Solid Phase versus Solution Phase Synthesis of Heterocyclic Macrocycles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
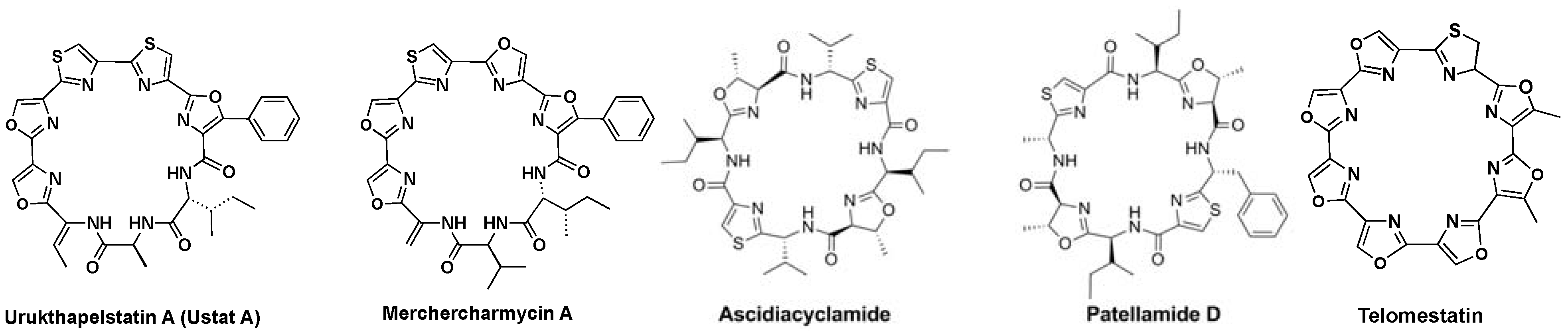
:1. Introduction
2. Results and Discussion
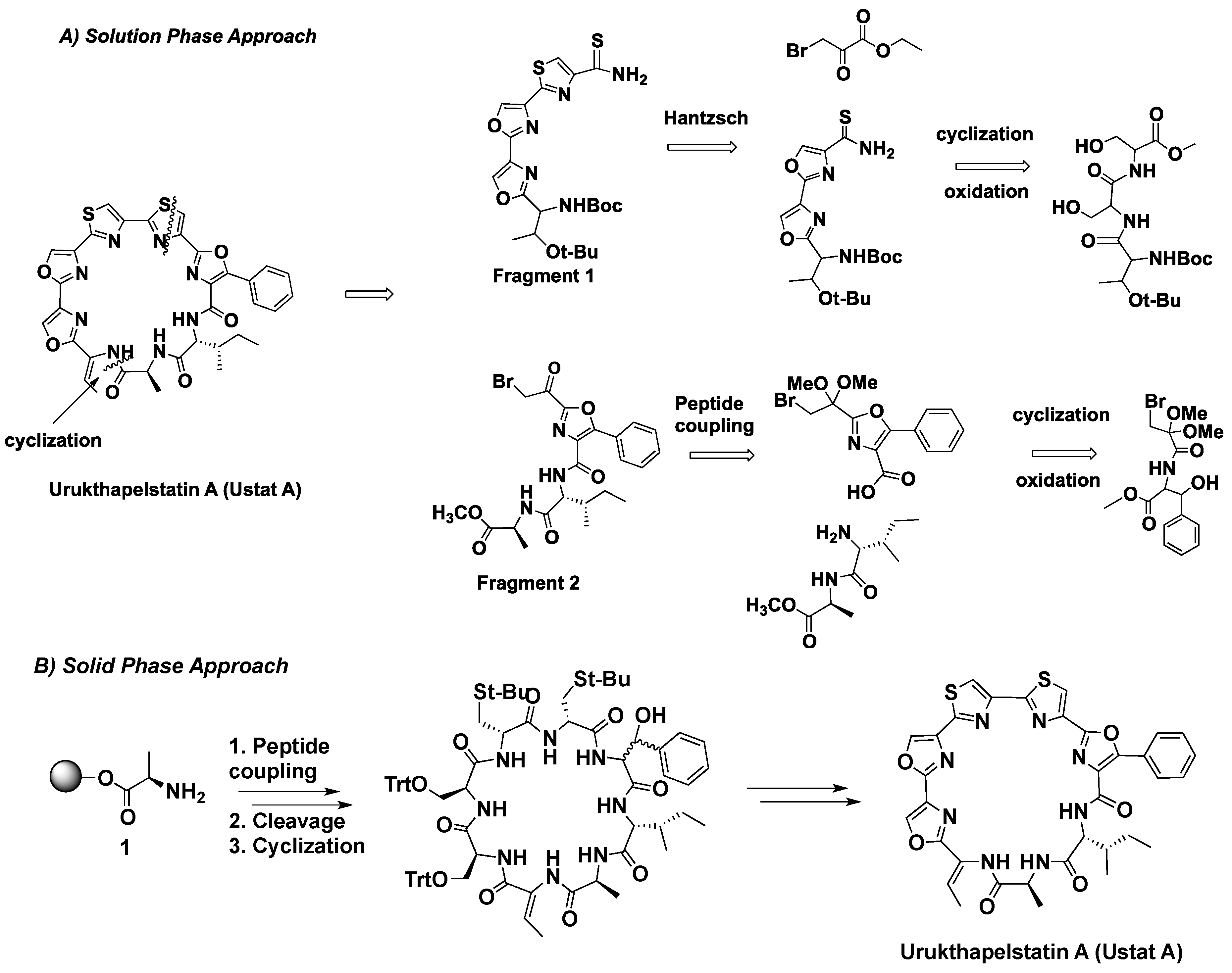
2.1. Synthetic Approach
2.2. Solution Phase Synthesis
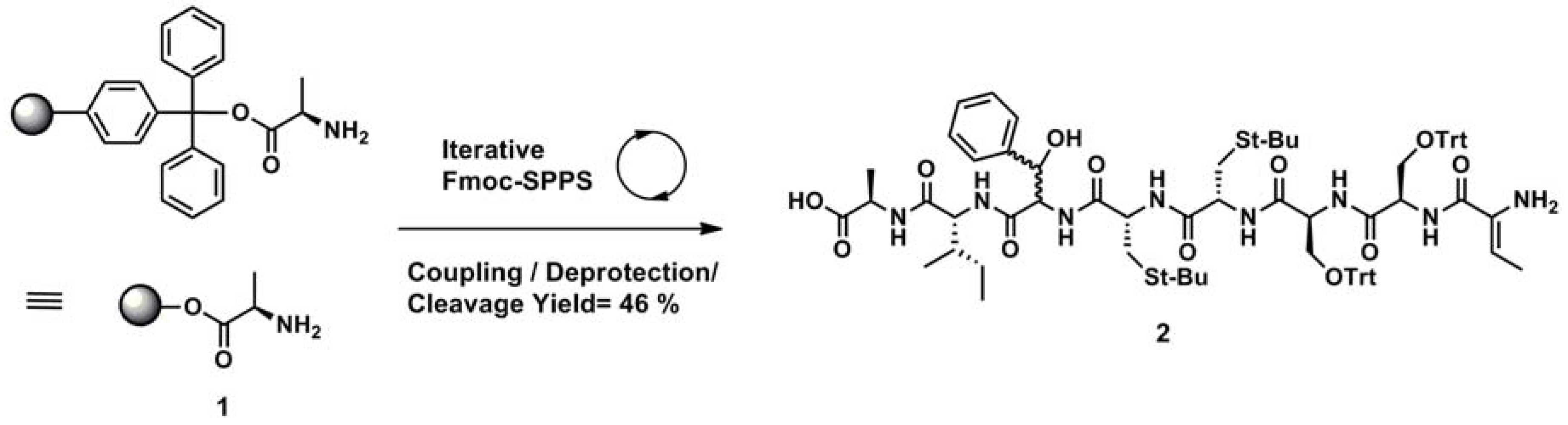
2.3. Solid Phase Synthesis
3. Experimental
3.1. Materials
3.2. Synthesis
3.2.1. General Procedure for Solid Couplings
3.2.2. Solid Phase Synthesis of Linear Precursor 2
3.2.3. Synthesis of the Cyclized Compound 3
3.2.4. Synthesis of Oxazole-based Macrocycle 4
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Jarvis, L.M. Breakthroughs in manufacturing are making large-scale synthesis of peptides a viable proposition. C E News 2006, 84, 23–25. [Google Scholar]
- Chatterjee, J.; Gilon, C.; Hoffman, A.; Kessler, H. N-Methylation of peptides: A new perspective in medicinal chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Danho, W.; Swistok, J.; Khan, W.; Chu, X.; Cheung, A.; Fry, D.; Sun, H.; Kurylko, G.; Rumennik, L.; Cefalu, J.; et al. Opportunities and challenges of developing peptide drugs in the pharmaceutical industry. In Peptides for Youth, Proceedings of the 20th American Peptide Symposium, Montreal, Canada, 23–28 June 2007; Valle, S.D., Escher, E., Lubell, W.D., Eds.; Springer: New York, NY, USA, 2009; Volume 611, pp. 467–469. [Google Scholar]
- Fletcher, J.M.; Hughes, R.A. Modified low molecular weight cyclic peptides as mimetics of BDNF with improved potency, proteolytic stability and transmembrane. Bioorg. Med. Chem. 2009, 17, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides, the ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef] [PubMed]
- Fairlie, D.P.; Abbenante, G.; March, D.R. Macrocyclic peptidomimetics—Forcing peptides into bioactive conformations. Curr. Med. Chem. 1995, 2, 654–686. [Google Scholar]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Loffet, A. Peptides as Drugs: Is there a Market? Eur. Pept. Soc. 2002, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Starzl, T.E.; Klintmalm, G.B.; Porter, K.A.; Iwatsuki, S.; Schroter, G.P. Clinical trials of cyclosporin A. N. Engl. J. Med. 1981, 305, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Chieze, S.; Delbaldo, C.; Ady-Vago, N.; Guzman, C.; Lopez-Lazaro, L.; Lozahic, S.; Jimeno, J.; Pico, F.; Armand, J.P.; et al. Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 7871–7880. [Google Scholar] [CrossRef] [PubMed]
- Maroun, J.A.; Belanger, K.; Seymour, L.; Matthews, S.; Roach, J.; Dionne, J.; Soulieres, D.; Stewart, D.; Goel, R.; Charpentier, D.; et al. Phase I study of Aplidine in a daily35 one-hour infusion every 3 weeks in patients with solid tumors refractory to standard therapy. A National Cancer Institute of Canada Clinical Trials Group study: NCIC CTG IND 115. Ann. Oncol. 2006, 17, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Raymond, E.; Faivre, S. Aplidine: A paradigm of how to handle the activity and toxicity of a novel marine anticancer poison. Curr. Pharm. Des. 2007, 13, 3427–3429. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kanoh, K.; Imanaka, H.; Adachi, K.; Nishizawa, M.; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenes YM11-542 II physcico-chemical properties and structural elucidation. J. Antibiot. 2007, 60, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kanoh, K.; Yamori, T.; Kasai, H.; Katsuta, A.; Adachi, K.; Shin-ya, K.; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenes YM11-542 I fermentation, isolation, and biological properties. J. Antibiot. 2007, 60, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Irschik, H.; Reichenbach, H.; Hofle, G.; Jansen, R. The thuggacins, novel antibacterial macrolides from sorangium cellulosum acting against selected gram-positive bacteria. J. Antibiot. 2007, 60, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Riego, E.; Hernández, D.; Albericio, F.; Álvarez, M. Directly linked polyazoles: Important moieties in natural products. Synthesis 2005, 2005, 1907–1922. [Google Scholar] [CrossRef]
- Ying, Y.; Taori, K.; Kim, H.; Hong, J.; Luesch, H. Total synthesis and molecular target of Largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Malet, L.; Canedo, L.; Maria, C.C.; Fernando, R.J. New cytotoxic depsipeptides. WO 2005/000880 A2, 2005. [Google Scholar]
- Kanoh, K.; Matsuo, Y.; Adachi, K.; Imagawa, H.; Nishizawa, M.; Shizuri, Y. Mechercharmycins A and B, cytotoxic substances from marine-derived thermoactinomyces sp. YM3-251. J. Antibiot. 2005, 58, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Inoue, M.; Hamada, Y.; Kato, S.; Shioiri, T. X-ray crystal structure of ascidiacyclamide, a cytotoxic cyclic peptide from ascidian. J. Chem. Soc. Chem. Commun. 1987, 370–371. [Google Scholar] [CrossRef]
- Asano, A.; Minoura, K.; Yamada, T.; Numata, A.; Ishida, T.; Doi, M.; Katsuya, Y.; Mezaki, Y.; Sasaki, M.; Taniguchi, T.; et al. Effect of asymmetric modification on the conformation of ascidiacyclamide analogs. J. Pept. Res. 2002, 60, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Jacobs, R.S. A marine natural product, patellamide D, reverses multidrug resistance in a human leukemic cell line. Cancer Lett. 1993, 71, 97–102. [Google Scholar] [CrossRef]
- Miyazaki, T.; Pan, Y.; Joshi, K.; Purohit, D.; Hu, B.; Demir, H.; Mazumder, S.; Okabe, S.; Yamori, T.; Viapiano, M.S.; et al. Telomestatin impairs glioma stem cell survival and growth through the disruption of telomeric G-quadruplex and inhibition of the proto-oncogene, c-Myb. Clin. Cancer Res. 2012, 18, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Telomestatin. Available online: http://www.clinicaltrials.gov (accessed on 1 October 2012).
- Hernandez, D.; Riego, E.; Albericio, F.; Alvarez, M. Synthesis of natural product derivatives containing 2,4-concatenated oxazoles. Eur. J. Org. Chem. 2008, 3389–3396. [Google Scholar] [CrossRef]
- Hernandez, D.; Vilar, G.; Riego, E.; Canedo, L.M.; Cuevas, C.; Albericio, F.; Alvarez, M. Synthesis of IB-01211, a cyclic peptide containing 2,4-concentrated thia- and oxazoles, vix Hantzsch macrocyclization. Org. Lett. 2007, 9, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Shin-ya, K.; Wierzba, K.; Matsuo, K.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. Synthesis of Telomerastatin. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Riego, E.; Francesch, A.; Cuevas, C.; Albericio, F.; Alvarez, M. Preparation of penta-azole containing cyclopeptides: challenges in macrocyclization. Tetrahedron 2007, 63, 9862–9870. [Google Scholar] [CrossRef]
- Ward, D.E.; Gai, Y.; Lazny, R.; Pedras, M.S.C. Probing Host-selective Phytoxicity: Synthesis of Destruxin B and several natural analogues. J. Org. Chem. 2001, 66, 7832–7840. [Google Scholar] [CrossRef] [PubMed]
- Tarver, J.; Pfizenmayer, A.J.; Joullié, M.M. Total synthesis of comformationally constrained Didemnin B analogues. Replacement of N,O-dimethyltyrosine with L-1,2,3,4-tetrahydroisoquinoline and L-1,2,3,4-tetrahydro-7-methoxyisoquinoline. J. Org. Chem. 2001, 66, 7575–7587. [Google Scholar] [CrossRef] [PubMed]
- Li, W.R.; Ewing, W.R.; Harris, B.D.; Joullié, M.M. Total synthesis and structural investigations of didemnins A, B, and C. J. Am. Chem. Soc. 1990, 112, 7659–7672. [Google Scholar] [CrossRef]
- Mayer, S.C.; Ramanjulu, J.; Vera, M.D.; Pfizenmayer, A.; Joullié, M.M. Synthesis of new didemnin B analogs for investigations of structure/biological activity relationships. J. Org. Chem. 1994, 59, 5192–5205. [Google Scholar] [CrossRef]
- Vera, M.D.; Pfizenmayer, A.J.; Ding, X.; Ahuja, D.; Toogood, P.L.; Joullié, M.M. Synthesis and biological evaluation of didemnin photoaffinity analogues. Bioorg. Med. Chem. Lett. 2001, 11, 1871–1874. [Google Scholar] [CrossRef]
- Vera, M.D.; Pfizenmayer, A.J.; Ding, X.; Xiao, D.; Joullié, M.M. [Lys3]didemnins as potential affinity ligands. Bioorg. Med. Chem. Lett. 2001, 11, 13–16. [Google Scholar] [CrossRef]
- Xiao, D.; Vera, M.D.; Liang, B.; Joullié, M.M. Total synthesis of a conformationally constrained didemnin B analog. J. Org. Chem. 2001, 66, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Ramanjulu, J.; Ding, X.; Li, W.R.; Joullié, M.M. Synthesis of a reduced ring analog of Didemnin B. J. Org. Chem. 1997, 62, 4961–4969. [Google Scholar] [CrossRef]
- Kopp, F.; Stratton, C.F.; Akella, L.B.; Tan, D.S. A diversity-oriented synthesis approach to macrocycles via oxidative ring expansion. Nat. Chem. Biol. 2012, 8, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.; Kilner, C.A.; Wilson, A.J. Expedient synthesis of benzene tricarboxamide macrocycles derived from p-aminobenzoic acid. Tetrahedron Lett. 2010, 51, 1361–1363. [Google Scholar] [CrossRef]
- Kim, S.J.; Lin, C.-C.; Pan, C.-M.; Rananaware, D.P.; Ramsey, D.M.; McAlpine, S.R. A structure-activity relationship study of compounds containing sequential oxazoles and thiazoles. Med. Chem. Commun. 2012, in press. [Google Scholar]
- Carroll, C.L.; Johnston, J.V.C.; Kekec, A.; Brown, J.D.; Parry, E.; Cajica, J.; Medina, I.; Cook, K.M.; Corral, R.; Pan, P.-S.; et al. Synthesis and cytotoxicity of novel Sansalvamide A derivatives. Org. Lett. 2005, 7, 3481–3484. [Google Scholar] [CrossRef] [PubMed]
- Styers, T.J.; Kekec, A.; Rodriguez, R.A.; Brown, J.D.; Cajica, J.; Pan, P.-S.; Parry, E.; Carroll, C.L.; Medina, I.; Corral, R.; et al. Synthesis of Sansalvamide A derivatives and their cytotoxicity in the colon cancer cell line HT-29. Bioorg. Med. Chem. 2006, 14, 5625–5631. [Google Scholar] [CrossRef] [PubMed]
- Styers, T.J.; Rodriguez, R.A.; Pan, P.-S.; McAlpine, S.R. High-yielding macrocyclization conditions used in the synthesis of novel Sansalvamide A derivatives. Tetrahedron Lett. 2006, 47, 515–517. [Google Scholar] [CrossRef]
- Rodriguez, R.A.; Pan, P.-S.; Pan, C.-M.; Ravula, S.; Lapera, S.A.; Singh, E.K.; Styers, T.J.; Brown, J.D.; Cajica, J.; Parry, E.; et al. Synthesis of second generation Sansalvamide A derivatives: Novel templates as potent anti-tumor agents. J. Org. Chem. 2007, 72, 1980–2002. [Google Scholar] [CrossRef] [PubMed]
- Singh, E.K.; Ravula, S.; Pan, C.-M.; Pan, P.S.; Vasko, R.C.; Lapera, S.A.; Weerasinghe, S.V.W.; Pflum, M.K.H.; McAlpine, S.R. Synthesis and biological evaluation of histone deacetylase inhibitors that are based on FR235222: A cyclic tetrapeptide scaffold. Bioorg. Med. Chem. Lett. 2008, 18, 2549–2554. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Styers, T.J.; Rodriguez, R.A.; Pan, P.-S.; Vasko, R.C.; McAlpine, S.R. Synthesis and cytotoxicity of a new class of potent decapeptide macrocycles. Org. Lett. 2008, 10, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Singh, E.K.; Nazarova, L.A.; Lapera, S.A.; Alexander, L.D.; McAlpine, S.R. Histone deacetylace inhibitors: Synthesis of cyclic tetrapeptides and their triazole analogs. Tetrahedron Lett. 2010, 51, 4357–4360. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Singh, E.K.; Wahyudi, H.; Alexander, L.D.; Kunicki, J.; Nazarova, L.A.; Fairweather, K.A.; Giltrap, A.M.; Jolliffe, K.A.; McAlpine, S.R. Synthesis of Sansalvamide A peptiodmimetics: Trizaole, oxazole, thiazole, and pseudoproline containing compounds. Tetrahedron 2012, 68, 1029–1051. [Google Scholar] [CrossRef] [PubMed]
- Singh, E.; Ramsey, D.M.; McAlpine, S.R. Total synthesis of natural product trans,trans- Sanguinamide B and its structurally related conformational analogs. Org. Lett. 2012, 14, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Alexander, L.D.; Sellers, R.P.; Davis, M.E.; Ardi, V.C.; Johnson, V.A.; Vasko, R.C.; McAlpine, S.R. Evaluation of Di-sansalvmide A derivatives: Synthesis, structure-activity relationship, and mechanism of action. J. Med. Chem. 2009, 52, 7927–7930. [Google Scholar] [CrossRef] [PubMed]
- Sellers, R.P.; Alexander, L.D.; Johnson, V.A.; Lin, C.-C.; Savage, J.; Corral, R.; Moss, J.; Slugocki, T.S.; Singh, E.K.; Davis, M.R.; et al. A third generation of Sansalvamide A derivatives: Design and synthesis of Hsp90 Inhibitors. Bioorg. Med. Chem. 2010, 18, 6822–6856. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, D.M.; McConnell, J.R.; Alexander, L.D.; Tanaka, K.W.; Vera, C.M.; Mcalpine, S.R. A new Hsp90 inhibitorthat exhibits a novel biological profile. Bioorg. Med. Chem. Lett. 2012, 22, 3287–3290. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.-M.; Lin, C.-C.; Kim, S.J.; Sellers, R.P.; McAlpine, S.R. Progress toward the synthesis of Urukthapelstatin A and two analogues. Tetrahedron Lett. 2012, 53, 4065–4069. [Google Scholar] [CrossRef] [PubMed]
- Joullié, M.M.; Lassen, K.M. Evolution of amide bond formation. ARKIVOC 2010, 8, 189–250. [Google Scholar]
- Barlos, K.; Gatos, D.; Koutsogianni, S. Fmoc/Trt-amino acids: Comparison to Fmoc/tBu-amino acids in peptide synthesis. J. Pept. Res. 1998, 51, 194–200. [Google Scholar] [CrossRef] [PubMed]
- All AA’s were commercially available from Chem-impex, polypeptide, and GL Biochem.
Sample Availability: Samples of the compounds are available from the authors. |




© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, S.J.; McAlpine, S.R. Solid Phase versus Solution Phase Synthesis of Heterocyclic Macrocycles. Molecules 2013, 18, 1111-1121. https://doi.org/10.3390/molecules18011111
Kim SJ, McAlpine SR. Solid Phase versus Solution Phase Synthesis of Heterocyclic Macrocycles. Molecules. 2013; 18(1):1111-1121. https://doi.org/10.3390/molecules18011111
Chicago/Turabian StyleKim, Seong Jong, and Shelli R. McAlpine. 2013. "Solid Phase versus Solution Phase Synthesis of Heterocyclic Macrocycles" Molecules 18, no. 1: 1111-1121. https://doi.org/10.3390/molecules18011111
APA StyleKim, S. J., & McAlpine, S. R. (2013). Solid Phase versus Solution Phase Synthesis of Heterocyclic Macrocycles. Molecules, 18(1), 1111-1121. https://doi.org/10.3390/molecules18011111