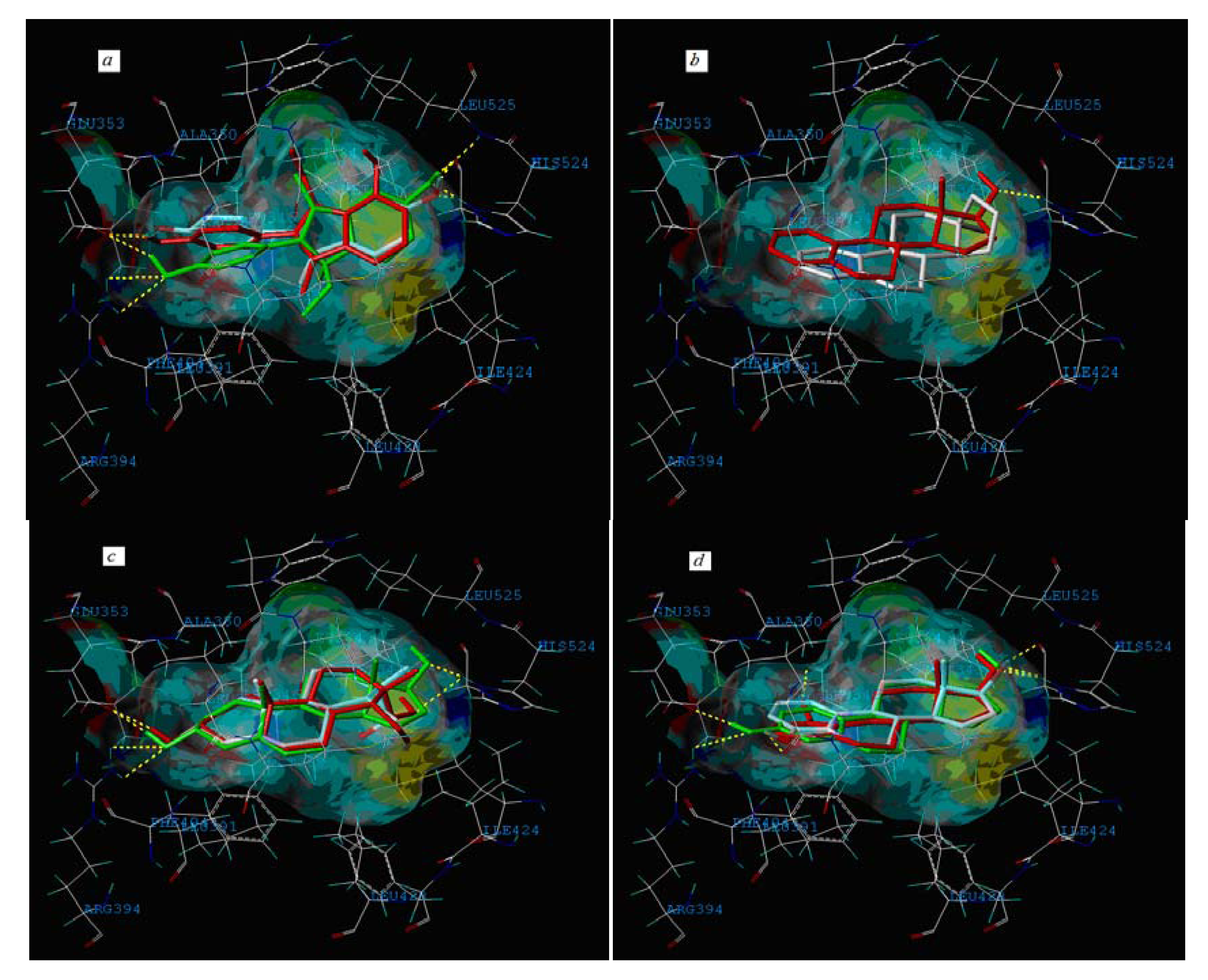
2.3. Interpretation of the Built Model
2.4. Comparison with Other Models
 = 0.61 and r2ext = 0.6718 (calculated by ourselves). However, the deficiency of large r2- gap (0.97–0.61), which is often observed for CoMFA and similar models, may indicate the lower stability of this model. Compared to classical QSAR, the CoMFA method has obtained great success through employing the biological environment surrounding the molecules to interpret the mechanisms of action. However, there also present drawbacks for CoMFA, the main of which is that the complexity of the models is increased as a result of the requirement of 3D conformations, a suitable alignment rule of compounds and a large number of variables. It can make the method more difficult to reproduce a model or at least to apply it to new compounds if the alignment rules are too specific or not suitable for other chemical classes, and the range of chemicals that can be analyzed is limited. A PLS-HQSAR model, based on the use of fragment descriptors, was also derived from the same set of molecules. But the modeling performances are reduced compared to the CoMFA model (r2 = 0.93, = 0.53), and a large gap between r2 and can be observed. The third model was constructed using 365 CODESSA descriptors and the best PLS model computed with these descriptors, including three components (r2 = 0.68, = 0.54). Afterward, Asikainen et al. [37] constructed a QSAR model for the same complete data set using the kNN method and for the whole data set was 0.75. The performance of this QSAR model seems the better one. However, a large amount of descriptors (176) were used as model input, the statistic parameters of model were average result, and then the built model was found to be very difficult to interpret.
= 0.61 and r2ext = 0.6718 (calculated by ourselves). However, the deficiency of large r2- gap (0.97–0.61), which is often observed for CoMFA and similar models, may indicate the lower stability of this model. Compared to classical QSAR, the CoMFA method has obtained great success through employing the biological environment surrounding the molecules to interpret the mechanisms of action. However, there also present drawbacks for CoMFA, the main of which is that the complexity of the models is increased as a result of the requirement of 3D conformations, a suitable alignment rule of compounds and a large number of variables. It can make the method more difficult to reproduce a model or at least to apply it to new compounds if the alignment rules are too specific or not suitable for other chemical classes, and the range of chemicals that can be analyzed is limited. A PLS-HQSAR model, based on the use of fragment descriptors, was also derived from the same set of molecules. But the modeling performances are reduced compared to the CoMFA model (r2 = 0.93, = 0.53), and a large gap between r2 and can be observed. The third model was constructed using 365 CODESSA descriptors and the best PLS model computed with these descriptors, including three components (r2 = 0.68, = 0.54). Afterward, Asikainen et al. [37] constructed a QSAR model for the same complete data set using the kNN method and for the whole data set was 0.75. The performance of this QSAR model seems the better one. However, a large amount of descriptors (176) were used as model input, the statistic parameters of model were average result, and then the built model was found to be very difficult to interpret.| Model | Method | Descriptors | r2 | | r2ext |
|---|---|---|---|---|---|
| Tong [30] | PLS | CoMFA | 0.97 | 0.61 | 0.6712 |
| Tong [30] | PLS | CODESSA | 0.68 | 0.54 | 0.0217 |
| Tong [36] | PLS | HQSAR | 0.93 | 0.53 | - |
| Asikainen [37] | KNN-QSAR | Dragon | 0.86 | 0.73 | - |
| This paper | MLR | Dragon | 0.7540 | 0.6909 | 0.5308 |
of 0.6909 and r2 of 0.7540 based on a simple and unambiguous MLR method. Although the value of r2 is less than those of CoMFA and HQSAR models and the r2ext value is less than that of CoMFA, the value of is greater than that of CoMFA and HQSAR methods and a small r2- gap. In addition, our model has a explicit functional form, which makes it easy to use for other researcher. The comprehensive assessment (LOO and LMO cross validation, external validation and y-randomization test) provides satisfactory results. Therefore, our model may have certain advantages according to the above-mentioned discussion.3. Experimental
3.1. Data Set
3.2. Descriptor Generation and Preprocessing
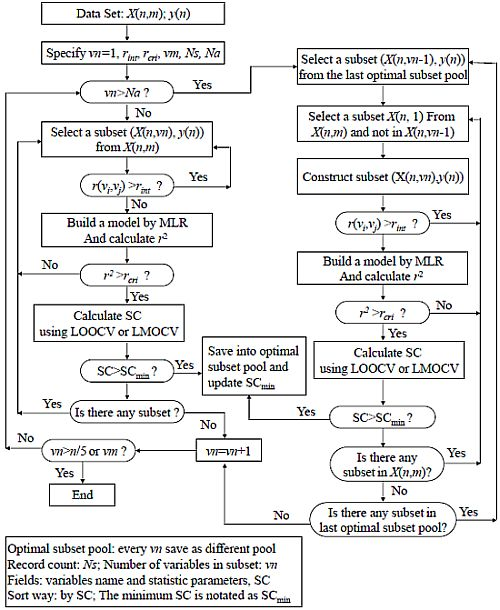
2.3. Variable Selection Method Based on Variable Interaction (VSMVI)
 . They were incorporated into the ASR procedure to accelerate the variable screening speed and control the quality of model, respectively. In fact, The VSMVI method has adopted the ideas of forward selection, VSMP and Group Method of Data Handling (GMDH) [41], and thus it has a high speed for screening variable. The two-order interaction is critically important, so the two-variable combination is the start of the screening. The VSMVI procedure includes two parts. One is the specified (Ns) optimal single variable and two variables subsets; these are selected using a similar VSMP technique and saved into an optimal subset pool. The other part is every subset of the last Ns optimal subset pool combined with one variable that is not in the selected subset, just in order to create a new subset. The new Ns optimal subsets are also saved into a new optimal subset pool. The speed of variable selection will be much quicker than VSMP, because the calculations necessary for variable combination are much fewer than those for the VSMP method. For example, for selecting a five-descriptor optimal subset from 53 descriptors, there are 2869685 combinations that need to be calculated. For VSMVI with 1,000 optimal subsets saved (Ns = 1,000), this drops to only 53,000 combinations.
. They were incorporated into the ASR procedure to accelerate the variable screening speed and control the quality of model, respectively. In fact, The VSMVI method has adopted the ideas of forward selection, VSMP and Group Method of Data Handling (GMDH) [41], and thus it has a high speed for screening variable. The two-order interaction is critically important, so the two-variable combination is the start of the screening. The VSMVI procedure includes two parts. One is the specified (Ns) optimal single variable and two variables subsets; these are selected using a similar VSMP technique and saved into an optimal subset pool. The other part is every subset of the last Ns optimal subset pool combined with one variable that is not in the selected subset, just in order to create a new subset. The new Ns optimal subsets are also saved into a new optimal subset pool. The speed of variable selection will be much quicker than VSMP, because the calculations necessary for variable combination are much fewer than those for the VSMP method. For example, for selecting a five-descriptor optimal subset from 53 descriptors, there are 2869685 combinations that need to be calculated. For VSMVI with 1,000 optimal subsets saved (Ns = 1,000), this drops to only 53,000 combinations.- (a) A subset, X(n,vn), is systematically selected from the initial data set, X(n,m). All correlation coefficients, r(vi,vj), between all pairs of variables in the subset are calculated. If the value of any a r(vi,vj) is larger than the rint specified above, then the selection of the next subset is initiated. If rint remains larger than r(vi,vj), variable screening becomes quicker, because the cross validation procedure, which consumes the most time in variable selection, is avoided.
- (b) If all values of all r(vi,vj)s are smaller than the rint, a multiple linear regression (MLR) model is built between the independent variable subset, X(n,vn), and the dependent variable set, y(n), and the correlation coefficient r2 of the model is calculated. If the value of r2 is smaller than the rcri, the next subset is selected continuously according to step (a).
- (c) If the value of r2 is larger than rcri, a stop criterion (SC) is calculated. The SC can be q2, RMSEV (root-mean-square errors of validation) and so on, and in this paper, the correlation coefficient in LMOCV process,
![Molecules 17 06126 i014]() is used. If the value of SC is larger than SCmin (minimum SC in the optimal subset pool), the subset is plunged into the optimal subset pool. At the same time, the subset having the smallest value of SC (SCmin) is replaced, and the SCmin is updated.
is used. If the value of SC is larger than SCmin (minimum SC in the optimal subset pool), the subset is plunged into the optimal subset pool. At the same time, the subset having the smallest value of SC (SCmin) is replaced, and the SCmin is updated. - (d) If any subset still exists that has not been selected in the whole subset space, the process will return to step (a) to continue the selection of the next subset; otherwise vn is increased by one. If vn is not greater than n/5 or vm, the process will return to step (2), where vm is the maximum value of variables in the optimal subset.

- (a) A subset, X(n,vn − 1), is systematically selected from the last optimal subset pool, in which the number of variables is vn − 1.
- (b) A subset, X(n,1), is also systematically selected from the initial data set, X(n,m), and the variable in X(n,1) is not included in the selected X(n,vn − 1). A new subset, X(n,vn), is constructed from X(n,1) and X(n,vn-1). All correlation coefficients, r(vi,vj), between all pairs of the variables in the subset are calculated.
- (c) If the value of any r(vi,vj) is larger than the rint specified above, then the process returns to (b) to continue the selection of the next subset. If all values of all r(vi,vj)s are smaller than the rint, a MLR model is built between the independent variable subset, X(n,vn), and the dependent variable set, y(n), and the correlation coefficient r2 of model is calculated. If the value of r2 is smaller than the rcri, the next subset is selected continuously according to step (b).
- (d) If the value of r2 is larger than rcri, the SC is calculated. If the value of SC is larger than SCmin, the subset is plunged into optimal subset pool and the subset that has the smallest value of SC (SCmin) is replaced. At the same time, the SCmin is updated. If any subset still exists that has not been selected in the X(n,m), the process will return to step (b) to continue the selection of the next subset, or will go to step (e).
- (e) If any subset still exists that was not selected in the last optimal subset pool, the process will return to step (b) to continue the selection of the next subset, or will go to step (a); otherwise vn will be increased by one. If vn is not greater than n/5 or vm, the process will return to step (3) or the optimal process will be ended.
3.4. Leave-Multiple-Out Cross Validation
 of test set (external set). They can be calculated by the following equations:
of test set (external set). They can be calculated by the following equations:


 are the experimental values (logRBA in present study) of the ith compound in the training and test sets;
are the experimental values (logRBA in present study) of the ith compound in the training and test sets;  and
and  represent the estimators of the ith compound obtained via the linear model;
represent the estimators of the ith compound obtained via the linear model;  and test are corresponding average values; and n and ns denote the number of compounds of the training and test sets, respectively.
and test are corresponding average values; and n and ns denote the number of compounds of the training and test sets, respectively.  or RMSEVLMOCV, is employed to assess the average prediction error of a model. Corresponding standard deviation STD or STDRMSEVLMOCV is used to evaluate the robustness of a model. They are defined as follows:
or RMSEVLMOCV, is employed to assess the average prediction error of a model. Corresponding standard deviation STD or STDRMSEVLMOCV is used to evaluate the robustness of a model. They are defined as follows:



 is the estimator of the dependent variable value obtained via ith iteration in LMOCV; N is the number of iterations; and
is the estimator of the dependent variable value obtained via ith iteration in LMOCV; N is the number of iterations; and  and
and  are the correlation coefficient and RMSEV of ith iteration, respectively.
are the correlation coefficient and RMSEV of ith iteration, respectively.3.5. Application Domain
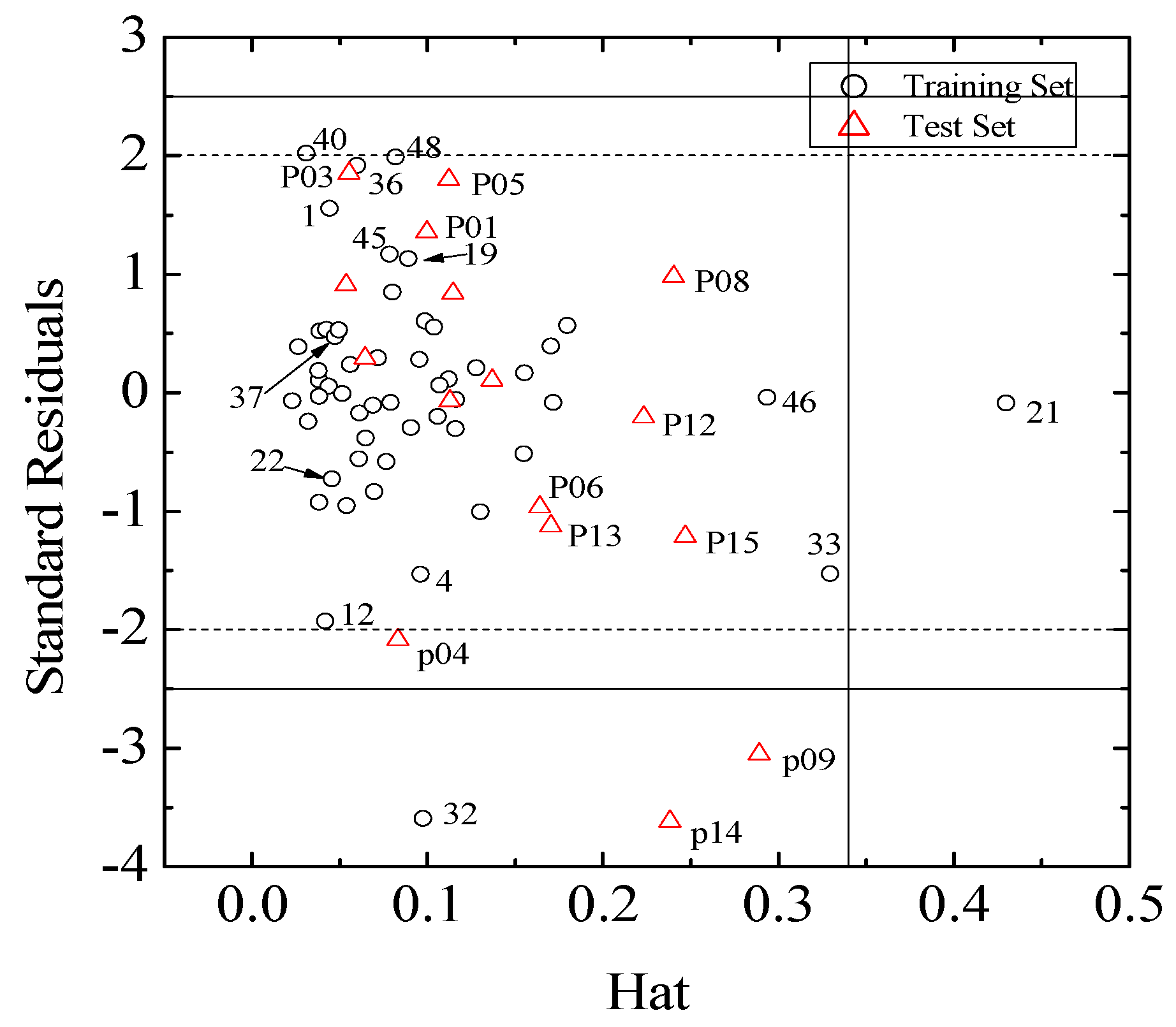
 (I = 1, 2… n), where xi is the descriptor row vector of the query compound, and X is the n × (k-1) of k model descriptor values for n training set. Control leverage h* is generally fixed at 3k/n, where k is the number of model parameters (including the constant term of the MLR model), and n is the number of compounds used to construct the model. A leverage greater than control value h* means that the predicted logRBA is the result of substantial extrapolation of the model, and therefore may be unreliable [46]. The compounds with leverage greater than control value h* are identified as X outliers, which affect model performance, whereas those with standardized residuals greater than 2.5 standard deviation units are identified as Y outliers.
(I = 1, 2… n), where xi is the descriptor row vector of the query compound, and X is the n × (k-1) of k model descriptor values for n training set. Control leverage h* is generally fixed at 3k/n, where k is the number of model parameters (including the constant term of the MLR model), and n is the number of compounds used to construct the model. A leverage greater than control value h* means that the predicted logRBA is the result of substantial extrapolation of the model, and therefore may be unreliable [46]. The compounds with leverage greater than control value h* are identified as X outliers, which affect model performance, whereas those with standardized residuals greater than 2.5 standard deviation units are identified as Y outliers.3.6. Chance Correlation Validation
 () [51] was also calculated to check the distance of QSAR models from chance models, where the means the squared mean correlation coefficient of random models.
() [51] was also calculated to check the distance of QSAR models from chance models, where the means the squared mean correlation coefficient of random models. 4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Wolohan, P.; Reichert, D.E. CoMFA and docking study of novel estrogen receptor subtype selective ligands. J. Comput. Aided Mol. Des. 2003, 17, 313–328. [Google Scholar] [CrossRef]
- Sonnenschein, C.; Soto, A.M. An updated review of environmental estrogen and androgen mimics and antagonists. J. Steroid Biochem. Mol. Biol. 1998, 65, 143–150. [Google Scholar] [CrossRef]
- Lintelmann, J.; Katayama, A.; Kurihara, N.; Shore, L.; Wenzel, A. Endocrine disruptors in the environment. Pure Appl. Chem 2003, 75, 631–681. [Google Scholar] [CrossRef]
- Devillers, J. Endocrine Disruption Modeling; CRC Press: New York, NY, USA, 2009. [Google Scholar]
- Bolger, R.; Wiese, T.E.; Ervin, K.; Nestich, S.; Checovich, W. Rapid Screening of environmental chemicals for estrogen receptor binding capacity. Environ. Health Perspect. 1998, 106, 551–557. [Google Scholar] [CrossRef]
- Devillers, J.; Marchand-Geneste, N.; Carpy, A.; Porcher, J.M. SAR and QSAR modeling of endocrine disruptors. SAR QSAR Environ. Res. 2006, 17, 393–412. [Google Scholar] [CrossRef]
- Fang, H.; Tong, W.; Welsh, W.; Sheehan, D. QSAR models in receptor-mediated effects: The nuclear receptor superfamily. J. Mol. Struc. Theochem 2003, 622, 113–125. [Google Scholar] [CrossRef]
- Schmieder, P.; Ankley, G.; Mekenyan, O.; Walker, J.; Bradbury, S. Quantitative structure-activity relationship models for prediction of estrogen receptor binding affinity of structurally diverse chemicals. Environ. Toxicol. Chem. 2003, 22, 1844–1854. [Google Scholar] [CrossRef]
- Wolpert, D. The relationship between Occam’s razor and convergent guessing. Complex Syst. 1990, 4, 319–368. [Google Scholar]
- Bell, D.; Wang, H. A formalism for relevance and its application in feature subset selection. Mach. Learn. 2000, 41, 175–195. [Google Scholar] [CrossRef]
- González, M.P.; Terán, C.; Saíz-Urra, L.; Teijeir, M. Variable selection methods in QSAR: An overview. Curr. Top. Med. Chem. 2008, 8, 1606–1627. [Google Scholar] [CrossRef]
- Tsygankova, I.G. Variable selection in QSAR models for drug design. Curr. Comput. Aided Drug Des. 2008, 4, 132–142. [Google Scholar] [CrossRef]
- Abraham, B.; Chipman, H.; Vijayan, K. Some risks in the construction and analysis of supersaturated designs. Technometrics 1999, 41, 135–141. [Google Scholar] [CrossRef]
- Smith, J.S.; Macina, O.T.; Sussman, N.B.; Luster, M.I.; Karol, M.H. A robust structure-activity relationship (SAR) model for esters that cause skin irritation in humans. Toxicol. Sci. 2000, 55, 215–222. [Google Scholar] [CrossRef]
- Liu, S.; Lu, J.-C.; Kolpin, D.W.; Meeker, W.Q. Analysis of environmental data with censored observations. Environ. Sci. Technol. 1997, 31, 3358–3362. [Google Scholar] [CrossRef]
- Liu, S.S.; Liu, H.L.; Yin, C.S.; Wang, L.S. VSMP: A novel variable selection and modeling method based on the prediction. J. Chem. Inf. Comput. Sci. 2003, 43, 964–969. [Google Scholar] [CrossRef]
- Whitley, D.; Ford, M.; Livingstone, D. Unsupervised forward selection: a method for eliminating redundant variables. J. Chem. Inf. Comput. Sci. 2000, 40, 1160–1168. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. Roy. Stat. Soc. Ser. B (Methodological) 1996, 58, 267–288. [Google Scholar]
- Zheng, W.F.; Tropsha, A. Novel variable selection quantitative structure-property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci. 2000, 40, 185–194. [Google Scholar] [CrossRef]
- Kubinyi, H. Variable selection in QSAR studies. I. An evolutionary algorithm. QSAR Comb. Sci. 1994, 13, 285–294. [Google Scholar] [CrossRef]
- Agrafiotis, D.; Cedeno, W. Feature selection for structure-activity correlation using binary particle swarms. J. Med. Chem. 2002, 45, 1098–1107. [Google Scholar] [CrossRef]
- Shen, Q.; Jiang, J.H.; Tao, J.C.; Shen, G.L.; Yu, R.Q. Modified ant colony optimization algorithm for variable selection in QSAR modeling: QSAR studies of cyclooxygenase inhibitors. J. Chem. Inf. Model. 2005, 45, 1024–1029. [Google Scholar] [CrossRef]
- Martens, H.A.; Dardenne, P. Validation and verification of regression in small data sets. Chemometr. Intell. Lab. Syst. 1998, 44, 99–121. [Google Scholar] [CrossRef]
- Næs, T. Leverage and influence measures for principal component regression. Chemometr. Intell. Lab. Syst. 1989, 5, 155–168. [Google Scholar] [CrossRef]
- Höskuldsson, A. Dimension of linear models. Chemometr. Intell. Lab. Syst. 1996, 32, 37–55. [Google Scholar] [CrossRef]
- Efron, B. How biased is the apparent error rate of a prediction rule? J. Am. Stat. Assoc. 1986, 81, 461–470. [Google Scholar] [CrossRef]
- Shao, J. Linear model selection by cross-validation. J. Am. Stat. Assoc. 1993, 88, 486–494. [Google Scholar] [CrossRef]
- Stone, M. An asymptotic equivalence of choice of model by cross-validation and Akaike’s criterion. J. Roy. Stat. Soc. Ser. B (Methodological) 1977, 39, 44–47. [Google Scholar]
- Zhang, P. Model selection via multifold cross validation. Ann. Stat. 1993, 21, 299–313. [Google Scholar]
- Tong, W.D.; Perkins, R.; Strelitz, R.; Collantes, E.R.; Keenan, S.; Welsh, W.J.; Branham, W.S.; Sheehan, D.M. Quantitative structure-activity relationships (QSARs) for estrogen binding to the estrogen receptor: Predictions across species. Environ. Health Perspect. 1997, 105, 1116–1124. [Google Scholar]
- Brzozowski, A.; Pike, A.; Dauter, Z.; Hubbard, R.; Bonn, T.; Engström, O.; Öhman, L.; Greene, G.; Gustafsson, J.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Fang, H.; Tong, W.; Shi, L.M.; Blair, R.; Perkins, R.; Branham, W.; Hass, B.S.; Xie, Q.; Dial, S.L.; Moland, C.L.; et al. Structure-activity relationships for a large diverse set of natural, synthetic, and environmental estrogens. Chem. Res. Toxicol. 2001, 14, 280–294. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley VCH: New York, NY, USA, 2000. [Google Scholar]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Wiley VCH: New York, NY, USA, 2009. [Google Scholar]
- Tong, W.; Lowis, D.R.; Perkins, R.; Chen, Y.; Welsh, W.J.; Goddette, D.W.; Heritage, T.W.; Sheehan, D.M. Evaluation of quantitative structure-activity relationship methods for large-scale prediction of chemicals binding to the estrogen receptor. J. Chem. Inf. Comput. Sci. 1998, 38, 669–677. [Google Scholar] [CrossRef]
- Asikainen, A.H.; Ruuskanen, J.; Tuppurainen, K.A. Consensus kNN QSAR: A versatile method for predicting the estrogenic activity of organic compounds in silico. A comparative study with five estrogen receptors and a large, diverse set of ligands. Environ. Sci. Technol. 2004, 38, 6724–6729. [Google Scholar] [CrossRef]
- Tetko, I.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.; Radchenko, E.; Zefirov, N.; Makarenko, A.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef]
- VCCLAB Virtual Computational Chemistry Laboratory Home Page. Available online: http://www.vcclab.org (accessed on 20 November 2011).
- Liu, S.S.; Liu, H.L.; Yin, C.S.; Wang, L.S. VSMP: A novel variable selection and modeling method based on the prediction. J. Chem. Inf. Comput. Sci. 2003, 43, 964–969. [Google Scholar] [CrossRef]
- Farlow, S.J. The GMDH algorithm of ivakhnenko. Am. Stat. 1981, 35, 210–215. [Google Scholar]
- Hawkins, D.; Basak, S.; Mills, D. Assessing model fit by cross-validation. J. Chem. Inf. Comput. Sci. 2003, 43, 579–586. [Google Scholar] [CrossRef]
- Cruciani, G.; Baroni, M.; Clementi, S.; Costantino, G.; Riganelli, D.; Skagerberg, B. Predictive ability of regression models. Part I: Standard deviation of prediction errors (SDEP). J. Chemom. 1992, 6, 335–346. [Google Scholar] [CrossRef]
- Baumann, K. Cross-validation as the objective function for variable-selection techniques. Trac-Trends Anal. Chem. 2003, 22, 395–406. [Google Scholar] [CrossRef]
- Xu, Q.-S.; Liang, Y.-Z. Monte carlo cross validation. Chemometr. Intell. Lab. Syst. 2001, 56, 1–11. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Netzeva, T.; Worth, A.; Aldenberg, T.; Benigni, R.; Cronin, M.; Gramatica, P.; Jaworska, J.; Kahn, S.; Klopman, G.; Marchant, C. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. Altern. Lab. Anim. 2005, 33, 1–19. [Google Scholar]
- Jaworska, J.; Nikolova-Jeliazkova, N.; Aldenberg, T. QSAR applicability domain estimation by projection of the training set in descriptor space: A review. Altern. Lab. Anim. 2005, 33, 445–459. [Google Scholar]
- Héberger, K. Quantitative structure-(chromatographic) retention relationships. J. Chromatogr. A 2007, 1158, 273–305. [Google Scholar] [CrossRef]
- Wold, S.; Eriksson, L. Statistical Validation of QSAR Results. In Chemometric Methods in Molecular Design; Waterbeemd, H.V.D., Ed.; Wiley VCH Publishers, Inc.: New York, NY, USA, 1995; Volume 2, pp. 309–318. [Google Scholar]
- Mitra, I.; Saha, A.; Roy, K. Exploring quantitative structure-activity relationship studies of antioxidant phenolic compounds obtained from traditional Chinese medicinal plants. Mol. Simul. 2010, 36, 1067–1079. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yi, Z.; Zhang, A. A QSAR Study of Environmental Estrogens Based on a Novel Variable Selection Method. Molecules 2012, 17, 6126-6145. https://doi.org/10.3390/molecules17056126
Yi Z, Zhang A. A QSAR Study of Environmental Estrogens Based on a Novel Variable Selection Method. Molecules. 2012; 17(5):6126-6145. https://doi.org/10.3390/molecules17056126
Chicago/Turabian StyleYi, Zhongsheng, and Aiqian Zhang. 2012. "A QSAR Study of Environmental Estrogens Based on a Novel Variable Selection Method" Molecules 17, no. 5: 6126-6145. https://doi.org/10.3390/molecules17056126