Design and Synthesis of New Chacones Substituted with Azide/Triazole Groups and Analysis of Their Cytotoxicity Towards HeLa Cells

Abstract
:1. Introduction
2. Results and Discussion



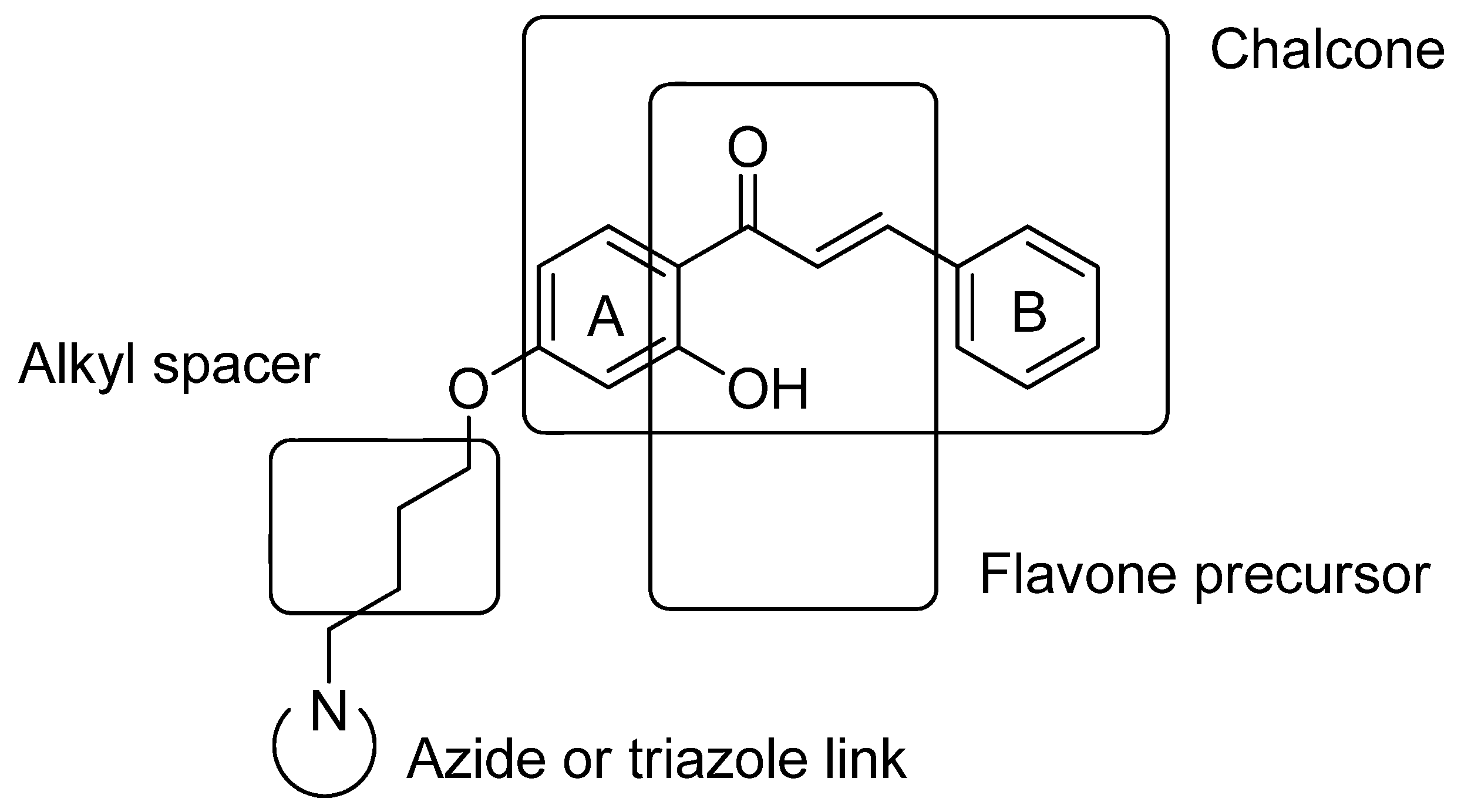{kind=link}
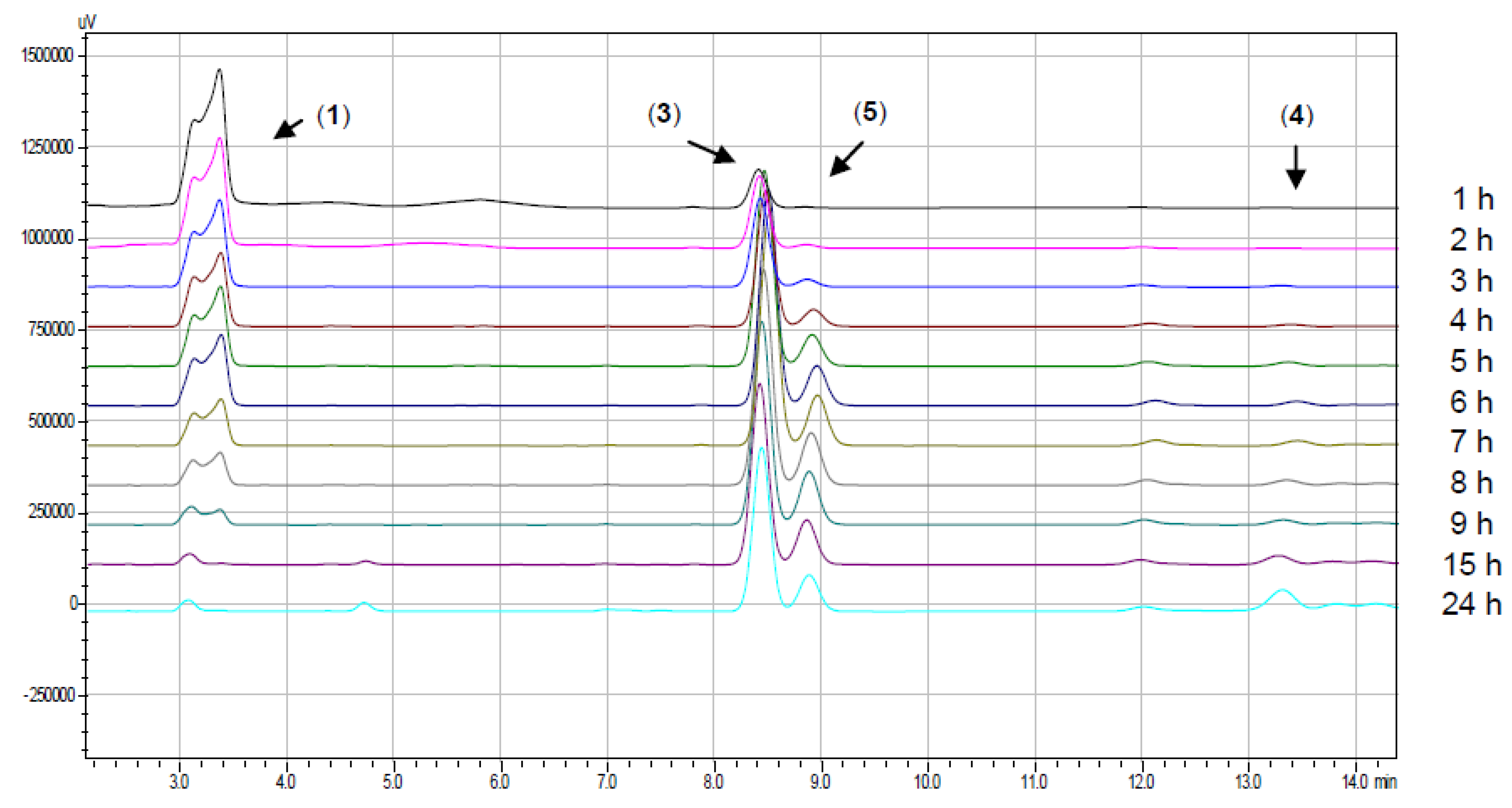{kind=link}
{kind=link}
{kind=link}
| Entry | Condition | 3 | 4 | 5 |
|---|---|---|---|---|
| 1 | DMF/r.t./2 h | 21.8 | 74.4 | 3.8 |
| 2 | DMSO/r.t./2 h | 15.8 | 82.0 | 2.2 |
| 3 | ACN/r.t./48 h | 71.1 | 3.2 | 10.0 |
| 4 | ACN/40 °C/15 h | 71.6 | 2.1 | 13.1 |

| Compound | R1 | Yield (%) | IC50 (µmol L−1) |
|---|---|---|---|
| 7a | H | 50 | >100 |
| 7b | 4-OMe | 52 | >100 |
| 7c | 4-OEt | 45 | >100 |
| 7d | 4-Cl | 81 | >100 |
| 7e | 2,3-Cl | 68 | >100 |
| Compound | R1 | R2 | Yield (%) | IC50 (µmol L−1) |
|---|---|---|---|---|
| 8a | H | CH2OH | 88 | >100 |
| 8b | 4-OMe | CH2OH | 81 | >100 |
| 8c | 4-OEt | CH2OH | 89 | 13.03 ± 2.51 |
| 8d | 4-Cl | CH2OH | 87 | >100 |
| 8e | 2,3-Cl | CH2OH | 91 | >100 |
| 8f | H | Ph | 96 | >100 |
| 8g | 4-OMe | Ph | 92 | >100 |
| 8h | 4-OEt | Ph | 83 | >100 |
| 8i | 4-Cl | Ph | 81 | >100 |
| 8j | 2,3-Cl | Ph | 89 | >100 |
| Cisplatin | - | - | - | 7.37 ± 2.42 |
3. Experimental
3.1. Chemistry
3.2. General Procedure of O-Alkylation of 1-(2,4-Dihydroxyphenyl)ethan-1-one (resacetophenone, 1) [24,27]
3.3. Preparation of 1-[4-(4-Azidobutoxy)-2-hydroxyphenyl]ethan-1-one (6)
3.4. General Procedure for the Synthesis and Purification of Chalcones
3.5. General Cycloaddition Procedure
3.6. Biological Assays
Cytotoxic Assay
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Instituto do Câncer (INCA) home page. Incidência de Câncer no Brasil Estimativa 2012. Available online: http://www.inca.gov.br/estimativa/2012/ (accessed on 9 August 2012).
- Frigato, S.; Hoga, L.A.K. Assistência à mulher com câncer de colo uterino: O papel da enfermagem. Rev. Bras. Cancerol. 2003, 49, 209–214. [Google Scholar]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef]
- Lai, C.H.; Huang, K.G.; Hong, J.H.; Lee, C.L.; Chou, H.H.; Chang, T.C.; Hsueh, S.; Huang, H.J.; Ng, K.K.; Tsai, C.S. Randomized trial of surgical staging (extraperitoneal or laparoscopic) versus clinical staging in locally advanced cervical cancer. Gynecol. Oncol. 2003, 89, 160–167. [Google Scholar]
- Zdzisława, N. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007, 42, 125–137. [Google Scholar] [CrossRef]
- Gutteridge, C.E.; Thota, D.S.; Curtis, S.M.; Kozar, M.P.; Li, Q.G.; Xie, L.S.; Zhang, J.; Melendez, V.; Asher, C.; Luong, T.T.; Gerena, L.; Nichols, D.A.; Montip, G. In vitro biotransformation, in vivo efficacy and pharmacokinetics of antimalarial chalcones. Pharmacology 2011, 87, 96–104. [Google Scholar]
- Kumar, R.; Mohanakrishnan, D.; Sharma, A.; Kaushik, N.K.; Kalia, K.; Sinha, A.K.; Sahal, D. Reinvestigation of structure-activity relationship of methoxylated chalcones as antimalarials: Synthesis and evaluation of 2,4,5-trimethoxy substituted patterns as lead candidates derived from abundantly available natural beta-asarone. Eur. J. Med. Chem. 2010, 45, 5292–5301. [Google Scholar]
- Bello, M.L.; Chiaradia, L.D.; Dias, L.R.S.; Pacheco, L.K.; Stumpf, T.R.; Mascarello, A.; Steindel, M.; Yunes, R.A.; Castro, H.C.; Nunes, R.J.; Rodrigues, C.R. Trimethoxy-chalcone derivatives inhibit growth of Leishmania braziliensis: Synthesis, biological evaluation, molecular modeling and structure-activity relationship (SAR). Bioorg. Med. Chem. 2011, 19, 5046–5052. [Google Scholar]
- Scotti, L.; Ferreira, E.I.; da Silva, M.S.; Scotti, M.T. Chemometric studies on natural products as potential inhibitors of the NADH oxidase from trypanosoma cruzi using the volsurf approach. Molecules 2010, 15, 7363–7377. [Google Scholar] [CrossRef]
- Cabrera, M.; Simoens, M.; Falchi, G.; Lavaggi, M.L.; Piro, O.E.; Castellano, E.E.; Vidal, A.; Azqueta, A.; Monge, A.; de Cerain, A.L.; et al. Synthetic chalcones, flavanones, and flavones as antitumoral agents: Biological evaluation and structure-activity relationships. Bioorg. Med. Chem. 2007, 15, 3356–3367. [Google Scholar] [CrossRef]
- Xiao, X.Y.; Hao, M.A.; Yang, X.Y.; Ba, Q.A.; Li, M.A.; Ni, S.J.; Wang, L.S.; Du, X.A. Licochalcone A inhibits growth of gastric cancer cells by arresting cell cycle progression and inducing apoptosis. Cancer Lett. 2011, 302, 69–75. [Google Scholar] [CrossRef]
- Kim, J.K.; Shin, E.K.; Park, J.H.; Kim, Y.H.; Park, J.H.Y. Antitumor and antimetastatic effects of licochalcone A in mouse models. J. Mol. Med. 2010, 88, 829–838. [Google Scholar] [CrossRef]
- Szliszka, E.; Czuba, Z.P.; Mazur, B.; Sedek, L.; Paradysz, A.; Krol, W. Chalcones enhance TRAIL-induced apoptosis in prostate cancer cells. Int. J. Mol. Sci. 2010, 11, 1–13. [Google Scholar]
- Mishra, N.; Arora, P.; Kumar, B.; Mishra, L.C.; Bhattacharya, A.; Awasthi, S.K.; Bhasin, V.K. Synthesis of novel substituted 1,3-diaryl propenone derivatives and their antimalarial activity in vitro. Eur. J. Med. Chem. 2008, 43, 1530–1535. [Google Scholar]
- Guantai, E.M.; Ncokazi, K.; Egan, T.J.; Gut, J.; Rosenthal, P.J.; Smith, P.J.; Chibale, K. Design, synthesis and in vitro antimalarial evaluation of triazole-linked chalcone and dienone hybrid compounds. Bioorg. Med. Chem. 2010, 18, 8243–8256. [Google Scholar]
- Awasthi, S.K.; Mishra, N.; Kumar, B.; Sharma, M.; Bhattacharya, A.; Mishra, L.C.; Bhasin, V.K. Potent antimalarial activity of newly synthesized substituted chalcone analogs in vitro. Med. Chem. Res. 2009, 18, 407–420. [Google Scholar] [CrossRef]
- Sharma, N.; Mohanakrishnan, D.; Shard, A.; Sharma, A.; Sinha, A.K.; Sahal, D. Stilbene-Chalcone hybrids: Design, synthesis, and evaluation as a new class of antimalarial scaffolds that trigger cell death through stage specific apoptosis. J. Med. Chem. 2012, 55, 297–311. [Google Scholar]
- Kamal, A.; Prabhakar, S.; Ramaiah, M.J.; Reddy, P.V.; Reddy, C.R.; Mallareddy, A.; Shankaraiah, N.; Reddy, T.L.N.; Pushpavalli, S.N.C.V.L.; Pal-Bhadra, M. Synthesis and anticancer activity of chalcone-pyrrolobenzodiazepine conjugates linked via 1,2,3-triazole ring side-armed with alkane spacers. Eur. J. Med. Chem. 2011, 46, 3820–3831. [Google Scholar]
- Aufort, M.; Herscovici, J.; Bouhours, P.; Moreau, N.; Girard, C. Synthesis and antibiotic activity of a small molecules library of 1,2,3-triazole derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 1195–1198. [Google Scholar]
- Sangshetti, J.N.; Lokwani, D.K.; Sarkate, A.P.; Shinde, D.B. Synthesis, Antifungal Activity, and Docking Study of Some New 1,2,4-triazole Analogs. Chem. Biol. Drug Des. 2011, 78, 800–809. [Google Scholar] [CrossRef]
- Melo, J.O.F.; Donnici, C.L.; Augusti, R.; Lopes, M.T.P.; Mikhailovskii, A.G. Synthesis of novel andhardly-obtainable 1,2,3-triazoles with potential antitumoral activity by a diazo-transfer reaction from 5,7-dinitro-3-diazo-1,3-dihydro-2H-indol-2-one to enaminones. Heterocycl. Commun. 2003, 9, 235–238. [Google Scholar]
- Deshpande, S.J.; Leger, P.R.; Sieck, S.R. Microwave synthesis of alpha-cyano chalcones. Tetrahedron Lett. 2012, 53, 1772–1775. [Google Scholar]
- Alvarez, S.G.; Alvarez, M.T. A practical procedure for the synthesis of alkyl azides at ambient temperature in dimethyl sulfoxide in high purity and yield. Synthesis 1997, 4, 413–414. [Google Scholar] [CrossRef]
- Aponte, J.C.; Castillo, D.; Estevez, Y.; Gonzalez, G.; Arevalo, J.; Hammond, G.B.; Sauvain, M. In vitro and in vivo anti-Leishmania activity of polysubstituted synthetic chalcones. Bioorg. Med. Chem. Lett. 2010, 20, 100–103. [Google Scholar]
- Carroll, J.B.; Jordan, B.J.; Xu, H.; Erdogan, B.; Lee, L.; Cheng, L.; Tiernan, C.; Cooke, G.; Rotello, V.M. Model systems for flavoenzyme activity: Site-isolated redox behavior in flavin-functionalized random polystyrene copolymers. Org. Lett. 2005, 7, 2551–2554. [Google Scholar]
- Still, W.C.; Kahn, M.; Mitra, A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar]
- Pisco, L.; Kordian, M.; Peseke, K.; Feist, H.; Michalik, D.; Estrada, E.; Carvalho, J.; Hamilton, G.; Rando, D.; Quincoces, J. Synthesis ofcompounds with antiproliferative activity asanalogues ofprenylated natural products existing inBrazilian propolis. Eur. J. Med. Chem. 2006, 41, 401–407. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Da Silva, G.D.; Da Silva, M.G.; Souza, E.M.P.V.E.; Barison, A.; Simões, S.C.; Varotti, F.P.; Barbosa, L.A.; Viana, G.H.R.; Villar, J.A.F.P. Design and Synthesis of New Chacones Substituted with Azide/Triazole Groups and Analysis of Their Cytotoxicity Towards HeLa Cells. Molecules 2012, 17, 10331-10343. https://doi.org/10.3390/molecules170910331
Da Silva GD, Da Silva MG, Souza EMPVE, Barison A, Simões SC, Varotti FP, Barbosa LA, Viana GHR, Villar JAFP. Design and Synthesis of New Chacones Substituted with Azide/Triazole Groups and Analysis of Their Cytotoxicity Towards HeLa Cells. Molecules. 2012; 17(9):10331-10343. https://doi.org/10.3390/molecules170910331
Chicago/Turabian StyleDa Silva, Graziele D., Marina G. Da Silva, Estrela M. P. V. E. Souza, Andersson Barison, Sarah C. Simões, Fernando P. Varotti, Leandro A. Barbosa, Gustavo H. R. Viana, and José A. F. P. Villar. 2012. "Design and Synthesis of New Chacones Substituted with Azide/Triazole Groups and Analysis of Their Cytotoxicity Towards HeLa Cells" Molecules 17, no. 9: 10331-10343. https://doi.org/10.3390/molecules170910331