3.2. Synthesis
3.2.1. Obtention of the Aminochloroiodopyridines
2,3-Dichloro-4-iodopyridine [
8,
15]. To a solution of
n-butyllithium (27.6 mL, 69 mmol, 2.5 M in hexanes) in dry Et
2O (153 mL) cooled at −78 °C, was added dropwise 2,2,6,6-tetramethylpiperidine (11.64 mL, 69 mmol). The mixture was stirred at −78 °C for 10 min and a solution of 2,3-dichloropyridine (10 g, 67.57 mmol) in dry THF (75 mL) was added dropwise. The resulting mixture was stirred at −78 °C for 30 min and a solution of I
2 (25.38 g, 100 mmol) in dry THF (75 mL) was added. The reaction was cooled to room temperature overnight, quenched with a saturated aqueous solution of Na
2S
2O
3 and extracted with EtOAc (3 × 50 mL). The combined organic layer was washed with a saturated aqueous solution of NaHCO
3, dried with anhydrous MgSO
4 and evaporated
in vacuo. The residue was purified on column chromatography [silica gel, cyclohexane/EtOAc (7:3)] to give a pale cream solid. M.p. = 110 °C (Ref. [
8] 113 °C). 67% yield.
1H-NMR (CDCl
3) δ 7.89 (d, 1H,
J = 5.1 Hz, H-6), 7.73 (d, 1H,
J = 5.1 Hz, H-5).
13C-NMR (CDCl
3) δ 148.5, 146.6, 135.4, 134.0, 111.2.
2-Amino-3-chloro-4-iodopyridine [
8]. A mixture of 2,3-dichloro-4-iodopyridine (4 g, 14.71 mmol) in NH
4OH 28% (75 mL) was heated in a Parr bomb at 129 °C for 16 h. The reaction was cooled to room temperature and CH
2Cl
2 (100 mL) was added. The mixture was extracted with CH
2Cl
2 (3 × 100 mL), dried with anhydrous MgSO
4 and evaporated
in vacuo. The residue was purified by flash chromatography [silica gel, cyclohexane/EtOAc (7:3)] and a white solid was obtained. M.p. = 110 °C. 38% yield.
1H-NMR (CDCl
3) δ 7.54 (d, 1H,
J = 5.3 Hz, H-6), 7.08 (d, 1H,
J = 5.3 Hz, H-5), 5.32 (s, 2H, NH
2).
13C-NMR (CDCl
3) δ 154.6, 145.6, 124.7, 119.4, 109.8.
t-Butyl-(4-chloropyridin-2-yl)carbamate [
16,
17]. To a solution of 2-amino-4-chloropyridine (3 g, 23.34 mmol) in
t-BuOH (43 mL), di-
t-butyl dicarbonate (5.6 g, 25.67 mmol) was added. The mixture was stirred at 30 °C overnight. A solid appeared and was filtered off, washed with
n-hexane and diethyl ether to give a white solid. M.p. = 150 °C. 77% yield.
1H-NMR (DMSO-
d6) δ 10.14 (bs, 1H, NH), 8.22 (d, 1H,
J = 5.4 Hz, H-6), 7.88 (d, 1H,
J = 1.9 Hz, H-3), 7.14 (dd, 1H,
J = 5.4–1.9 Hz, H-5), 1.47 (s, 9H, CH
3).
13C-NMR (DMSO-
d6) δ 153.69, 152.66, 149.34, 143.84, 118.29, 111.65, 80.15, 27.95.
t-Butyl-(4-chloro-3-iodopyridin-2-yl)carbamate [
17]. To a solution of
t-butyl-(4-chloropyridin-2-yl)carbamate (9 g, 39.5 mmol) in anhydrous THF (250 mL), TMEDA (14.4 mL) was added under nitrogen atmosphere and cooled to −70 °C. To the mixture, 2.5 M in hexane
n-BuLi (39.6 mL, 98 mmol) was added dropwise over a period of 30 min. The mixture was stirred at −70 °C for 1 h and then treated dropwise with a solution of I
2 (50 g, 198 mmol) in anhydrous THF (30 mL) at −70 °C. After the addition was complete, the reaction was stirred at −70 °C for 30 min and then allowed to warm to room temperature. The mixture was treated with an aqueous saturated solution of Na
2S
2O
3 and stirred for 30 min. The solution was extracted with EtOAc (3 × 200 mL). The combined organic layer was washed with brine, dried with anhydrous MgSO
4 and evaporated under reduce pressure. The product was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)] to give a white solid. M.p. = 183 °C. 75% yield.
1H-NMR (DMSO-
d6) δ 9.48 (s, 1H, NH), 8.30 (d, 1H,
J = 5.1 Hz, H-6), 7.47 (d, 1H,
J = 5.1 Hz, H-5), 1.45 (s, 9H, CH
3).
13C-NMR (DMSO-
d6) δ 154.9, 152.6, 149.0, 148.4, 122.0, 99.2, 79.4, 28.1.
2-Amino-4-chloro-3-iodopyridine [
7,
8,
17]. A suspension of
t-butyl-(4-chloro-3-iodopyridin-2-yl)carbamate (6 g, 23.5 mmol) in 48% hydrobromic acid (12 mL) was heated at 100 °C for 10 min to give a clear solution. The mixture was cooled, treated with ice and made basic with 10 M NaOH solution. The precipitated product was filtered off, washed with H
2O to give a cream solid. M.p. = 111 °C. 97% yield.
1H-NMR (DMSO-
d6) δ 7.84 (d, 1H,
J = 5.2 Hz, H-6), 6.72 (d, 1H,
J = 5.2 Hz, H-5), 6.44 (bs, 2H, NH
2).
13C-NMR (DMSO-
d6) δ 161.0, 148.4, 147.7, 113.0, 81.4.
3.2.2. General Procedure for Cyclization
8-Chloro-7-iodo-2-phenylimidazo[1,2-a]pyridine (1a). Method A. To a solution of 2-amino-3-chloro-4-iodopyridine (1 g, 3.94 mmol) in EtOH (10 mL), was added bromoacetophenone (1.56 g, 7.87 mmol). The mixture was stirred at 65 °C overnight. The solution was then evaporated to dryness in vacuo. The residue was dissolved in CH2Cl2 (50 mL) and the resulting solution made basic by the addition of a saturated aqueous solution of Na2CO3. The solution was extracted with CH2Cl2 (3 × 100 mL), the combined organic layers were dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude product was purified by flash chromatography [silica gel, cyclohexane/EtOAc (7:3)]. M.p. = 101 °C. 97% yield. 1H-NMR (CDCl3) δ 7.96 (dd, 2H, J = 8.2–1.3 Hz, Ph-2,6), 7.88 (s, 1H, H-3), 7.80 (d, 1H, J = 7.0 Hz, H-5), 7.44 (m, 2H, Ph-3,5), 7.35 (m, 1H, Ph-4), 7.12 (d, 1H, J = 7.0 Hz, H-6). 13C-NMR (CDCl3) δ 146.8, 143.0, 132.8, 128.7, 128.5, 126.4, 123.9, 121.7, 110.0, 92.1(1C not found).
Ethyl 8-chloro-7-iodoimidazo[1,2-a]pyridine-2-carboxylate (1b). Method B. To a solution of 2-amino-3-chloro-4-iodopyridine (1 g, 3,94 mmol) in DME (18 mL), was added ethyl bromopyruvate (1.16 g, 5.91 mmol). The mixture was stirred at room temperature overnight. The solution was evaporated in vacuo to dryness and EtOH (18 mL) was added. The mixture was stirred at 78 °C for 2 h. After cooling to room temperature, the solution was evaporated to dryness under vacuum. The residue was dissolved in CH2Cl2 (50 mL) and the resulting solution made basic by the addition of a saturated solution of Na2CO3. The solution was extracted with CH2Cl2 (3 × 100 mL), the combined organic layers were dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude product was washed with diethyl ether and n-hexane to give a pale cream solid. M.p. = 249 °C. 95% yield. 1H-NMR (CDCl3) δ 8.66 (s, 1H, H-3), 8.33 (d, 1H, J = 6.9 Hz, H-5), 7.41 (d, 1H, J = 6.9 Hz, H-6), 4.32 (q, 2H, J = 7.2 Hz, CH2), 1.32 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (CDCl3) δ 162.6, 142.3, 136.1, 127.1, 123.0, 120.8, 97.4, 61.0, 14.7 (1C not found).
8-Chloro-7-iodo-2-methylimidazo[1,2-a]pyridine (1c). Method A. 2-Amino-3-chloro-4-iodopyridine (1 g, 3.94 mmol) and chloroacetone (1.46 g, 15.75 mmol) were used. M.p. = 136 °C. Quantitative yield. 1H-NMR (CDCl3) δ 7.72 (d, 1H, J = 6.9 Hz, H-5), 7.38 (s, 1H, H-3), 7.07 (d, 1H, J = 6.9 Hz, H-6), 2.48 (s, 3H, CH3). 13C-NMR (CDCl3) δ 144.6, 131.9, 127.5, 123.6, 121.1, 111.6, 91.5, 14.4.
7-Chloro-8-iodo-2-phenylimidazo[1,2-a]pyridine (1d). Method A. 2-Amino-4-chloro-3-iodopyridine (1 g, 3.94 mmol) and bromoacetophenone (1.56 g, 7.87 mmol) were used. M.p. = 197 °C. 72% yield. 1H-NMR (CDCl3) δ 8.03–7.94 (m, 4H, Ph-2,6, H-3, H-5), 7.44 (m, 2H, Ph-3,5), 7.36 (m, 1H, Ph-4), 6.84 (d, 1H, J = 7.0 Hz, H-6). 13C-NMR (DMSO-d6) δ 145.5, 145.2, 135.1, 133.1, 128.8, 128.1, 127.2, 125.7, 113.1, 111.5, 88.6.
Ethyl 7-chloro-8-iodoimidazo[1,2-a]pyridine-2-carboxylate (1e). Method B. 2-Amino-4-chloro-3-iodopyridine (1 g, 3.94 mmol) was used as starting material. M.p. = 208 °C. 71% yield. 1H-NMR (DMSO-d6) δ 8.71 (s, 1H, H-3), 8.55 (d, 1H, J = 7.3 Hz, H-5), 7.16 (d, 1H, J = 7.3 Hz, H-6), 4.33 (q, 2H, J = 7.0 Hz, CH2), 1.32 (t, 3H, J = 7.0 Hz, CH3). 13C-NMR (DMSO-d6) δ 162.2, 145.4, 137.0, 136.2, 127.9, 120.4, 114.5, 89.9, 60.5, 14.3.
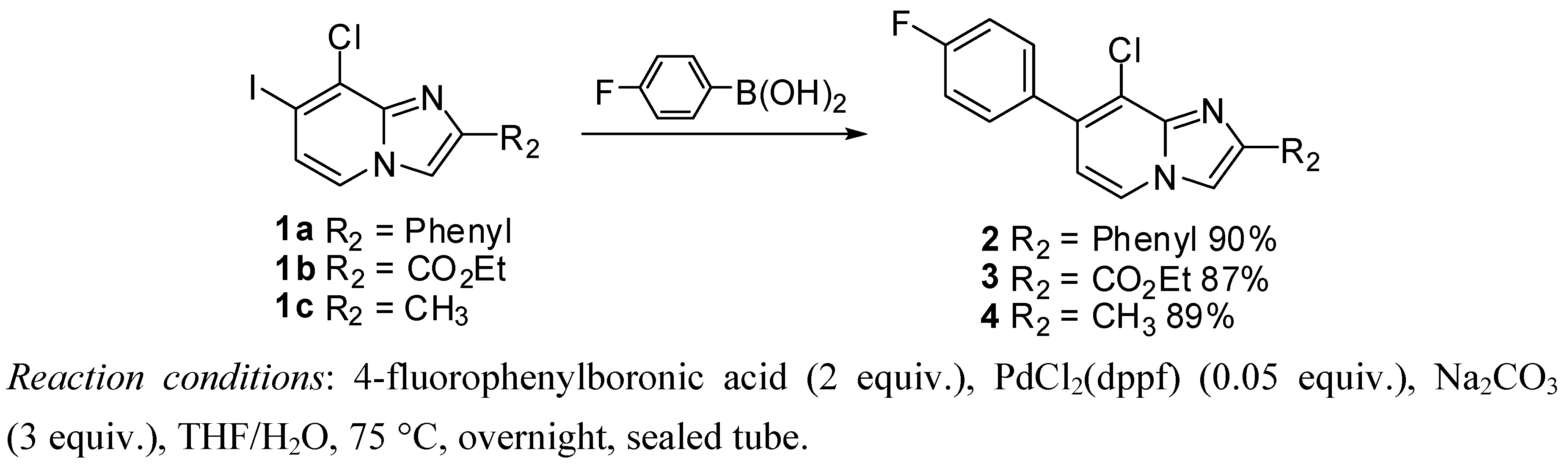
3.2.3. Suzuki Cross-Coupling on Compounds 1a–c
8-Chloro-7-(4-fluorophenyl)-2-phenylimidazo[1,2-a]pyridine (2). Method C. To a solution of 1a (500 mg, 1.41 mmol) in a mixture of THF (2.7 mL) and water (0.3 mL) in a sealed tube, were added 4-fluorophenylboronic acid (296 mg, 2.11 mmol), Na2CO3 (449 mg, 4.24 mmol) and PdCl2(dppf) (58 mg, 0.07 mmol, 5 mol%). The mixture was stirred at 75 °C for 1 h 30 min, 4-fluorophenylboronic acid (99 mg, 0.5 equiv.) and a few amount of PdCl2(dppf) were added. After overnight stirring at 75 °C and cooling to room temperature, CH2Cl2 (50 mL) and water (50 mL) were added and the solution was extracted with CH2Cl2 (3 × 50 mL). The combined organic layer was dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residues were purified by column chromatography (alumina, CH2Cl2). M.p. = 211 °C. 90% yield. 1H-NMR (CDCl3) δ 8.09 (d, 1H, J = 6.9 Hz, H-5), 8.02 (m, 2H, Ph-2,6), 7.92 (s, 1H, H-3), 7.52 (dd, 2H, J = 8.8–5.2 Hz, F-Ph-2,6), 7.45 (m, 2H, Ph-3,5), 7.37 (m, 1H, Ph-4), 7.18 (t, 2H, J = 8,8 Hz, F-Ph-3,5), 6.80 (d, 1H, J = 6.9 Hz, H-6). 13C-NMR (CDCl3) δ 162.6, 147.1, 134.6, 133.2, 133.1, 131.1, 128.7, 128.3, 126.4, 123.4, 115.5, 114.8, 109.4. (2C not found) Anal. Calcd for C19H12ClFN2: C, 70.70; H, 3.75; N, 8.68. Found: C, 70.94; H, 3.67; N, 8.73.
Ethyl 8-chloro-7-(4-fluorophenyl)imidazo[1,2-a]pyridine-2-carboxylate (3). Method C: 1b (500 mg, 1.43 mmol), 4-fluorophenylboronic acid (300 mg, 2.14 mmol then 100 mg, 0.71 mmol), Na2CO3 (454 mg, 4.38 mmol) and PdCl2(dppf) (58 mg, 0.07 mmol, 5 mol%) were used. M.p. = 199 °C. 87% yield. 1H-NMR (CDCl3) δ 8.27 (s, 1H, H-3), 8.15 (d, 1H, J = 7.2 Hz, H-5), 7.50 (dd, 2H, J = 8.7–5.4 Hz, F-Ph-2,6), 7.17 (t, 2H, J = 8.7 Hz, F-Ph-3,5), 6.90 (d, 1H, J = 7.2 Hz, H-6), 4.46 (q, 2H, J = 7.2 Hz, CH2), 1.42 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (CDCl3) δ 162.7, 162.7, 143.1, 137.7, 136.0, 132.6, 131.1, 124.1, 121.5, 118.3, 116.4, 115.5, 61.3, 14.3. Anal. Calcd for C16H12ClFN2O2: C, 60.29; H, 3.79; N, 8.79. Found: C, 60.51; H, 3.57; N, 8.81.
8-Chloro-7-(4-fluorophenyl)-2-methylimidazo[1,2-a]pyridine (4). Method C. 1c (500 mg, 1.71 mmol), 4-fluorophenylboronic acid (359 mg, 2.56 mmol then 120 mg, 0.85 mmol), Na2CO3 (545 mg, 5.14 mmol) and PdCl2(dppf) (70 mg, 0.09 mmol, 5 mol%) were used. M.p. = 164 °C. 89% Yield. 1H-NMR (CDCl3) δ 8.06 (d, 1H, J = 6.9 Hz, H-5), 7.52 (dd, 2H, J = 8.7–5.5 Hz, F-Ph-2,6), 7.46 (s, 1H, H-3), 7.19 (t, 2H, J = 8.7 Hz, F-Ph-3,5), 6.81 (d, 1H, J = 6.9 Hz, H-6), 2.56 (s, 3H, CH3). 13C-NMR (CDCl3) δ 162.7, 144.0, 133.0, 131.1, 123.3, 115.5, 114.7, 111.1, 14.13. (3C not found) Anal. Calcd for C14H10ClFN2: C, 64.50; H, 3.87; N, 10.75. Found: C, 64.36; H, 3.91; N, 10.83.
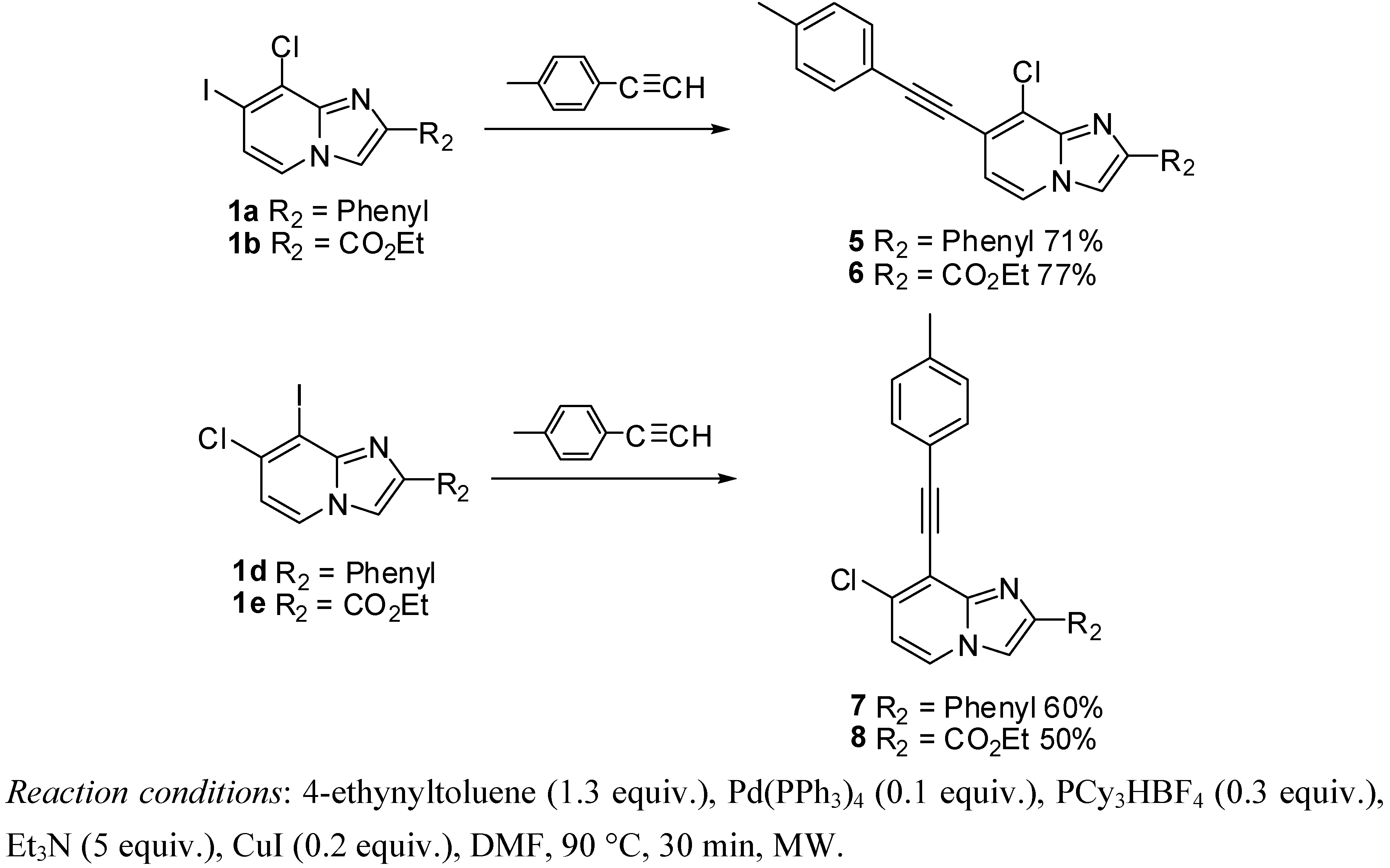
3.2.4. General Procedure for Sonogashira Cross-Coupling Reaction
8-Chloro-7-(p-tolylethynyl)-2-phenylimidazo[1,2-a]pyridine (5). Method D. To a solution of 1a (500 mg, 1.41 mmol) in DMF (6 mL), were added Et3N ((1 mL, 7.06 mmol), PCy3HBF4 (156 mg, 0.42 mmol), p-tolylacetylene (213 mg, 1.84 mmol), Pd(PPh3)4 (163 mg, 0.14 mmol) and CuI (50 mg, 0.26 mmol). The mixture was heated at 90 °C by microwave irradiation for 30 min. After cooling to room temperature, water (50 mL) was added and the solution was extracted with EtOAc (3 × 50 mL). The combined organic layers were dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residues were purified by flash chromatography (silica, cyclohexane/EtOAc (7:3)). M.p. = 176 °C. 71% yield. 1H-NMR (CDCl3) δ 8.04–7.95 (m, 3H, Ph-2,6, H-5), 7.88 (s, 1H, H-3), 7.55–7.40 (m, 4H, Ph-3,5, CH3-Ph-2,6), 7.35 (m, 1H, Ph-4), 7.20 (d, 2H, J = 7.9 Hz, CH3-Ph-3,5), 6.90 (d, 1H, J = 6.8 Hz, H-6), 2.39 (s, 3H, CH3). 13C-NMR (CDCl3) δ 147.4, 139.4, 132.9, 131.7, 129.2, 128.7, 128.6, 128.4, 126.4, 123.2, 119.3, 118.6, 115.0, 110.2, 98.1, 84.6, 21.6 (1C not found). Anal. Calcd for C22H15ClN2: C, 77.08; H, 4.41; N, 8.17. Found: C, 77.24; H, 4.46; N, 8.07.
Ethyl 8-chloro-7-(p-tolylethynyl)imidazo[1,2-a]pyridine-2-carboxylate (6). Method D. 1b (500 mg, 1.43 mmol), Et3N (1 mL, 7.14 mmol), PCy3HBF4 (158 mg, 0.43 mmol), p-tolylacetylene (215 mg, 1.86 mmol), Pd(PPh3)4 (165 mg, 0.14 mmol) and CuI (50 mg, 0.26 mmol) were used. M.p. = 211 °C. 77% yield. 1H-NMR (CDCl3) δ 8.21 (s, 1H, H-3), 8.04 (d, 1H, J = 7.2 Hz, H-5), 7.49 (d, 2H, J = 8.1 Hz, CH3-Ph-2,6), 7.19 (d, 2H, J = 8.1 Hz, CH3-Ph-3,5), 6.97 (d, 2H, J = 7.2 Hz, H-6), 4.46 (q, 2H, J = 7.5 Hz, CH2), 2.39 (s, 3H, CH3), 1.43 (t, 3H, J = 7.5 Hz, CH3). 13C-NMR (CDCl3) δ 162.5, 142.7, 139.8, 137.7, 131.8, 129.3, 126.0, 124.0, 120.6, 119.0, 116.5, 99.5, 84.0, 61.4, 21.6, 14.32 (1C not found). Anal. Calcd for C19H15ClN2O2: C, 67.36; H, 4.46; N, 8.27. Found: C, 67.49; H, 4.41; N, 8.38.
7-Chloro-8-(p-tolylethynyl)-2-phenylimidazo[1,2-a]pyridine (7). Method D. 1d (250 mg, 0.71 mmol), Et3N (0.5 mL, 3.53 mmol), PCy3HBF4 (78 mg, 0.21 mmol), p-tolylacetylene (107 mg, 0.92 mmol), Pd(PPh3)4 (81 mg, 0.07 mmol) and CuI (25 mg, 0.13 mmol) were used. M.p. = 118 °C. 60% yield. 1H-NMR (CDCl3) δ 8.00 (m, 3H, H-5, Ph-2,6), 7.84 (s, 1H, H-3), 7.62 (d, 2H, J = 8.0 Hz, CH3-Ph-2,6), 7.44 (m, 2H, Ph-3,5), 7.34 (m, 1H, Ph-4), 7.20 (d, 2H, J = 8.0 Hz, CH3-Ph-3,5), 6.85 (d, 1H, J = 7.4 Hz, H-6), 2.39 (s, 3H, CH3). 13C-NMR (CDCl3) δ 146.9, 139.3, 133.1, 132.1, 129.1, 128.9, 128.6, 128.3, 126.3, 126.2, 124.5, 119.6, 115.8, 114.2, 108.9, 81.4, 21.6 (1C not found). Anal. Calcd for C22H15ClN2: C, 77.08; H, 4.41; N, 8.17. Found: C, 77.25; H, 4.43; N, 8.19.
Ethyl 7-chloro-8-(p-tolylethynyl)imidazo[1,2-a]pyridine-2-carboxylate (8). Method D. 1e (250 mg, 0.71 mmol), Et3N (0.5 mL, 3.53 mmol), PCy3HBF4 (78 mg, 0.21 mmol), p-tolylacetylene (107 mg, 0.92 mmol), Pd(PPh3)4 (81 mg, 0.07 mmol) and CuI (25 mg, 0.13 mmol) were added. M.p. = 175 °C. 50% yield. 1H-NMR (CDCl3) δ 8.20 (s, 1H, H-3), 8.10 (d, 1H, J = 7.2 Hz, H-5), 7.55 (d, 2H, J = 8.0 Hz, CH3-Ph-2,6), 7.17 (d, 2H, J = 8.0 Hz, CH3-Ph-3,5), 6.95 (d, 1H, J = 7.2 Hz, H-6), 4.44 (q, 2H, J = 7.1 Hz, CH2), 2.37 (s, 3H, CH3), 1.42 (t, 3H, J = 7.1 Hz, CH3). 13C-NMR (CDCl3) δ 162.7, 144.8, 139.5, 137.6, 135.0, 132.0, 129.0, 125.2, 119.2, 118.0, 115.9, 113.8, 102.8, 80.9, 61.3, 21.6, 14.3. Anal. Calcd for C19H15ClN2O2: C, 67.36; H, 4.46; N, 8.27. Found: C, 67.21; H, 4.47; N, 8.46.
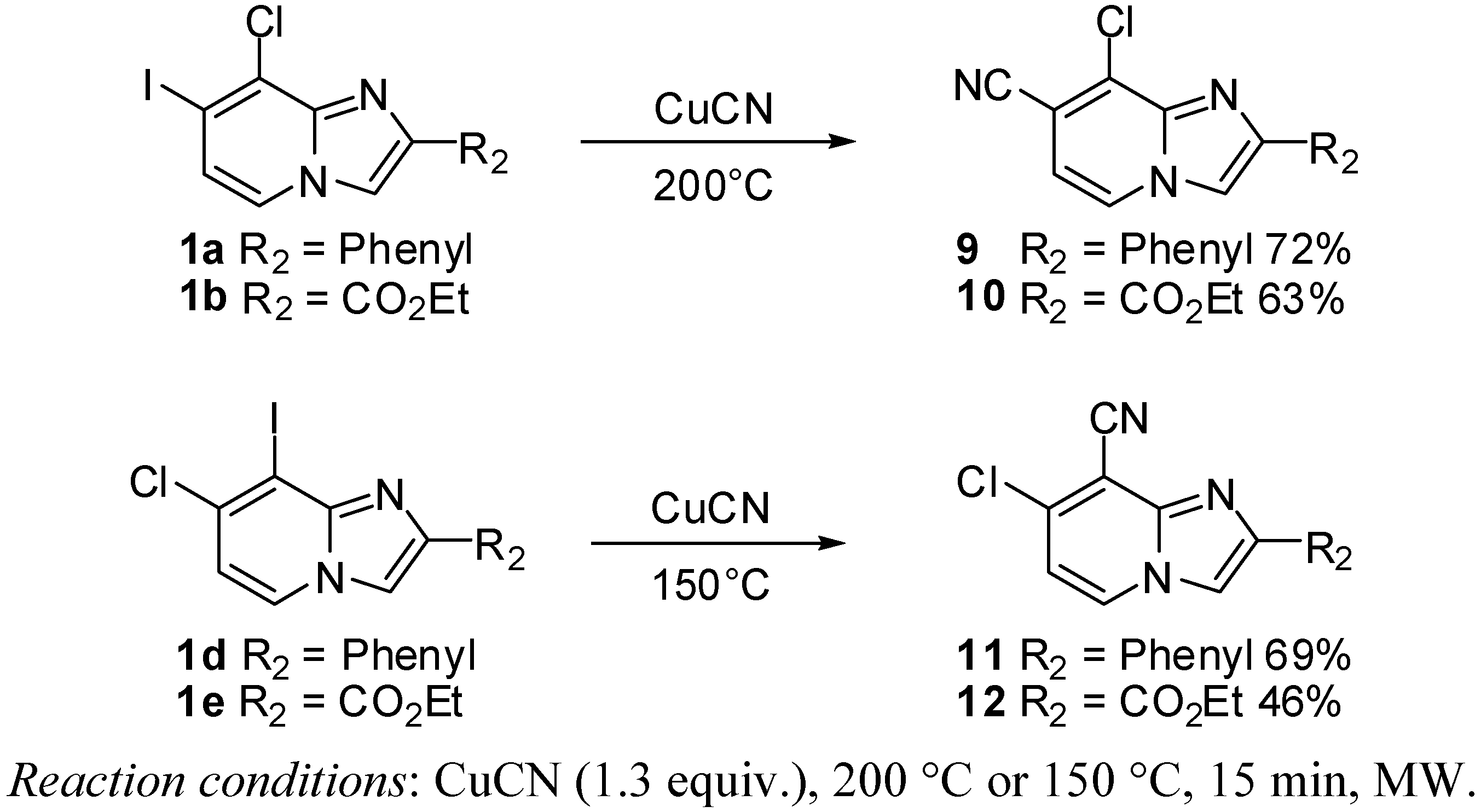
3.2.5. General Procedure for Cyanation Reaction
8-Chloro-2-phenylimidazo[1,2-a]pyridine-7-carbonitrile (9). Method E. To a solution of 1a (400 mg, 1.13 mmol) in DMF (793 µL), CuCN (132 mg, 1.47 mmol) was added. The tube was evacuated and back filled with nitrogen. Then the reaction mixture was heated at 150 °C or 200 °C by microwave irradiation for 15 min. After cooling to room temperature, an aqueous solution of NH4OH 10% (50 mL) and CH2Cl2 (50 mL) were added and the organic layer was washed with an aqueous solution of NH4OH (3 × 50 mL), dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residues were purified on column chromatography (silica gel, CH2Cl2). The reaction mixture was heated at 200 °C by microwave irradiation. M.p. = 200 °C. 72% yield. 1H-NMR (CDCl3) δ 8.71 (s, 1H, H-3), 8.67 (d, 1H, J = 7.0 Hz, H-5), 8.01 (m, 2H, Ph-2,6), 7.48 (m, 2H, Ph-3,5), 7.39 (m, 1H, Ph-4), 7.30 (d, 1H, J = 7.0 Hz, H-6). 13C-NMR (CDCl3) δ 147.4, 140.7, 132.4, 128.9, 128.8, 127.0, 126.7, 125.9, 115.6, 113.6, 112.6, 106.2. Anal. Calcd for C14H8ClN3: C, 66.28; H, 3.18; N, 16.56. Found: C, 65.94; H, 3.33; N, 16.72.
Ethyl 8-chloro-7-cyanoimidazo[1,2-a]pyridine-2-carboxylate (10). Method E. 1b (400 mg, 1.14 mmol) was used as starting material. The reaction mixture was heated at 200 °C by microwave irradiation. M.p. = 229 °C. 63% yield. 1H-NMR (CDCl3) δ 8.35 (s, 1H, H-3), 8.20 (d, 1H, J = 7.0 Hz, H-5), 7.05 (d, 1H, J = 7.0 Hz, H-6), 4.50 (q, 2H, J = 7.5 Hz, CH2), 1.45 (t, 3H, J = 7.5 Hz, CH3). 13C-NMR (CDCl3) δ 161.9, 141.3, 139.8, 131.4, 125.3, 120.0, 114.4, 114.0, 109.3, 61.8, 14.3. Anal. Calcd for C11H8ClN3O2: C, 52.92; H, 3.23; N, 16.83. Found: C, 53.16; H, 3.48; N, 16.97.
7-Chloro-2-phenylimidazo[1,2-a]pyridine-8-carbonitrile (11). Method E. 1d (400 mg, 1.13 mmol) was used as starting material. The reaction mixture was heated at 150 °C by microwave irradiation. M.p. = 197 °C. 69% yield. 1H-NMR (CDCl3) δ 8.22 (d, 1H, J = 7.2 Hz, H-5), 7.96 (m, 2H, Ph-2,6), 7.91 (s, 1H, H-3), 7.45 (m, 2H, Ph-3,5), 7.37 (m, 1H, Ph-4), 6.90 (d, 1H, J = 7.2 Hz, H-6). 13C-NMR (CDCl3) δ 148.4, 143.3, 137.3, 132.2, 129.0, 128.8, 126.4, 113.5, 112.6, 109.3, 102.0 (1 C not found). Anal. Calcd for C14H8ClN3: C, 66.28; H, 3.18; N, 16.56. Found: C, 66.34; H, 3.35; N, 16.51.
Ethyl 7-chloro-8-cyanoimidazo[1,2-a]pyridine-2-carboxylate (12). Method E. 1e (400 mg, 1.14 mmol) was used as starting material. The reaction mixture was heated at 150 °C by microwave irradiation. M.p. = 227 °C. 46% yield. 1H-NMR (CDCl3) δ 8.31 (d, 1H, J = 7.2 Hz, H-5), 8.28 (s, 1H, H-3), 7.06 (d, 1H, J = 7.4 Hz, H-6), 4.48 (q, 2H, J = 7.1 Hz, CH2), 1.45 (t, 3H, J = 7.1 Hz, CH3). 13C-NMR (CDCl3) δ 162.1, 139.8, 139.2, 129.6, 118.4, 115.3, 111.7, 61.8, 14.3 (2C not found). Anal. Calcd for C11H8ClN3O2: C, 52.92; H, 3.23; N, 16.83. Found: C, 53.17; H, 3.32; N, 16.79.
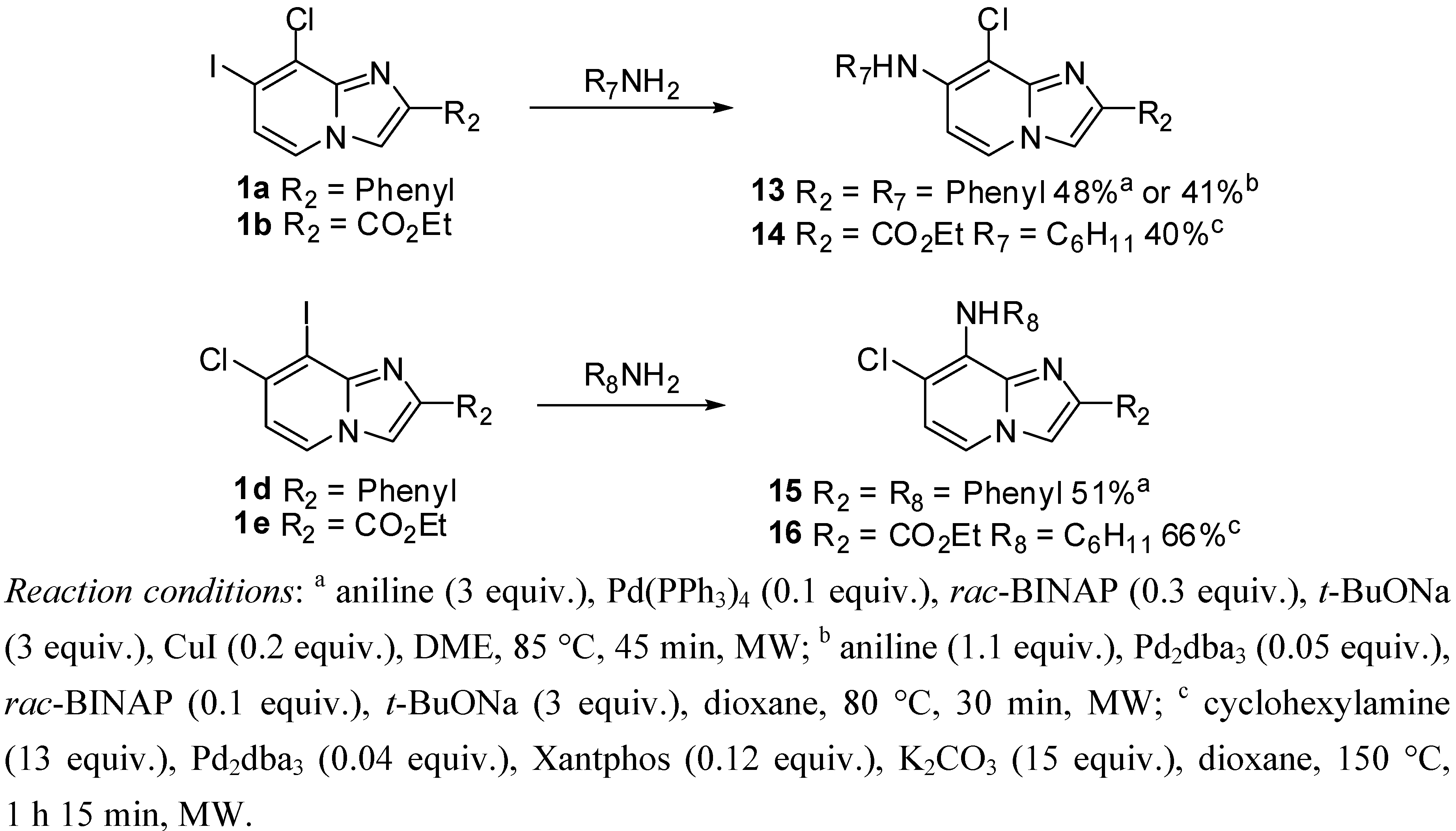
3.2.6. General Procedure for Buchwald-Hartwig Cross-Coupling Reaction
N-(8-Chloro-2-phenylimidazo[1,2-a]pyridin-7-yl)aniline (13). Method F. To a solution of 1a (100 mg, 0.28 mmol) in DME (3 mL) under argon, were added successively rac-BINAP (53 mg, 0.085 mmol), t-BuONa (81 mg, 0.85 mmol), Pd(PPh3)4 (33 mg, 0.03 mmol), CuI (10 mg, 0.053 mmol) and aniline (77 µL, 0.85 mmol). The mixture was heated at 85 °C by microwave irradiation for 45 min. After cooling to room temperature, water (50 mL) was added and the solution was extracted with EtOAc (3 × 50 mL), dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residues were purified by flash chromatography (silica, cyclohexane/EtOAc (7:3)). M.p. = 170 °C. 48% yield. 1H-NMR (CDCl3) δ 7.93 (m, 2H, Ph-2,6), 7.82 (d, 1H, J = 7.4 Hz, H-5), 7.67 (s, 1H, H-3), 7.47–7.28 (m, 5H, Ph-3,4,5, Ph-NH-3,5), 7.15 (m, 3H, Ph-NH-2,4,6), 6.75 (d, 1H, J = 7.4 Hz, H-6), 6.42 (s, 1H, NH). 13C-NMR (CDCl3) δ 139.7, 138.2, 133.4, 129.7, 128.6, 128.1, 126.2, 124.5, 124.3, 121.9, 120.1, 108.3, 104.3 (2C not found). Anal. Calcd for C19H14ClN3: C, 71.36; H, 4.41; N, 13.14. Found: C, 71.55; H, 4.39; N, 13.26.
Ethyl 8-chloro-7-(cyclohexylamino)imidazo[1,2-a]pyridine-2-carboxylate (14). Method G. To a solution of 1b (100 mg, 0.29 mmol) in dioxane (3 mL), were added Pd2dba3 (11 mg, 0.01 mmol), Xantphos (20 mg, 0.03 mmol), K2CO3 (591 mg, 4.29 mmol) and cyclohexylamine (0.42 mL, 3.71 mmol). The mixture was irradiated under microwaves at 150 °C during 1 h 15 min. After cooling to room temperature, EtOAc (50 mL) and water (50 mL) were added. The solution was extracted with EtOAc (3 × 50 mL), dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residues were purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 176 °C. 40% yield. 1H-NMR (CDCl3) δ 7.98 (s, 1H, H-3), 7.90 (d, 1H, J = 7.5 Hz, H-5), 6.56 (d, 1H, J = 7.5 Hz, H-6), 4.57 (m, 1H, NH), 4.41 (q, 2H, J = 7.0 Hz, CH2), 3.37 (m, 1H, CyHex), 2.02 (m, 2H, CyHex), 1.80 (m, 2H, CyHex), 1.66 (m, 1H, CyHex), 1.32 (m, 9H, CH3, CyHex). 13C-NMR (CDCl3) δ 163.3, 144.7, 141.3, 136.6, 124.8, 116.5, 104.2, 99.6, 60.9, 51.8, 33.6, 25.4, 24.6, 14.4. Anal. Calcd for C16H20ClN3O2: C, 59.72; H, 6.26; N, 13.06. Found: C, 59.88; H, 6.23; N, 12.97.
N-(7-Chloro-2-phenylimidazo[1,2-a]pyridin-8-yl)aniline (15). Method F. 1d (100 mg, 0.28 mmol) was used as starting material. M.p. = 167 °C. 51% yield. 1H-NMR (CDCl3) δ 7.92 (m, 2H, Ph-2,6), 7.82 (s, 1H, H-3), 7.76 (d, 1H, J = 7.2 Hz, H-5), 7.43 (m, 2H, Ph-3,5), 7.32 (m, 3H, Ph-4, Ph-NH-3,5), 7.00 (m, 4H, Ph-NH-2,6,4, NH), 6.79 (d, 1H, J = 7.2 Hz, H-6). 13C-NMR (CDCl3) δ 145.3, 142.2, 141.6, 133.2, 128.7, 128.6, 128.1, 127.3, 126.0, 121.9, 119.4, 119.2, 119.0, 115.7, 109.3. Anal. Calcd for C16H20ClN3O2: C, 59.72; H, 6.26; N, 13.06. Found: C, 59.88; H, 6.23; N, 12.97.
Ethyl 7-chloro-8-(cyclohexylamino)imidazo[1,2-a]pyridine-2-carboxylate (16). Method G. 1e (100 mg, 0.29 mmol) was used as starting material. M.p. = 90 °C. 66% yield. 1H-NMR (CDCl3) δ 8.03 (s, 1H, H-3), 7.49 (d, 1H, J = 7.2 Hz, H-5), 6.77 (d, 1H, J = 7.2 Hz, H-6), 4.43 (q, 2H, J = 7.0 Hz, CH2), 2.05 (m, 2H, CyHex), 1.77 (m, 2H, CyHex), 1.63 (m, 2H, NH, CyHex), 1.28 (m, 9H, CH3, CyHex). 13C-NMR (CDCl3) δ 162.0, 157.0, 144.9, 133.7, 126.7, 118.5, 117.8, 114.7, 61.3, 53.2, 34.5, 25.6, 24.9, 14.3. Anal. Calcd for C16H20ClN3O2: C, 59.72; H, 6.26; N, 13.06. Found: C, 59.95; H, 6.32; N, 13.01.
3.2.7. Substitution of the Chlorine Atom in Position 8 of Compounds 2–6, 9–10, 14
Ethyl 7-(4-fluorophenyl)-8-(p-tolyl)imidazo[1,2-a]pyridine-2-carboxylate (17). Method H. To a solution of 3 (100 mg, 0.31 mmol) in a mixture of dioxane (0.5 mL) and ethanol (0.3 mL), were added p-tolylboronic acid (60 mg, 0.44 mmol), K2CO3 (88 mg, 0.63 mmol) and Pd(PPh3)4 (36 mg, 0.031 mmol). The mixture was heated at 150 °C by microwave irradiation for 15 min. After cooling to room temperature, water (50 mL) was added and the solution was extracted with CH2Cl2 (3 × 50 mL). The organic layer was dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residue was purified by column chromatography (silica gel, CH2Cl2). M.p. = 241 °C. 95% yield. 1H-NMR (CDCl3) δ 8.25 (s, 1H, H-3), 8.19 (d, 1H, J = 7.2 Hz, H-5), 7.22 (d, 2H, J = 7.9 Hz, CH3-Ph-2,6), 7.12 (dd, J = 8.7–5.4 Hz, 2H, F-Ph-2,6), 7.03 (d, 2H, J = 7.9 Hz, CH3-Ph-3,5), 6.92 (m, 3H, F-Ph-3,5, H-6), 4.39 (q, 2H, J = 7.2 Hz, CH2), 2.30 (s, 3H, CH3), 1.38 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (CDCl3) δ 163.1, 161.9, 145.3, 137.6, 137.2, 135.9, 135.2, 131.3, 131.0, 129.3, 128.7, 124.5, 117.3, 117.2, 115.2, 61.0, 21.2, 14.3. (1C not found) Anal. Calcd for C23H19FN2O2: C, 73.78; H, 5.11; N, 7.48. Found: C, 74.03; H, 5.08; N, 7.56.
Ethyl 7-(4-fluorophenyl)-8-(4-methoxyphenyl)imidazo[1,2-a]pyridine-2-carboxylate (18). Method H. 4-Methoxyphenylboronic acid (96 mg, 0.63 mmol) was used. The crude residue was purified by column chromatography (silica gel, CH2Cl2). M.p. = 199 °C. 76% yield. 1H-NMR (CDCl3) δ 8.26 (s, 1H, H-3), 8.20 (d, 1H, J = 7.2 Hz, H-5), 7.28 (d, 2H, J = 8.7 Hz, CH3O-Ph-2,6), 7.12 (dd, 2H, J = 8.7–5.4 Hz, F-Ph-2,6), 6.96–6.91 (m, 3H, H-6, F-Ph-3,5), 6.78 (d, 2H, J = 8.7 Hz, CH3O-Ph-3,5), 4.40 (q, 2H, J = 7.2 Hz, CH2), 3.78 (s, 3H, CH3O), 1.39 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (CDCl3) δ 162.6, 161.9, 159.1, 144.9, 136.6, 136.3, 134.7, 132.3, 131.2, 128.3, 125.9, 124.9, 117.5, 117.4, 115.2, 113.3, 61.1, 55.2, 14.3. Anal. Calcd for C23H19FN2O3: C, 70.76; H, 4.91; N, 7.18. Found: C, 70.84; H, 5.11; N, 7.01.
7-(4-Fluorophenyl)-8-(p-tolyl)-2-phenylimidazo[1,2-a]pyridine (19). Method I. To a solution of 2 (100 mg, 0.31 mmol) in a mixture of DME (2 mL) and water (1 mL), were added p-tolylboronic acid (84 mg, 0.62 mmol), Na2CO3 (66 mg, 0.62 mmol) and Pd(PPh3)4 (18 mg, 0.016 mmol, 5 mol%). The mixture was heated at 120 °C for 30 min by microwave irradiation. After cooling to room temperature, water (50 mL) was added and the solution was extracted with CH2Cl2 (3 × 50 mL). The organic layer was dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residue was purified by column chromatography (silica gel, CH2Cl2). M.p. = 222 °C. 90% yield. 1H-NMR (CDCl3) δ 8.11 (d, 1H, J = 6.9 Hz, H-5), 7.95 (m, 2H, Ph-2,6), 7.91 (s, 1H, H-3), 7.42–7.28 (m, 5H, CH3-Ph-2,6, Ph-3,4,5), 7.17–7.11 (m, 4H, F-Ph-2,6, CH3-Ph-3,5), 6.94 (t, 2H, J = 8.7 Hz, F-Ph-3,5), 6.85 (d, 1H, J = 6,9 Hz, H-6), 2.36 (s, 3H, CH3). 13C-NMR (CDCl3) δ 161.8, 146.3, 145.4, 137.3, 135.9, 134.6, 133.7, 131.7, 131.4, 131.3, 128.6, 128.5, 127.8, 126.3, 123.9, 115.7, 115.1, 108.3, 21.3 (1C not found). Anal. Calcd for C26H19FN2: C, 82.52; H, 5.06; N, 7.40. Found: C, 82.77; H, 5.39; N, 7.28.
7-(4-Fluorophenyl)-2-methyl-8-(p-tolyl)imidazo[1,2-a]pyridine (20). Method I. 4 (100 mg, 0.38 mmol), p-tolylboronic acid (104 mg, 0.77 mmol), Na2CO3 (82 mg, 0.77 mmol, 2 eq) and Pd(PPh3)4 (22 mg, 0.02 mmol, 5 mol%) were used. The mixture was heated for 60 min. The crude residue was purified by flash chromatography (silica, CH2Cl2/MeOH (99:1)). M.p. = 195 °C. 84% yield. 1H-NMR (CDCl3) δ 8.11 (d, 1H, J = 7.2 Hz, H-5), 7.45 (s, 1H, H-3), 7.18 (d, 2H, J = 8.1 Hz, CH3-Ph-2,6), 7.11–7.08 (m, 4H, F-Ph-2,6, CH3-Ph-3,5), 6.90 (t, 2H, J = 8.7 Hz, F-Ph-3,5), 6.82 (d, 1H, J = 7.2 Hz, H-6), 2.44 (s, 3H, CH3), 2.29 (s, 3H, CH3). 13C-NMR (CDCl3) δ 161.7, 144.6, 143.7, 137.1, 135.7, 134.3, 131.8, 131.3, 130.7, 128.7, 127.3, 123.7, 114.9, 114.9, 109.9, 21.2, 14.4. Anal. Calcd for C21H17FN2: C, 79.72; H, 5.42; N, 8.85. Found: C, 79.95; H, 5.48; N, 8.75.
7-(4-Fluorophenyl)-8-(4-methoxyphenyl)-2-phenylimidazo[1,2-a]pyridine (21). Method I. 2 (100 mg, 0.31 mmol), 4-methoxyphenylboronic acid (94 mg, 0.62 mmol) were used. The mixture was heated for 30 min. The crude residue was purified by column chromatography (silica gel, CH2Cl2). M.p. = 201 °C. 82% yield. 1H-NMR (CDCl3) δ 8.15 (d, 1H, J = 7.0 Hz, H-5), 7.99–7.89 (m, 3H, Ph-2,6, H-3), 7.43–7.27 (m, 5H, Ph-3,4,5, CH3O-Ph-2,6), 7.12 (dd, 2H, J = 9–5.4 Hz, F-Ph-2,6), 6.94 (t, 2H, J = 8.7 Hz, F-Ph-3,5), 6.89–6.78 (m, 3H, CH3O-Ph-3,5, H-6), 3.81 (s, 3H, CH3O). 13C-NMR (CDCl3) δ 161.9, 159.0, 146.0, 145.3, 135.9, 134.8, 133.5, 132.7, 131.4, 128.6, 127.9, 126.9, 126.3, 124.0, 115.8, 115.2, 113.4, 108.5, 55.2 (1 C not found). Anal. Calcd for C26H19FN2O: C, 79.17; H, 4.86; N, 7.10. Found: C, 79.35; H, 5.91; N, 7.24.
7-(4-Fluorophenyl)-2-methyl-8-(pyridin-4-yl)imidazo[1,2-a]pyridine (22). Method I. 4 (100 mg, 0.38 mmol), pyridin-4-ylboronic acid (76 mg, 0.54 mmol), Na2CO3 (82 mg, 0.77 mmol) and Pd(PPh3)4 (22 mg, 0.02 mmol, 5 mol%) were used. The mixture was heated for 60 min. The crude residue was purified by flash chromatography (silica, CH2Cl2/MeOH (99:1)). M.p. = 188 °C. 80% yield. 1H-NMR (CDCl3) δ 8.54 (d, 2H, J = 6.0 Hz, Pyr-2,6), 8.15 (d, 1H, J = 6.9 Hz, H-5), 7.46 (s, 1H, H-3), 7.31 (dd, 2H, J = 6.0 Hz, Pyr-3,5), 7.08 (dd, 2H, J = 8.7–5.4 Hz, F-Ph-2,6), 6.94 (t, 2H, J = 8.7 Hz, F-Ph-3,5), 6.87 (d, 1H, J = 6.9 Hz, H-6), 2.47 (s, 3H, CH3). 13C-NMR (CDCl3) δ 162.2, 148.9, 144.1, 143.2, 135.7, 134.4, 131.3, 126.2, 125.0, 124.0, 115.7, 115.5, 110.3, 14.2 (1C not found). Anal. Calcd for C19H14FN3: C, 75.23; H, 4.65; N, 13.85. Found: C, 75.52; H, 4.56; N, 13.98.
8-(Fur-2-yl)-7-(p-tolylethynyl)-2-phenylimidazo[1,2-a]pyridine (23). Method I. 5 (100 mg, 0.29 mmol), fur-2-ylboronic acid (65 mg, 0.58 mmol), Na2CO3 (62 mg, 0.58 mmol) and Pd(PPh3)4 (17 mg, 0.02 mmol, 5 mol%) were used. The mixture was heated for 75 min. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. > 300 °C. 19% yield. 1H-NMR (300 MHz, CDCl3) δ 8.08–7.96 (m, 4H, H-5, Ph-2,6, Fur-3), 7.91 (s, 1H, H-3) 7.74 (d, 1H, J = 1.7–0.9 Hz, Fur-5), 7.53–7.40 (m, 4H, Ph-3,5, CH3-Ph-2,6), 7.34 (m, 1H, Ph-4), 7.21 (d, 2H, J = 7.9 Hz, CH3-Ph-3,5), 6.98 (d, 1H, J = 6.8 Hz, H-6), 6.70 (dd, 1H, J = 3.4–1.7 Hz, Fur-4), 2.41 (s, 3H, CH3). 13C-NMR (CDCl3) δ 148.6, 145.8, 143.0, 139.0, 133.0, 131.5, 129.2, 128.7, 128.2, 126.2, 123.0, 120.5, 120.2, 117.2, 115.2, 114.5, 111.9, 109.4, 96.0, 88.5, 21.6 (1C not found). Anal. Calcd for C26H18N2O: C, 83.40; H, 4.85; N, 7.48. Found: C, 83.56; H, 4.91; N, 7.34.
2-Phenyl-8-(thien-3-yl)imidazo[1,2-a]pyridine-7-carbonitrile (24). Method I. 9 (100 mg, 0.40 mmol), thien-3-ylboronic acid (71 mg, 0.55 mmol), Na2CO3 (84 mg, 0.79 mmol) and Pd(PPh3)4 (23 mg, 0.02 mmol, 5 mol%) were used. The mixture was heated for 15 min. The crude residue was purified by flash chromatography (silica, petroleum ether/EtOAc (100:0) to (20:80)). M.p. = 193 °C. 71% yield. 1H-NMR (CDCl3) δ 8.48 (dd, 1H, J = 3.0–1.3 Hz, Th-2), 8.11 (d, 1H, J = 7.0 Hz, H-5), 8.05 (dd, 1H, J = 5.1–1.3 Hz, Th-4), 7.99 (m, 3H, H-3, Ph-2,6), 7.52 (dd, 1H, J = 5.1–3.0 Hz, Th-5), 7.45 (m, 2H, Ph-3,5), 7.37 (m, 1H, Ph-4), 7.01 (d, 1H, J = 7.0 Hz, H-6). 13C-NMR (CDCl3) δ 148.3, 142.7, 132.7, 132.5, 130.8, 129.7, 129.0, 128.8, 128.7, 126.3, 125.3, 124.0, 118.6, 114.3, 110.6, 103.2. Anal. Calcd for C18H11N3S: C, 71.74; H, 3.68; N, 13.94. Found: C, 71.62; H, 3.77; N, 14.05.
2-Phenyl-8-(pyridin-4-yl)imidazo[1,2-a]pyridine-7-carbonitrile (25). Method I. 9 (100 mg, 0.40 mmol), pyridin-4-ylboronic acid (78 mg, 0.55 mmol), Na2CO3 (84 mg, 0.79 mmol), Pd(PPh3)4 (23 mg, 0.02 mmol, 5 mol%) were used. The mixture was heated for 15 min. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 278 °C. 42% yield. 1H-NMR (CDCl3) δ 8.84 (d, 2H, J = 5.8 Hz, Pyr-2,6), 8.78 (d, 1H, J = 7.0 Hz, H-5), 8.74 (s, 1H, H-3), 7.96 (m, 2H, Ph-2,6), 7.84 (m, 2H, Pyr-3,5), 7.47 (m, 2H, Ph-3,5), 7.36 (m, 2H, H-6, Ph-4). 13C-NMR (CDCl3) δ 149.8, 147.5, 141.8, 140.7, 132.6, 132.4, 128.9, 128.6, 127.8, 126.0, 124.7, 117.4, 113.3, 112.5, 104.9. Anal. Calcd for C19H12N4: C, 77.01; H, 4.08; N, 18.91. Found: C, 77.12; H, 3.97; N, 18.89.
Ethyl 7-(cyclohexylamino)-8-(p-tolyl)imidazo[1,2-a]pyridine-2-carboxylate (26). Method H. 14 (100 mg, 0.31 mmol), p-tolylboronic acid (84.3 mg, 0.62 mmol) were used. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 169 °C. 27% yield. 1H-NMR (CDCl3) δ 8.04 (d, 1H, J = 7.5 Hz, H-5), 8.00 (s, 1H, H-3), 7.35 (d, 2H, J = 8.0 Hz, CH3-Ph-2,6), 7.25 (d, 2H, J = 8.0 Hz, CH3-Ph-3,5), 6.65 (d, 1H, J = 7.5 Hz, H-6), 4.34 (q, 2H, J = 7.0 Hz, CH2), 4.15 (s, 1H, NH), 3.32 (m, 1H, CyHex), 2.39 (s, 3H, CH3), 1.94 (m, 2H, CyHex), 1.64 (m, 3H, CyHex), 1.20 (m, 8H, CH3, CyHex). 13C-NMR (CDCl3) δ 163.4, 146.9, 142.5, 137.7, 135.8, 130.4, 129.9, 129.6, 125.6, 115.7, 108.4, 105.3, 60.7, 52.1, 33.6, 25.5, 24.7, 21.3, 14.3. Anal. Calcd for C23H27FN3O2: C, 73.18; H, 7.21; N, 11.13. Found: C, 73.39; H, 7.18; N, 11.09.
Ethyl 8-(1H-indol-6-yl)-7-(p-tolylethynyl)imidazo[1,2-a]pyridine-2-carboxylate (27). Method H. 6 (100 mg, 0.3 mmol), indol-6-ylboronic acid (96.6 mg, 0.6 mmol), K2CO3 (83 mg, 0.59 mmol) and Pd(PPh3)4 (34 mg, 0.03 mmol) were used. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 242 °C. 35% yield. 1H-NMR (CDCl3) δ 10.17 (bs, 1H, NH), 8.17–8.09 (m, 2H, H-5, H-3), 7.78 (s, 1H, Indol-7), 7.39 (d, 1H, J = 8.1 Hz, Indol-4*), 7.24 (d, 1H, J = 8.1 Hz, Indol-5*), 7.19–6.92 (m, 6H, H-6, CH3-Ph, Indol-2), 6.33 (s, 1H, Indol-3), 4.27 (q, 2H, J = 7.1 Hz, CH2), 2.27 (s, 3H, CH3), 1.14 (t, 3H, J = 7.1 Hz, CH3). 13C-NMR (CDCl3) δ 161.6, 143.7, 139.2, 135.4, 134.6, 133.5, 131.6, 129.1, 128.1, 126.2, 125.6, 124.5, 121.2, 119.2, 119.0, 118.6, 118.0, 114.0, 101.1, 97.0, 87.0, 61.5, 21.5, 14.0. (1 C not found) Anal. Calcd for C27H21N3O2: C, 77.31; H, 5.05; N, 10.02. Found: C, 77.47; H, 4.98; N, 10.13.
Ethyl 7-cyano-8-(pyridin-4-yl)imidazo[1,2-a]pyridine-2-carboxylate (28). Method H. 10 (100 mg, 0.4 mmol), pyridin-4-ylboronic acid (80 mg, 0.56 mmol), K2CO3 (112 mg, 0.8 mmol), Pd(PPh3)4 (46 mg, 0.04 mmol) were used. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (100:0) to (10:90)]. M.p. = 185 °C. 31% yield. 1H-NMR (CDCl3) δ 8.86 (d, 2H, J = 6.0 Hz, Pyr-2,6), 8.40 (s, 1H, H-3), 8.35 (d, 1H, J = 7.0 Hz, H-5), 7.91 (d, 2H, J = 6.0 Hz, Pyr-3,5), 7.19 (d, 1H, J = 7.0 Hz, H-6), 4.45 (q, 2H, J = 7.1 Hz, CH2), 1.41 (t, 3H, J = 7.1 Hz, CH3). 13C-NMR (CDCl3) δ 162.2, 149.9, 142.2, 139.9, 126.8, 124.6, 119.3, 116.4, 115.0, 107.5, 61.7, 14.3. (2C not found) Anal. Calcd for C16H12N4O2: C, 65.75; H, 4.14; N, 19.17. Found: C, 65.92; H, 4.28; N, 19.04.
Ethyl 7-cyano-8-(fur-2-yl)imidazo[1,2-a]pyridine-2-carboxylate (29). Method H. 10 (100 mg, 0.4 mmol), fur-2-ylboronic acid (63 mg, 0.56 mmol), K2CO3 (112 mg, 0.8 mmol), Pd(PPh3)4 (46 mg, 0.04 mmol) were used. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 185 °C. 34% yield. 1H-NMR (CDCl3) δ 8.29–8.22 (m, 2H, H-3, Fur-3), 8.07 (d, 1H, J = 7.2 Hz, H-5), 7.78 (dd, 1H, J = 1.7–0.8 Hz, Fur-5), 7.08 (d, 1H, J = 7.2 Hz, H-6), 6.71 (dd, 1H, J = 3.6–1.7 Hz, Fur-4), 4.48 (q, 2H, J = 7.2 Hz, CH2), 1.46 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (CDCl3) δ 162.4, 148.8, 145.3, 140.1, 138.5, 123.7, 122.2, 119.3, 118.9, 117.9, 116.2, 113.0, 61.5, 14.3. (1 C not found) Anal. Calcd for C15H11N3O3: C, 64.05; H, 3.94; N, 14.94. Found: C, 63.92; H, 4.08; N, 15.02.
Ethyl 7-(cyclohexylamino)-8-(pyridin-4-yl)imidazo[1,2-a]pyridine-2-carboxylate (30). Method H. 14 (100 mg, 0.31 mmol), pyridin-4-ylboronic acid (76.2 mg, 0.62 mmol) were used. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (6:4)]. M.p. = 182 °C. 28% yield. 1H-NMR (CDCl3) δ 8.72 (d, 2H, J = 5.1 Hz, Pyr-2,6), 7.98 (m, 2H, H-5, H-3), 7.57 (d, 2H, J = 5.1 Hz, Pyr-3,5), 6.65 (d, 1H, J = 7.7 Hz, H-6), 4.35 (q, 2H, J = 7.0 Hz, CH2), 4.26 (m, 1H, NH), 3.35 (m, 1H, CyHex), 1.91 (m, 2H, CyHex), 1.66 (m, 3H, CyHex), 1.40–1.05 (m, 8H, CH3, CyHex). 13C-NMR (CDCl3) δ 163.4, 150.6, 149.9, 146.0, 142.3, 136.4, 126.6, 125.8, 121.4, 115.9, 105.0, 60.9, 52.0, 33.5, 25.4, 24.7, 14.3. Anal. Calcd for C21H24N4O2: C, 69.21; H, 6.64; N, 15.37. Found: C, 69.38; H, 6.71; N, 15.24.
7-(4-Methoxyphenyl)-2-phenyl-8-(p-tolylethynyl)imidazo[1,2-a]pyridine (31). Method I. 7 (100 mg, 0.29 mmol), 4-methoxyphenylboronic acid (88 mg, 0.58 mmol), Na2CO3 (62 mg, 0.58 mmol), Pd(PPh3)4 (17 mg, 0.014 mmol, 5 mol%) were used. The mixture was heated for 30 min. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 84 °C. 56% yield. 1H-NMR (CDCl3) δ 8.12–8.01 (m, 3H, H-5, Ph-2,6), 7.87 (s, 1H, H-3), 7.75 (m, 2H, CH3O-Ph-2,6), 7.46 (m, 4H, Ph-3,5, CH3-Ph-2,6), 7.34 (m, 1H, Ph-4), 7.16 (d, 2H, J = 7.7 Hz, CH3-Ph-3,5), 7.03 (m, 2H, CH3O-Ph-3,5), 6.89 (d, 1H, J = 7.0 Hz, H-6), 3.89 (s, 3H, CH3O), 2.38 (s, 3H, CH3). 13C-NMR (CDCl3) δ 159.7, 146.3, 145.8, 140.2, 138.7, 133.5, 131.7, 131.0, 130.5, 129.0, 128.6, 128.0, 126.3, 124.4, 120.2, 114.5, 113.6, 109.6, 108.4, 98.6, 84.3, 55.4, 21.6. Anal. Calcd for C29H22N2O: C, 84.03; H, 5.35; N, 6.76. Found: C, 84.13; H, 5.34; N, 6.81.
Ethyl 7-(4-methoxyphenyl)-8-(p-tolylethynyl)imidazo[1,2-a]pyridine-2-carboxylate (32). Method H. 8 (100 mg, 0.30 mmol), 4-methoxyphenylboronic acid (65 mg, 0.42 mmol) were used. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 240 °C. 69% yield. 1H-NMR (CDCl3) δ 8.22 (s, 1H, H-3), 8.13 (d, 1H, J = 7.2 Hz, H-5), 7.75 (d, 2H, J = 9 Hz, CH3O-Ph-2,6), 7.41 (d, 2H, J = 8.1 Hz, CH3-Ph-2,6), 7.12 (d, 2H, J = 8.1 Hz, CH3O-Ph-3,5), 7.08–6.98 (m, 3H, H-6, CH3-Ph-3,5), 4.47 (q, 2H, J = 7.2 Hz, CH2), 3.89 (s, 3H, CH3O), 2.35 (s, 3H, CH3), 1.46 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (CDCl3) δ 163.0, 160.0, 145.5, 141.8, 138.9, 137.1, 131.8, 130.6, 130.4, 129.0, 124.8, 119.9, 117.6, 116.3, 113.7, 110.9, 99.6, 83.6, 61.3, 55.4, 21.6, 14.4. Anal. Calcd for C26H22N2O3: C, 76.08; H, 5.40; N, 6.82. Found: C, 76.24; H, 5.29; N, 6.64.
7-(4-Methoxyphenyl)-2-phenylimidazo[1,2-a]pyridine-8-carbonitrile (33). Method I. 11 (100 mg, 0.40 mmol), 4-methoxyphenylboronic acid (84 mg, 0.55 mmol), Na2CO3 (84 mg, 0.79 mmol) and Pd(PPh3)4 (23 mg, 0.02 mmol, 5 mol%) were used. The mixture was heated for 15 min. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 185 °C. 61% yield. 1H-NMR (CDCl3) δ 8.26 (d, 1H, J = 7.0 Hz, H-5), 8.00 (m, 2H, Ph-2,6), 7.88 (s, 1H, H-3), 7.62 (d, 2H, J = 9.0 Hz, CH3O-Ph-2,6), 7.44 (m, 2H, Ph-3,5), 7.35 (m, 1H, Ph-4), 7.02 (d, 2H, J = 9.0 Hz, CH3O-Ph-3,5), 6.90 (d, 1H, J = 7.0 Hz, H-6), 3.86 (s, 3 H, CH3O). 13C-NMR (CDCl3) δ 160.8, 147.5, 144.6, 144.1, 132.7, 130.0, 128.7, 128.5, 128.4, 128.3, 126.3, 115.5, 114.4, 113.5, 108.8, 98.1, 55.4. Anal. Calcd for C21H15N3O: C, 77.52; H, 4.65; N, 12.91. Found: C, 77.68; H, 4.53; N, 12.94.
7-(4-Methylphenyl)-N,2-diphenylimidazo[1,2-a]pyridin-8-amine (34). Method I. 15 (100 mg, 0.53 mmol), p-tolylboronic acid (362 mg, 2.66 mmol), Na2CO3 (112.4 mg, 1.06 mmol) and Pd(PPh3)4 (30.6 mg, 0.03 mmol, 5 mol%) were used. The mixture was heated for 30 min. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (70:30)] to give a white solid. M.p. = 237 °C. 44% yield. 1H-NMR (CDCl3) δ 7.98 (m, 2H, Ph-3,5), 7.87 (m, 2H, H-3, H-5), 7.45 (m, 2H, Ph-2,6), 7.34 (m, 3H, Ph-4, CH3-Ph-2,6), 7.00 (m, 4H, CH3-Ph-3,5, Ph-NH-3,5), 6.88 (d, 1H, J = 7.0 Hz, H-6), 6.72 (m, 3H, Ph-NH-2,4,6), 2.27 (s, 3H, CH3). 13C-NMR (CDCl3) δ 141.8, 136.9, 135.4, 131.3, 130.0, 129.6, 128.9, 128.7, 128.5, 128.1, 127.9, 126.1, 120.7, 118.6, 118.2, 116.8, 108.8, 21.1 (2C not found). Anal. Calcd for C26H21N3: C, 83.17; H, 5.64; N, 11.19. Found: C, 83.34; H, 5.77; N, 11.18.
3.2.8. General Procedures for Double-Coupling Approach
3-[8-(4-Methoxyphenyl)-2-phenylimidazo[1,2-a]pyridin-7-yl]aniline (35). Method J. To a solution of 1a (100 mg, 0.28 mmol) in DME (2 mL) and water (1 mL), were added 3-aminophenylboronic acid (39 mg, 0.28 mmol), Na2CO3 (90 mg, 0.85 mmol) and Pd(PPh3)4 (16 mg, 0.014 mmol). After irradiation under microwaves for 30 min at 95 °C, 4-methoxyphenylboronic acid (60 mg, 0.40 mmol) and Pd(PPh3)4 (16 mg, 0.014 mmol) were added. The mixture was irradiated under microwaves during 30 min at 120 °C. After cooling to room temperature, CH2Cl2 (50 mL) and water (50 mL) were added and the solution was extracted with CH2Cl2 (3 × 50 mL). The combined organic layer was dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residue was purified by flash chromatography (alumina, CH2Cl2) to give a yellow solid. M.p. = 230 °C. 44% yield. 1H-NMR (CDCl3) δ 8.10 (d, 1H, J = 7.0 Hz, H-5), 7.94 (m, 3H, H-3, Ph-3,5), 7.42 (m, 4H, Ph-2,6, CH3O-Ph-2,6), 7.31 (m, 1H, Ph-4), 7.02 (t, 1H, J = 7.7 Hz, H2N-Ph-5), 6.86 (m, 3H, H-6, CH3O-Ph-3,5), 6.55 (m, 3H, H2N-Ph-4,6,2), 3.81 (s, 3H, CH3O), 3.23 (bs, 2H, NH2). 13C-NMR (CDCl3) δ 158.8, 146.1, 146.0, 145.5, 141.0, 135.8, 133.7, 132.6, 130.6, 128.9, 128.5, 127.8, 127.3, 126.2, 123.6, 120.3, 116.4, 115.9, 113.8, 113.2, 108.2, 55.1. Anal. Calcd for C26H21N3O: C, 79.77; H, 5.41; N, 10.73. Found: C, 79.92; H, 5.38; N, 10.63.
2-Phenyl-8-(pyridin-4-yl)-7-(thien-3-yl)imidazo[1,2-a]pyridine (36). Method J. Thien-3-ylboronic acid (36 mg, 0.28 mmol, 1 eq) and pyridin-4-ylboronic acid (56 mg, 0.40 mmol, 1.4 eq) were used. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (99:1)] to give a white solid. M.p. = 273 °C. 43% yield. 1H-NMR (CDCl3) δ 8.62 (dd, 2 H, J = 4.5–1.5 Hz, Pyr-2,6), 8.16 (d, 1H, J = 7.0 Hz, H-5), 7.92 (m, 3 H, H-3, Ph-3,5), 7.49 (dd, 2 H, J = 4.5–1.5 Hz, Pyr-3,5), 7.40 (m, 2 H, Ph-2,6), 7.32 (m, 1 H, Ph-4), 7.21 (dd, 1H, J = 5.0–2.9 Hz, Th-5), 7.09 (dd, 1H, J = 2.9–1.4 Hz, Th-2), 6.97 (d, 1H, J = 7.0 Hz, H-6), 6.76 (dd, 1H, J = 5.0–1.4 Hz, Th-4). 13C-NMR (CDCl3) δ 149.0, 146.9, 144.6, 144.3, 139.1, 133.5, 130.9, 128.6, 128.5, 128.1, 126.2, 125.8, 125.0, 124.6, 124.5, 115.2, 108.5 (1C not found). Anal. Calcd for C22H15N3S: C, 74.76; H, 4.28; N, 11.89. Found: C, 74.89; H, 4.21; N, 11.78.
8-(4-Fluorophenyl)-2-phenyl-7-(p-tolyl)imidazo[1,2-a]pyridine (37). Method K. To a solution of 1d (100 mg, 0.28 mmol) in DME (2 mL) and water (1 mL), were added 4-fluorophenylboronic acid (79 mg, 0.56 mmol), Na2CO3 (90 mg, 0.85 mmol) and Pd(PPh3)4 (16 mg, 0.014 mmol). After irradiation under microwaves for 1 h 30 min at 95 °C, p-tolylboronic acid (77 mg, 0.56 mmol) and Pd(PPh3)4 (16 mg, 0.014 mmol) were added. The mixture was irradiated under microwaves for 30 min at 120 °C. After cooling to room temperature, CH2Cl2 (50 mL) and water (50 mL) were added and the solution was extracted with CH2Cl2 (3 × 50 mL). The organic layer was dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (99:1)] to give a white solid. M.p. = 215 °C. 47% yield. 1H-NMR (CDCl3) δ 8.14 (d, 1H, J = 7.0 Hz, H-5), 7.98–7.90 (m, 3H, H-3, Ph-3,5), 7.50–7.37 (m, 4H, Ph-2,6, F-Ph-3,5), 7.32 (m, 1H, Ph-4), 7.11–6.95 (m, 6H, F-Ph-2,6, CH3-Ph-2,3,5,6), 6.92 (d, 1H, J = 7.0 Hz, H-6), 2.35 (s, 3H, CH3). 13C-NMR (CDCl3) δ 162.1, 146.3, 145.4, 136.9, 136.5, 133.7, 133.2, 131.0, 129.8, 129.6, 128.9, 128.5, 127.9, 126.2, 124.1, 115.9, 115.1, 114.8, 108.3, 21.1.
2-Phenyl-7-(p-tolylethynyl)imidazo[1,2-a]pyridine-8-carbonitrile (38). Method L. To a solution of 1d (100 mg, 0.28 mmol) in DMF (3 mL), was added CuCN (31 mg, 0.34 mmol). The mixture was irradiated under microwaves at 90 °C during 4 h. Then, 4-ethynyltoluene (47 µL, 0.37 mmol), Et3N (198 µL, 1.41 mmol), Pd(PPh3)4 (33 mg, 0.03 mmol), PCy3HBF4 (31 mg, 0.08 mmol) and CuI (11 mg, 0.056 mmol) were added. The mixture was irradiated under microwaves at 130 °C during 1 h. After cooling to room temperature, a solution of NH4OH 10% (50 mL) and EtOAc (50 mL) were added. The organic layer was washed with the solution of NH4OH (3 × 50 mL), dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residues were purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 214 °C. 32% yield. 1H-NMR (CDCl3) δ 8.22 (d, J = 7.0 Hz, 1H, H-5), 8.02 (m, 2H, Ph-2,6), 7.94 (s, 1H, H-3), 7.55 (m, 2H, CH3-Ph-2,6), 7.46 (m, 2H, Ph-3,5), 7.38 (m, 1H, Ph-4), 7.21 (d, 2H, J = 7.9 Hz, CH3-Ph-3,5), 6.95 (d, 1H, J = 7.0 Hz, H-6), 2.41 (s, 3H, CH3). 13C-NMR (CDCl3) δ 148.5, 140.4, 132.4, 132.2, 129.3, 128.8, 128.8, 128.7, 128.0, 127.7, 126.5, 125.2, 118.3, 114.4, 114.2, 109.7, 100.5, 84.8, 21.7. Anal. Calcd for C23H15N3: C, 82.86; H, 4.54; N, 12.60. Found: C, 82.97; H, 4.56; N, 12.68.
7-[4-(4-Fluorophenyl)piperazin-1-yl]-2-phenylimidazo[1,2-a]pyridine-8-carbonitrile (39). Method M. To a solution of 1d (100 mg, 0.28 mmol) in DMF (3 mL), CuCN (31 mg, 0.34 mmol) was added. The mixture was stirred at 90 °C during 4 h under microwave irradiation. Then DIPEA (0.1 mL, 0.57 mmol) and 4-fluorophenylpiperidine (98 mg, 0.57 mmol) were added. The mixture was heated at 130 °C during 1 h under microwaves irradiation. After cooling to room temperature, EtOAc (50 mL) and a solution of NH4OH 10% (50 mL) were added. The solution was extracted with EtOAc (3 × 50 mL), dried with anhydrous MgSO4 and evaporated under reduced pressure. The crude residue was purified by flash chromatography [silica, cyclohexane/EtOAc (7:3)]. M.p. = 224 °C. 67% yield. 1H-NMR (CDCl3) δ 8.06 (d, 1H, J = 7.5 Hz, H-5), 7.97 (m, 2H, Ph-2,6), 7.71 (s, 1H, H-3), 7.42 (m, 2H, Ph-3,5), 7.33 (m, 1H, Ph-4), 7.06–6.89 (m, 4H, F-Ph), 6.55 (d, 1H, J = 7.5 Hz, H-6), 3.79–3.69 (m, 4H, pip), 3.36–3.27 (m, 4H, pip). 13C-NMR (CDCl3) δ 157.7, 154.0, 147.2, 146.6, 145.4, 133.0, 129.1, 128.7, 128.2, 126.2, 118.5, 116.0, 115.8, 107.6, 105.3, 85.3, 50.5, 50.2. Anal. Calcd for C24H20FN5: C, 72.53; H, 5.07; N, 17.62. Found: C, 72.49; H, 5.01; N, 18.73.
Ethyl 8-cyano-7-(p-tolylethynyl)imidazo[1,2-a]pyridine-2-carboxylate (40). Method L. 1e (100 mg, 0.29 mmol) was used as starting material. M.p. = 93 °C. 43% yield. 1H-NMR (CDCl3) δ 8.22 (s, 1H, H-3), 8.12 (d, 1H, J = 7.2 Hz, H-5), 7.54 (d, 2H, J = 8.0 Hz, CH3-Ph-2,6), 7.17 (d, 2H, J = 8.0 Hz, CH3-Ph-3,5), 6.96 (d, 1H, J = 7.2 Hz, H-6), 4.44 (q, 2H, J = 7.2 Hz, CH2), 2.38 (s, 3H, CH3), 1.42 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (CDCl3) δ 162.6, 144.6, 139.6, 137.3, 135.3, 132.0, 129.0, 125.3, 119.1, 118.1, 116.0, 113.7, 102.9, 80.8, 61.4, 21.6, 14.3 (1 C not found). Anal. Calcd for C20H15N3O2: C, 72.94; H, 4.59; N, 12.76. Found: C, 73.27; H, 4.68; N, 12.81.
Ethyl 8-cyano-7-[4-(4-fluorophenyl)piperazin-1-yl]imidazo[1,2-a]pyridine-2-carboxylate (41). Method M. 1e (100 mg, 0.29 mmol) was used as starting material. M.p. = 193 °C. 54% yield. 1H-NMR (CDCl3) δ 8.20–7.91 (m, 2H, H-3, H-5), 7.07–6.88 (m, 4H, F-Ph), 6.67 (d, 1H, J = 7.0 Hz, H-6), 4.43 (q, 2H, J = 7.0 Hz, CH2), 3.83 (bs, 4H, pip), 3.33 (bs, 4H, H-pip), 1.42 (t, 3H, J = 7.0 Hz, CH3). 13C-NMR (CDCl3) δ 162.8, 157.9, 154.4, 145.3, 137.5, 132.5, 129.5, 123.6, 118.7, 117.2, 115.9, 107.4, 84.5, 61.3, 50.7, 49.9, 14.3. Anal. Calcd for C21H20FN5O2: C, 64.11; H, 5.12; N, 17.80. Found: C, 64.49; H, 5.08; N, 17.94.























{kind=link}
{kind=link}
{kind=link}
{kind=link}











