3.2. Synthesis
Compounds were prepared according to the general method reported by Kieczykowski
et al. [
20], exemplified by the preparation of
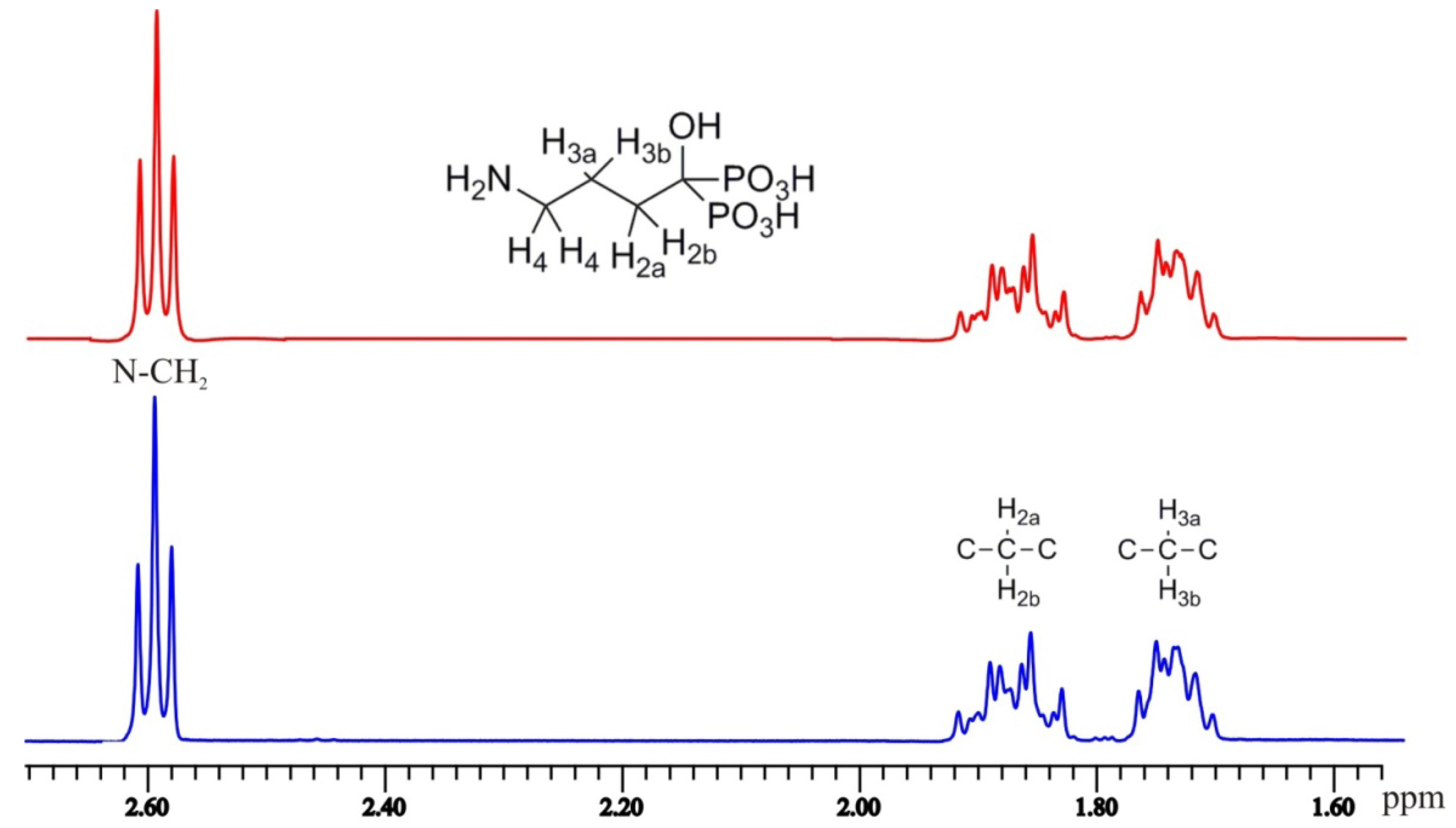
(4-amino-1-hydroxybutylidene)bisphosphonic acid (
2; CAS Registry Number 66376-36-1): 4-aminobutyric acid (2.5 g, 24.2 mmol), phosphorus acid (2.0 g, 24.2 mmol) and methanesulfonic acid (11 mL) were put in a flask with a reflux condenser and a CaCl
2-tube and the mixture was heated to 65 °C. PCl
3 (4.3 mL, 48.4 mmol) was added dropwise and the mixture was maintained at 65–70 °C for 18 h (generally 16–20 h). Water (25 mL) was added to the cooled mixture and the solution was refluxed for 5 h. Some of the water was evaporated, EtOH was added and the product was allowed to crystallize at 8 °C. The product was collected by filtration and washed with EtOH yielding 5.1 g of
2 (85%) as white solid, yield 85%;
1H-NMR (D
2O/NaOD, 500.1 MHz): δ 2.59 (2H, t,
J = 7.0 Hz), 1.93–1.82 (2H, m), 1.78–1.69 (2H, m);
13C-NMR (D
2O/NaOD, 125.8 MHz): 79.3 (t,
1JCP = 134.5 Hz), 44.6, 36.2, 30.2 (t,
2JCP = 5.0 Hz);
31P-NMR (D
2O/NaOD, 202.5 MHz) δ 18.89 (s); IR (KBr) 3275, 2262, 1630, 1498, 1472, 1400, 1374, 1319, 1212, 1165, 1130, 1056, 994, 934, 825, 751, 725, 665 cm
−1. Anal. Calcd. for C
4H
13NO
7P
2: C, 19.29; H, 5.26; N, 5.62; P, 24.9. Found: C, 19.19; H, 5.09; N, 5.51; P, 24.5.
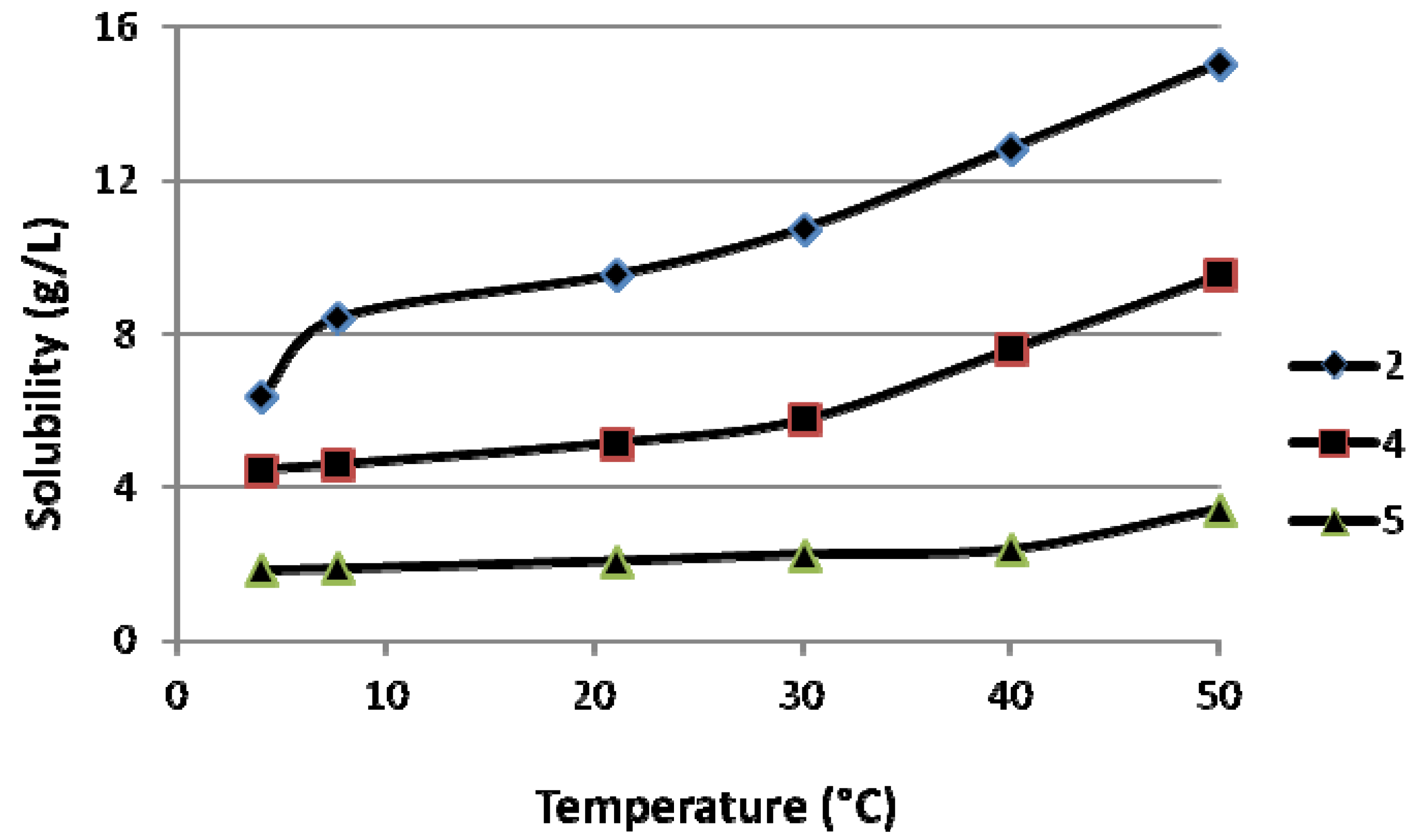
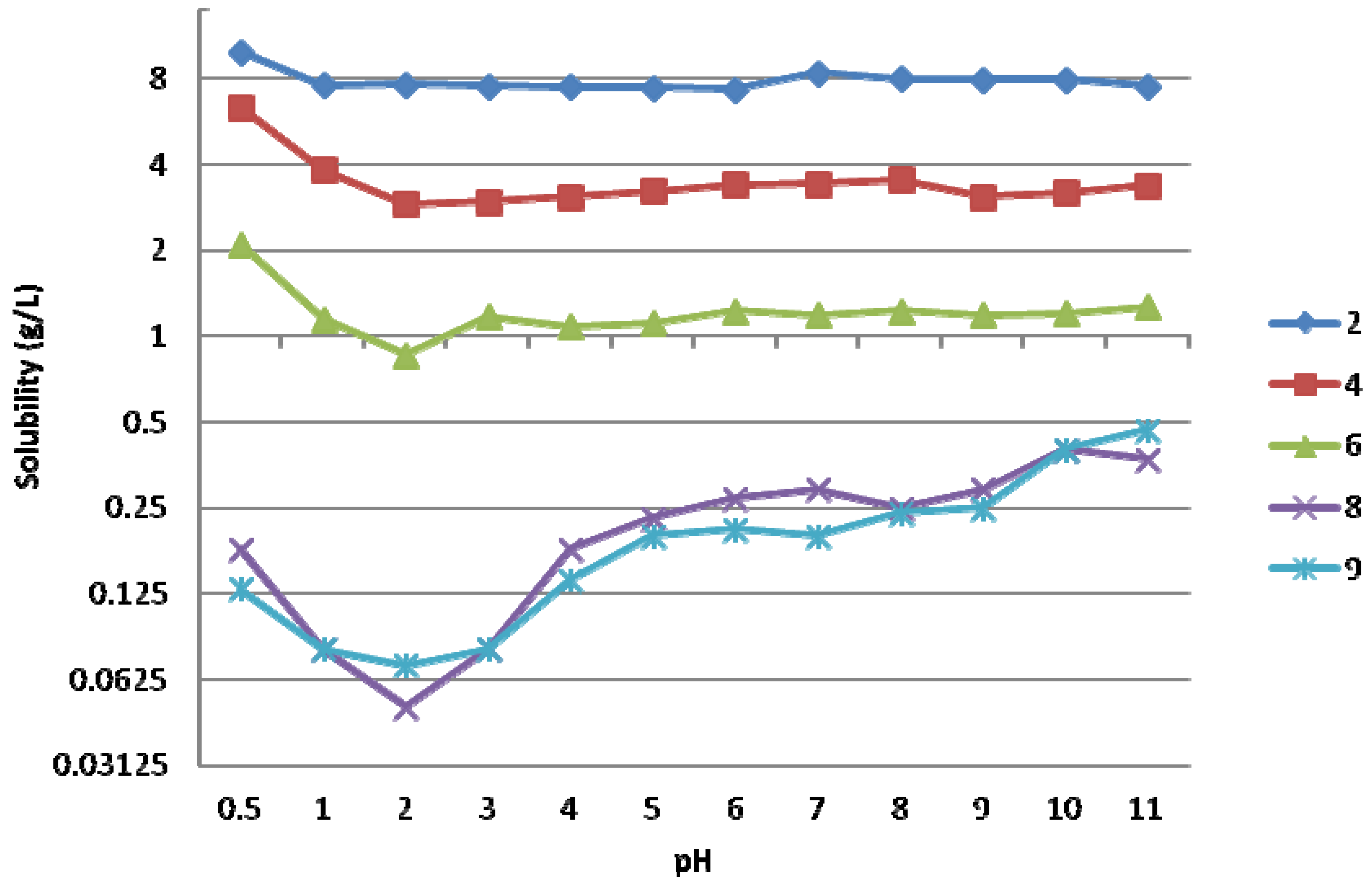
(3-Amino-1-hydroxypropylidene)bisphosphonic acid (1; CAS Registry Number 40391-99-9). The pH was adjusted to 4.5 to obtain the monosodium salt, yield 22%; 1H-NMR (D2O/NaOD, 500.1 MHz) δ 2.97–2.90 (2H, t, J = 7.8 Hz), 2.10–1.99 (2H, m); 13C-NMR (D2O/NaOD, 125.8 MHz): 78.7 (t, 1JCP = 133.8 Hz), 40.9, 40.0 (t, 2JCP = 5.7 Hz); 31P-NMR (D2O/NaOD, 202.5 MHz): δ 18.46 (s); IR (KBr) 3354, 3188, 2792, 2364, 1719, 1615, 1528, 1409, 1343, 1327, 1282, 1173, 1045, 984, 937, 895, 855, 775, 715, 683, 614 cm−1. Anal. Calcd. for C3H10NO7P2Na: C, 14.02; H, 3.92; N, 5.45; P, 24.1. Found: C, 14.48; H, 4.41; N, 5.64; P, 23.3.
(5-Amino-1-hydroxypentylidene)bisphosphonic acid (3; CAS Registry Number 89732-96-7). Yield 86%; 1H-NMR (D2O/NaOD, 500.1 MHz): δ 2.70 (2H, t, J = 7.0 Hz), 1.94–1.82 (2H, m), 1.66–1.57 (2H, m), 1.51–1.43 (2H, m); 13C-NMR (D2O/NaOD, 125.8 MHz): 79.2 (t, 1JCP = 133.5 Hz), 42.9, 38.1, 34.3, 24.1 (t, 2JCP = 5.3 Hz); 31P-NMR (D2O/NaOD, 202.5 MHz): δ 18.92 (s); IR (KBr) 3209, 2959, 2331, 1617, 1503, 1468, 1452, 1219–939, 824, 663 cm−1. Anal. Calcd. for C5H15NO7P2: C, 22.82; H, 5.75; N, 5.32; P, 23.5. Found: C, 22.59; H, 5.82; N, 5.30; P, 23.5.
(6-Amino-1-hydroxyhexylidene)bisphosphonic acid (4; CAS Registry Number 79778-41-9). Yield 88%; 1H-NMR (D2O/NaOD, 500.1 MHz): δ 2.61 (2H, t, J = 7.0 Hz), 1.94–1.82 (2H, m), 1.62–1.53 (2H, m), 1.51–1.43 (2H, m), 1.34–1.26 (2H, m); 13C-NMR (D2O/NaOD, 125.8 MHz): 79.6 (t, 1JCP = 134.4 Hz), 43.5, 39.0, 34.7, 30.4, 26.9 (t, 2JCP = 5.2 Hz); 31P-NMR (D2O/NaOD, 202.5 MHz): δ 19.12 (s); IR (KBr) 3178, 2953, 2296, 1648, 1582, 1496, 1476, 1441, 1112, 1034, 997, 939, 905, 824, 777, 724, 657 cm−1. Anal. Calcd. for C6H17NO7P2: C, 26.00; H, 6.18; N, 5.05; P, 22.4. Found: C, 25.97; H, 6.17; N, 4.97; P, 22.2.
(8-Amino-1-hydroxyoctylidene)bisphosphonic acid (5; CAS Registry Number: 144050-49-7). Yield 75%; 1H-NMR (D2O/NaOD, 500.1 MHz): δ 2.62 (2H, t, J = 7.0 Hz), 1.93–1.81 (2H, m), 1.61–1.52 (2H, m), 1.50–1.41 (2H, m), 1.39–1.24 (6H, m); 13C-NMR (D2O/NaOD, 125.8 MHz): 78.9 (t, 1JCP = 132.4 Hz), 43.2, 38.4, 33.6, 33.0, 31.5, 29.0, 27.0 (t, 2JCP = 5.6 Hz); 31P-NMR (D2O/NaOD, 202.5 MHz): δ 19.05 (s); IR (KBr) 3327, 3150, 2901, 2848, 2276, 1650, 1620, 1541, 1471, 1398, 1334, 1300, 1224, 1151, 1079, 967, 842, 813, 767, 729, 647 cm−1. Anal. Calcd. for C8H21NO7P2: C, 31.48; H, 6.93; N, 4.59; P, 20.3. Found: C, 31.44; H, 6.98; N, 4.69; P, 20.5.
(9-Amino-1-hydroxynonylidene)bisphosphonic acid (6; CAS Registry Number: 144050-48-6). As an exception to the general method 6 was refluxed for 5 h in 2 M HCl instead of water and pH was adjusted to 7 to obtain the disodium salt. The product was recrystallized from water-ethanol, yield 63%; 1H-NMR (D2O/NaOD, 500.1 MHz): δ 2.59 (2H, t, J = 7.0 Hz), 1.93–1.79 (2H, m), 1.59–1.50 (2H, m), 1.50–1.38 (2H, m), 1.37–1.21 (8H, m); 13C-NMR (D2O/NaOD, 125.8 MHz): 79.6 (t, 1JCP = 134.6 Hz), 43.4, 39.0, 34.7, 33.2, 31.9, 31.7, 28.9, 27.2 (t, 2JCP = 5.2 Hz); 31P-NMR (D2O/NaOD, 202.5 MHz): δ 19.30 (s); IR (KBr) 3195, 2910, 2850, 1637, 1558, 1475, 1168, 1040, 906, 720, 672 cm−1. Anal. Calcd. for C9H23NO7P2 H2O: C, 32.05; H, 7.47; N, 4.15; P, 18.4. Found: C, 31.55; H, 6.88; N, 3.92; P, 18.6.
(10-Amino-1-hydroxydecylidene)bisphosphonic acid (7). As an exception to the general method 7 was refluxed for 5 h in 2 M HCl instead of water and the product was washed with water, yield 66%; 1H-NMR (D2O/NaOD, 500.1 MHz): δ 2.58 (2H, t, J = 7.0 Hz), 1.93–1.80 (2H, m), 1.60–1.50 (2H, m), 1.47–1.38 (2H, m), 1.37–1.23 (10H, m); 13C-NMR (D2O/NaOD, 125.8 MHz): 79.7 (t, 1JCP = 134.5 Hz), 43.4, 39.1, 34.7, 33.3, 31.9, 31.9, 31.5, 28.9, 27.2 (t, 2JCP = 5.1 Hz); 31P-NMR (D2O/NaOD, 202.5 MHz): δ 19.25 (s); IR (KBr) 3327, 2913, 2849, 2274, 1648, 1542, 1472, 1338, 1228, 1082, 970, 921, 810, 772, 742, 728, 647 cm−1. Anal. Calcd. for C10H25NO7P2: C, 36.04; H, 7.56; N, 4.20; P, 18.6. Found: C, 35.87; H, 7.65; N, 3.94; P, 18.2.
(11-Amino-1-hydroxyundecylidene)bisphosphonic acid (8; CAS Registry Number: 97815-71-9). Yield 100%; 1H-NMR (D2O/NaOD, 500.1 MHz): δ 2.58 (2H, t, J = 7.0), 1.92–1.80 (2H, m), 1.58–1.49 (2H, m), 1.46–1.37 (2H, m), 1.36–1.23 (12H, m); 13C-NMR (D2O/NaOD, 125.8 MHz): 79.6 (t, 1JCP = 134.4 Hz), 43.4, 39.1, 34.6, 33.3, 31.9, 31.9, 31.6, 31.4, 28.9, 27.2 (t, 2JCP = 5.2 Hz); 31P-NMR (D2O/NaOD, 202.5 MHz): δ 19.33 (s); IR (KBr) 3566, 3198, 2914, 2847, 2664, 2340, 1635, 1521, 1475, 1380, 1300, 1206, 1155, 1097, 1042, 989–930, 722, 668 cm−1. Anal. Calcd. for C11H27NO7P2 H2O: C, 36.17; H, 8.00; N, 3.83; P, 17.0. Found: C, 36.20; H, 8.03; N, 3.73; P, 17.2.
(12-Amino-1-hydroxydodecylidene)bisphosphonic acid (9; CAS Registry Number: 724457-78-7). The product was washed with hot water, yield 85%; 1H-NMR (D2O/NaOD, 500.1 MHz): δ 2.58 (2H, t, J = 7.0), 1.92–1.80 (2H, m), 1.58–1.49 (2H, m), 1.46–1.37 (2H, m), 1.36–1.23 (14H, m); 13C-NMR (D2O/NaOD, 125.8 MHz): 79.6 (t, 1JCP = 134.4 Hz), 43.4, 39.1, 34.6, 33.3, 31.9 (2C), 31.6, 31.5, 31.4, 28.9, 27.2 (t, 2JCP = 5.2 Hz); 31P-NMR (D2O/NaOD, 202.5 MHz): δ 19.35 (s); IR (KBr) 3326, 2916, 2849, 2360, 2342, 1647, 1542, 1472, 1333, 1227, 1067, 969, 935, 808, 773, 749, 730, 668, 648 cm−1. Anal. Calcd. for C12H29NO7P2: C, 39.89; H, 8.09; N, 3.88; P, 17.1. Found: C, 40.31; H, 8.18; N, 3.85; P, 17.0.
(16-Amino-1-hydroxyhexadecylidene)bisphosphonic acid (10). As an exception to the general method 10 was stirred for two days at 70 °C and refluxed over night after addition of water. The product was washed with methanol and hot water, yield 78%, IR 2918, 2850, 1647, 1542, 1471, 1333, 1230, 1055, 970, 916, 804, 777, 757, 728, 667 cm−1. Anal. Calcd. for C16H37NO7P2: C, 46.04; H, 8.93; N, 3.36; P, 14.8. Found: C, 47.41; H, 9.08; N, 3.46; P, 14.5.



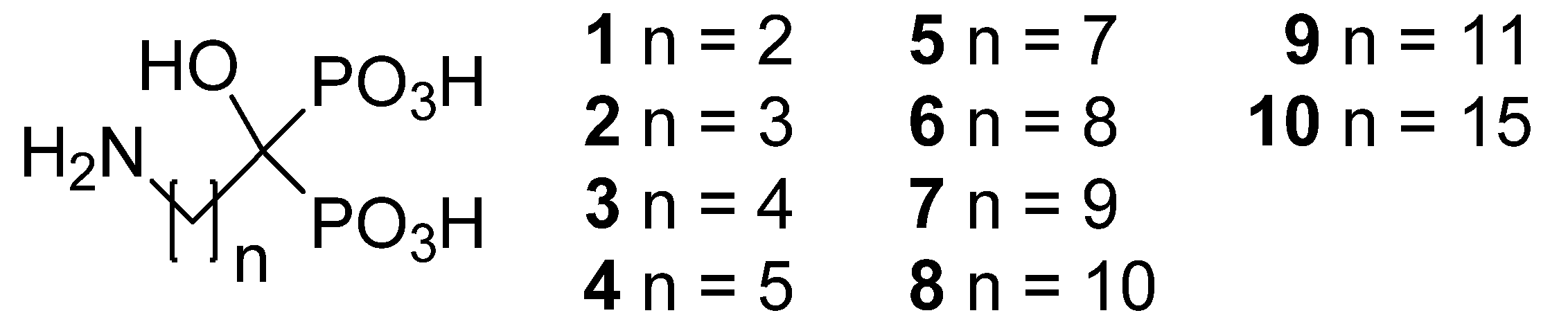{kind=link}
{kind=link}
{kind=link}
{kind=link}


