Strong Inhibition of Celastrol Towards UDP-Glucuronosyl Transferase (UGT) 1A6 and 2B7 Indicating Potential Risk of UGT-Based Herb-Drug Interaction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

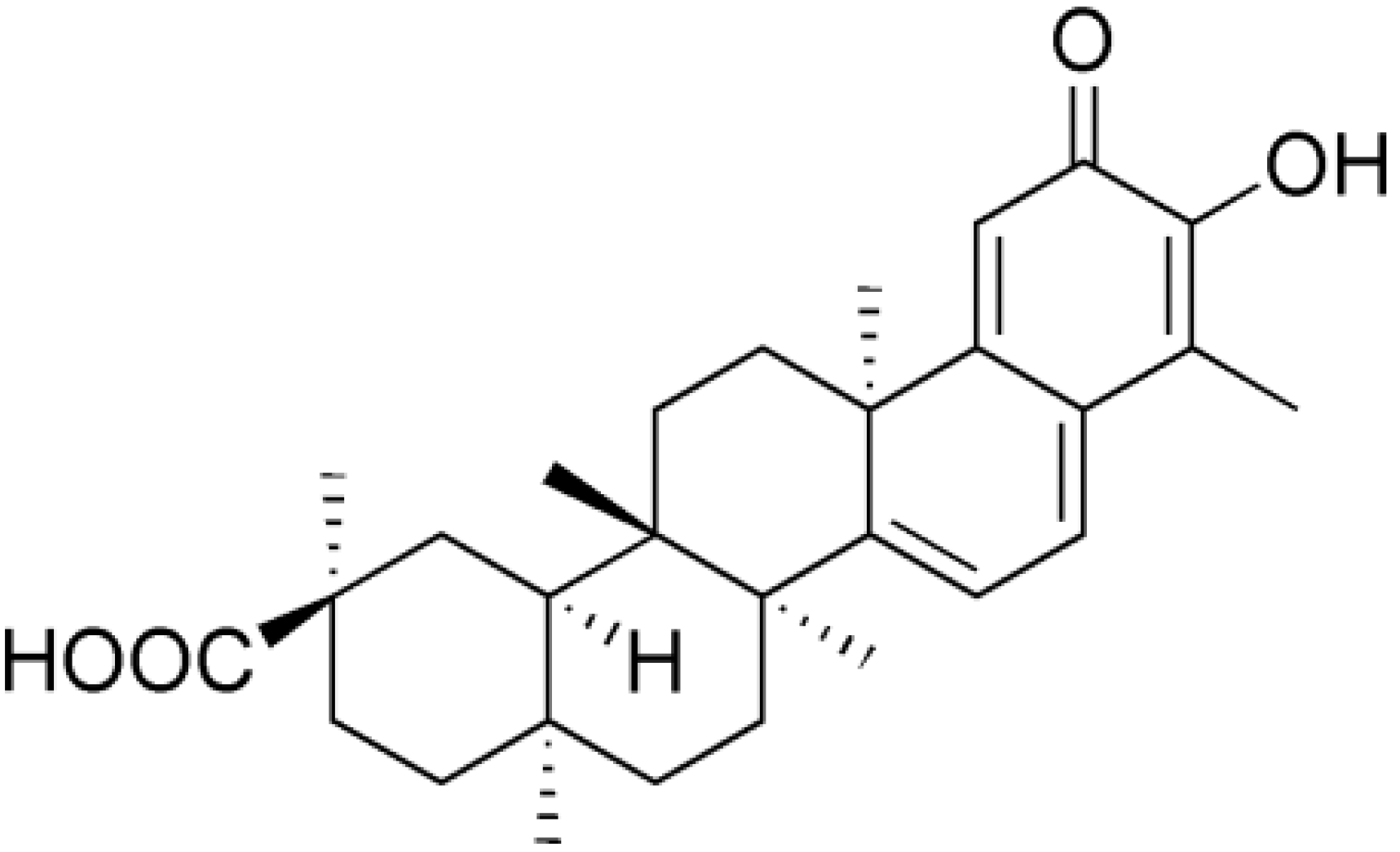
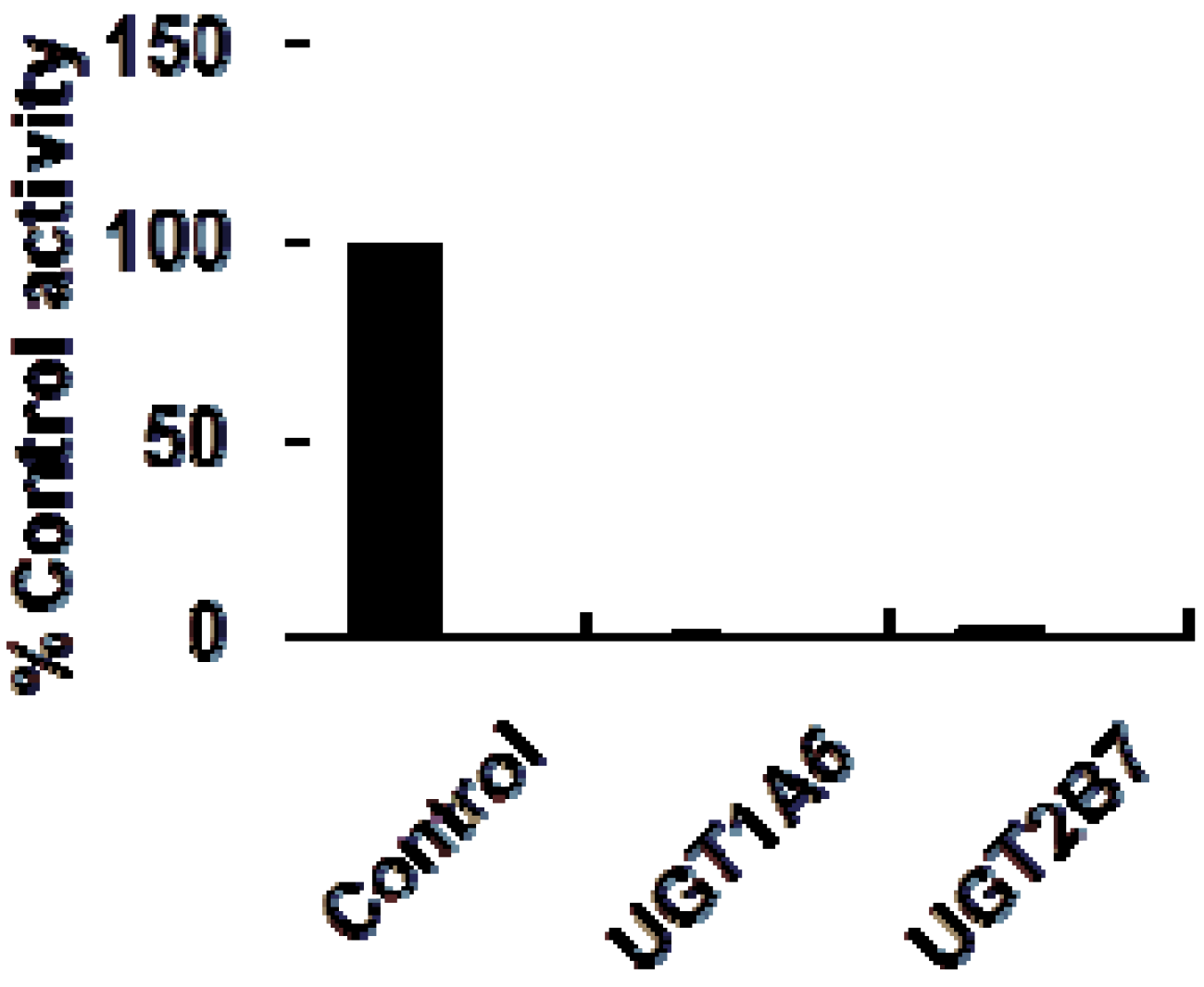
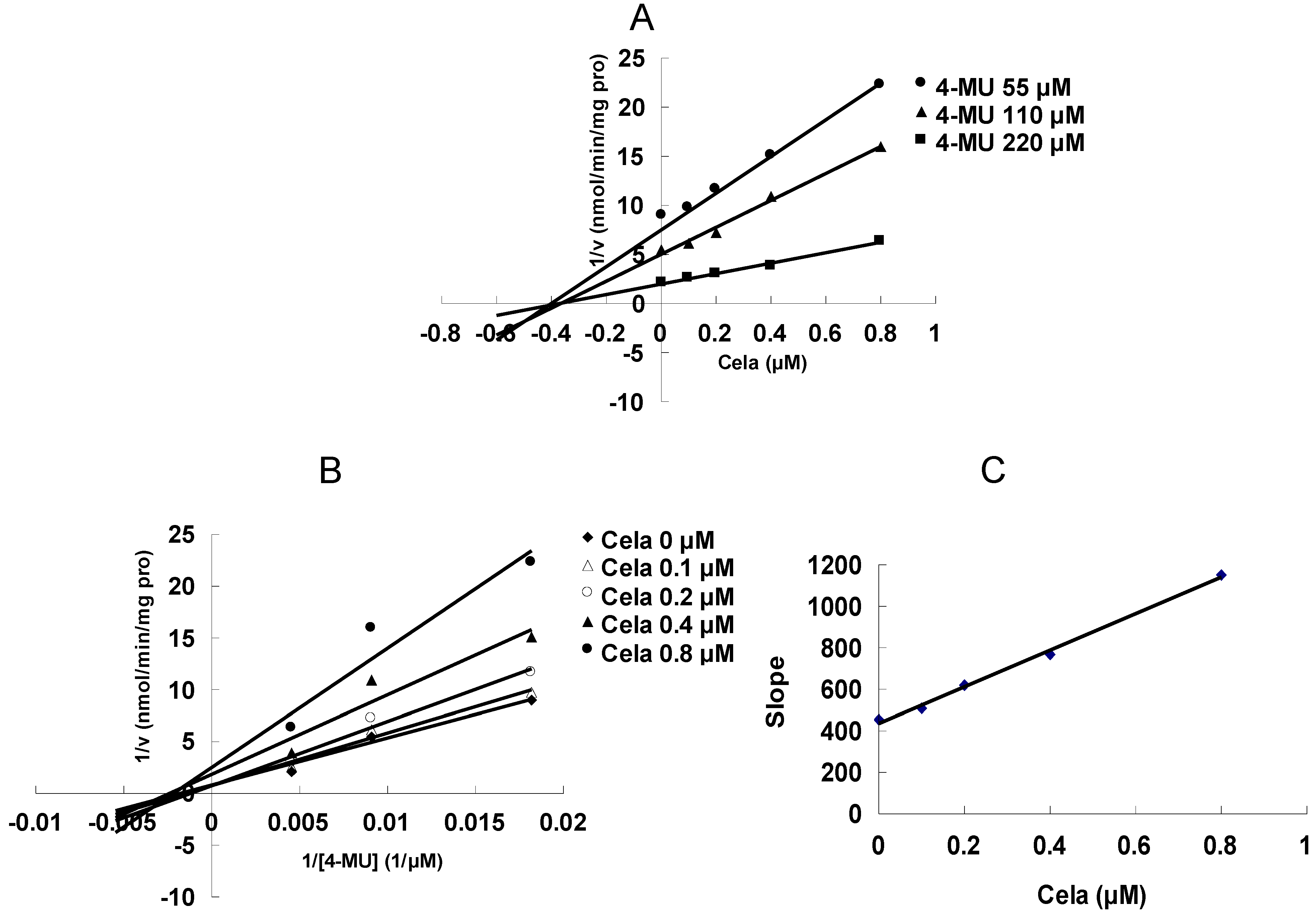
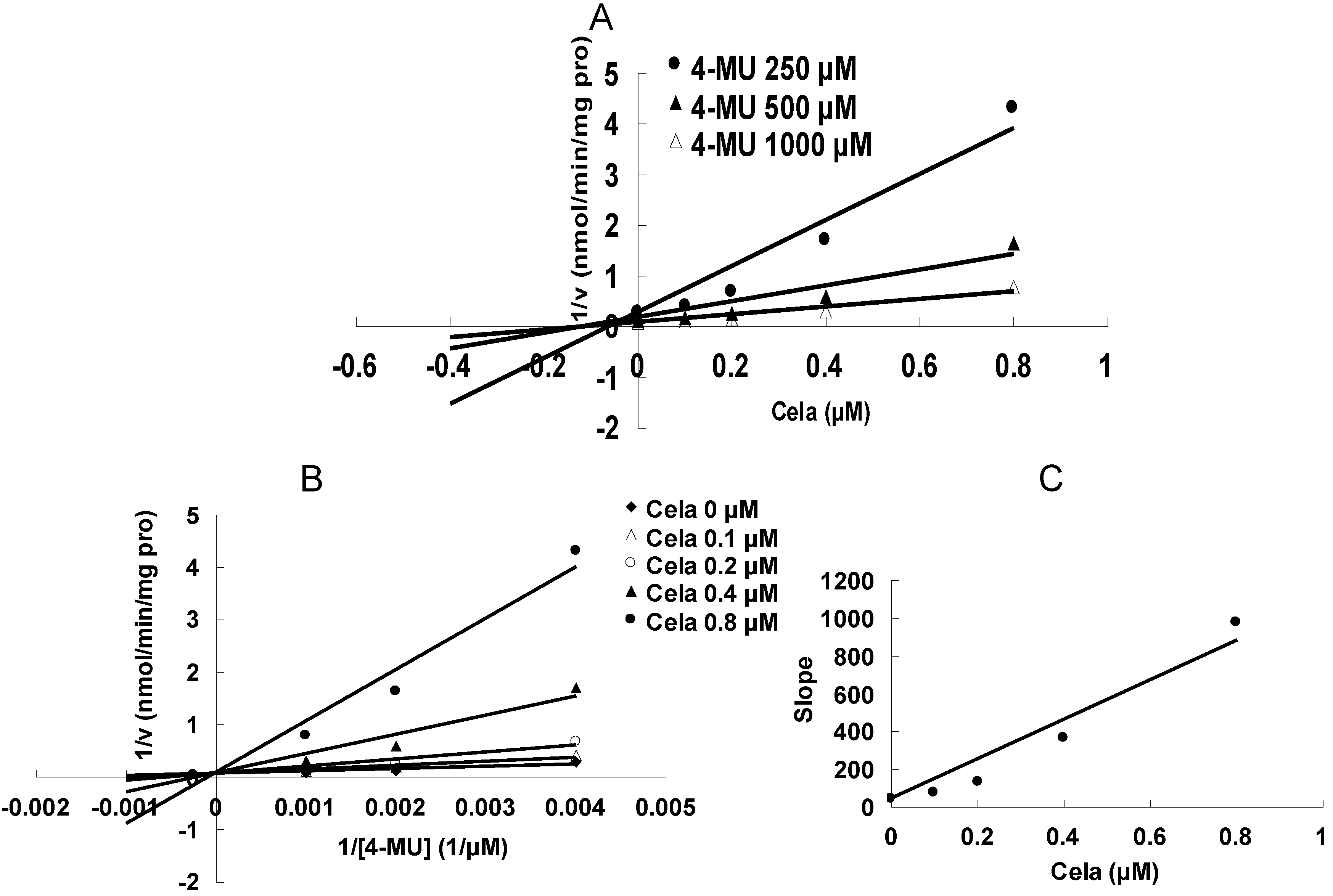
2. Results and Discussion



3. Experimental
3.1. Chemicals and Reagents
3.2. Enzyme Inhibition Experiment
4. Conclusions
- Sample Availability: Sample of the compound celastrol is available from the authors.
Reference and Notes
- Setty, A.R.; Sigal, L.H. Herbal medications commonly used in the practice of rheumatology: Mechanisms of action, efficacy, and side effects. Semin. Arthritis Rheum. 2005, 34, 773–784. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, X.W.; Cai, T.Y.; Cao, J.; Tu, C.X.; Lu, W.; He, Q.J.; Yang, B. Celastrol acts as a potent antimetastatic agent targeting β1 integrin and inhibiting cell-extracellular matrix adhesion, in part via the p38 mitogen-activated protein kinase pathway. J. Pharmacol. Exp. Ther. 2010, 334, 489–499. [Google Scholar] [CrossRef]
- Tao, X.; Younger, J.; Fan, F.Z.; Wang, B.; Lipsky, P.E. Benefit of an extract of Tripterygium wilfordii Hook F in patients with rheumatoid arthritis: A double-blind, placebocontrolled study. Arthritis Rheum. 2002, 46, 735–743. [Google Scholar] [CrossRef]
- Pinna, G.F.; Fiorucci, M.; Reimund, J.M.; Taquet, N.; Arondel, Y.; Muller, C.D. Celastrol inhibits pro-inflammatory cytokine secretion in Crohn’s disease biopsies. Biochem. Biophys. Res. Commun. 2004, 322, 778–786. [Google Scholar] [CrossRef]
- Cleren, C.; Calingasan, N.Y.; Chen, J.; Beal, M.F. Celastrol protects against MPTP- and 3-nitropropionic acidinduced neurotoxicity. J. Neurochem. 2005, 94, 995–1004. [Google Scholar] [CrossRef]
- Allison, A.C.; Cacabelos, R.; Lombardi, V.R.; Alvarez, X.A.; Vigo, C. Celastrol, a potent antioxidant and antiinflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 1341–1357. [Google Scholar] [CrossRef]
- Huang, Y.L.; Zhou, Y.X.; Fan, K.S.; Zhou, D. Celastrol inhibits the growth of human glioma xenografts in nude mice through suppressing VEGFR expression. Cancer Lett. 2008, 264, 101–106. [Google Scholar] [CrossRef]
- Dai, Y.; DeSano, J.; Tang, W.H.; Meng, X.J.; Meng, Y.; Burstein, E.; Lawrence, T.S.; Xu, L.A. Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One 2010, 5, e14153. [Google Scholar]
- Zhou, S.; Koh, H.L.; Gao, Y.; Gong, Z.Y.; Lee, E.J. Bioactivation of herbal constituents: Simple alerts in the complex system. Life Sci. 2004, 74, 935–968. [Google Scholar] [CrossRef]
- Lin, J.H.; Lu, A.Y.H. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–449. [Google Scholar]
- Hao, M.; Zhao, Y.; Chen, P.; Huang, H.; Liu, H.; Jiang, H.; Zhang, R.; Wang, H. Structure-activity relationship and substrate-dependent phenomena in effects of ginsenosides on activities of drug-metabolizing P450 enzymes. PLoS One 2008, 3, e2697. [Google Scholar]
- Fang, Z.Z.; Zhang, Y.Y.; Ge, G.B.; Liang, S.C.; Sun, D.X.; Zhu, L.L.; Dong, P.P.; Cao, Y.F.; Yang, L. Identification of cytochrome P450 (CYP) isoforms involved in metabolism of corynoline and assessment of its herb-drug interaction. Phytother. Res. 2011, 25, 256–263. [Google Scholar]
- Sun, D.X.; Fang, Z.Z.; Zhang, Y.Y.; Cao, Y.F.; Yang, L.; Yin, J. Inhibitory effects of curcumenol on human liver cytochrome P450 enzymes. Phytother. Res. 2010, 24, 1213–1216. [Google Scholar]
- Quintieri, L.; Palatini, P.; Moro, S.; Floreani, M. Inhibition of cytochrome P450 2C8-mediated drug metabolism by the flavonoid diosmetin. Drug Metab. Pharmacokinet. 2011, 26, 559–568. [Google Scholar]
- Miners, J.O.; Mackenzie, P.I. Drug glucuronidation in human. Pharmacol. Ther. 1991, 51, 347–369. [Google Scholar] [CrossRef]
- Kiang, T.K.; Ensom, M.H.; Chang, T.K. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol. Ther. 2005, 106, 97–132. [Google Scholar] [CrossRef]
- Ritter, J.K. Roles of glucuronidation and UDP—glucuronosyltransferasesin xenobiotic bioactivation reactions. Chem. Biol. Interact. 2000, 129, 171–193. [Google Scholar] [CrossRef]
- Tukey, R.H.; Strassburg, C.P. Human UDP-glucuronosyltransferases:metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef]
- Dong, R.H.; Fang, Z.Z.; Zhu, L.L.; Liang, S.C.; Ge, G.B.; Yang, L.; Liu, Z.Y. Investigation of UDP-glucuronosyltransferases (UGT) inhibitory properties of carvacrol. Phytother. Res. 2012, 26, 86–90. [Google Scholar] [CrossRef]
- Tsoutsikos, P.; Miners, J.O.; Stapleton, A.; Thomas, A.; Sallustio, B.C.; Knights, K.M. Evidence that unsaturated fatty acids are potent inhibitors of renal UDP-glucuronosyltransferases (UGT): kinetic studies using human kidney cortical microsomes and recombinant UGT1A9 and UGT2B7. Biochem. Pharmacol. 2010, 65, 919–921. [Google Scholar]
- Nishimura, Y.; Maeda, S.; Ikushiro, S.; Mackenzie, P.I.; Ishii, Y.; Yamada, H. Inhibitory effects of adenine nucleotides and related substances on UDP-glucuronosyltransferase: structure-effect relationships and evidence for an allosteric mechanism. Biochim. Biophys. Acta 2007, 1770, 1557–1566. [Google Scholar] [CrossRef]
- Bock, K.W.; Wiltfang, J.; Blume, R.; Ullrich, D.; Bircher, J. Paracetamol as a test drug to determine glucuronide formation in man. Effects of inducers and of smoking. Eur. J. Clin. Pharmacol. 1987, 31, 677–683. [Google Scholar] [CrossRef]
- Krishnaswamy, S.; Duan, S.X.; von Moltke, L.L.; Greenblatt, D.J.; Sudmeier, J.L.; Bachovchin, W.W.; Court, M.H. Serotonin (5-hydroxytryptamine) glucuronidation in vitro: Assay development, human liver microsome activities and species differences. Xenobiotica 2003, 33, 169–180. [Google Scholar] [CrossRef]
- Krishnaswamy, S.; Duan, S.X.; von Moltke, L.L.; Court, M.H. Validation of serotonin (5-hydroxtryptamine) as an in vitro substrate probe for human UDP-glucuronosyltransferase (UGT) 1A6. Drug. Metab. Dispos. 2003, 31, 133–139. [Google Scholar] [CrossRef]
- Uchaipichat, V.; Mackenzie, P.I.; Guo, X.H.; Gardner-Stephen, D.; Galetin, A.; Houston, J.B.; Miners, J.O. Human UDP- glucuronosyltransferases: Isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug. Metab. Dispos. 2004, 32, 413–423. [Google Scholar] [CrossRef]
- Du, W.; Huang, H. Correlation analysis of secondary metabolites and environmental factors in Tripterygium wilfordii. Acta Bot. Sin. 2008, 25, 707–713. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Y.-S.; Tu, Y.-Y.; Gao, X.-C.; Yuan, J.; Li, G.; Wang, L.; Deng, J.-P.; Wang, Q.; Ma, R.-M. Strong Inhibition of Celastrol Towards UDP-Glucuronosyl Transferase (UGT) 1A6 and 2B7 Indicating Potential Risk of UGT-Based Herb-Drug Interaction. Molecules 2012, 17, 6832-6839. https://doi.org/10.3390/molecules17066832
Zhang Y-S, Tu Y-Y, Gao X-C, Yuan J, Li G, Wang L, Deng J-P, Wang Q, Ma R-M. Strong Inhibition of Celastrol Towards UDP-Glucuronosyl Transferase (UGT) 1A6 and 2B7 Indicating Potential Risk of UGT-Based Herb-Drug Interaction. Molecules. 2012; 17(6):6832-6839. https://doi.org/10.3390/molecules17066832
Chicago/Turabian StyleZhang, Yong-Sheng, Yan-Yang Tu, Xing-Chun Gao, Jun Yuan, Gang Li, Liang Wang, Jian-Ping Deng, Qi Wang, and Ru-Meng Ma. 2012. "Strong Inhibition of Celastrol Towards UDP-Glucuronosyl Transferase (UGT) 1A6 and 2B7 Indicating Potential Risk of UGT-Based Herb-Drug Interaction" Molecules 17, no. 6: 6832-6839. https://doi.org/10.3390/molecules17066832