Access to Any Site Directed Stable Isotope (2H, 13C, 15N, 17O and 18O) in Genetically Encoded Amino Acids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
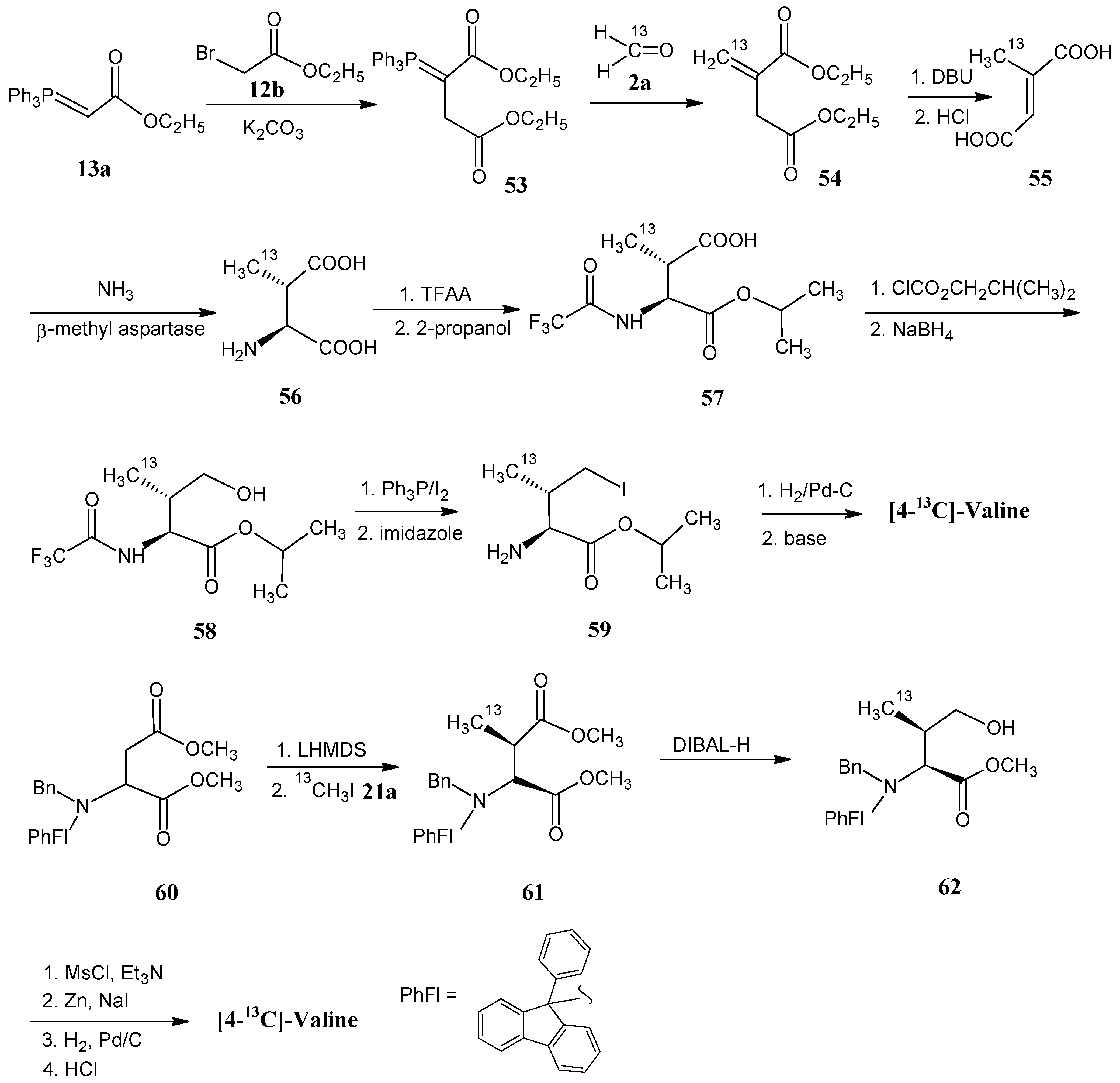{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Synthetic Schemes
3. General Methods to Synthesize L-Amino Acids
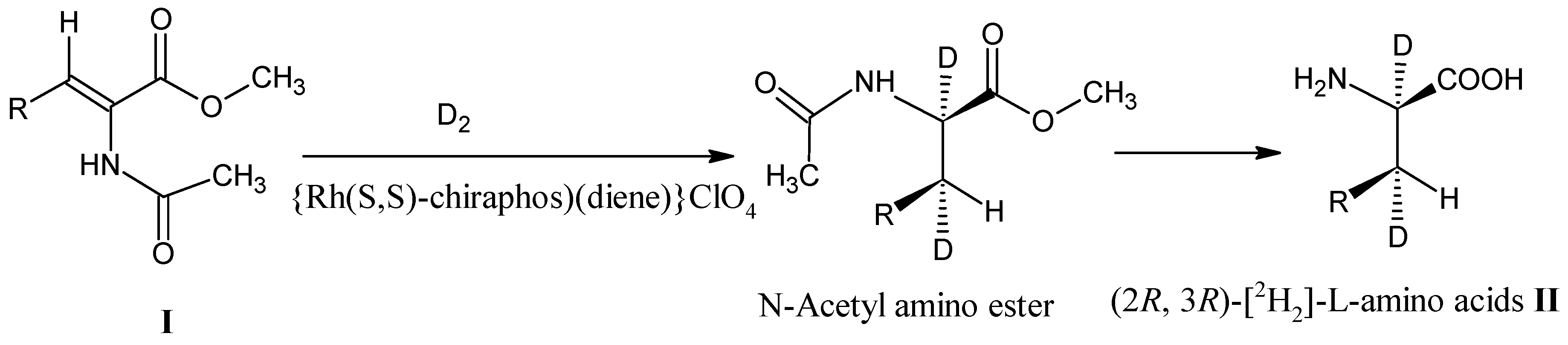
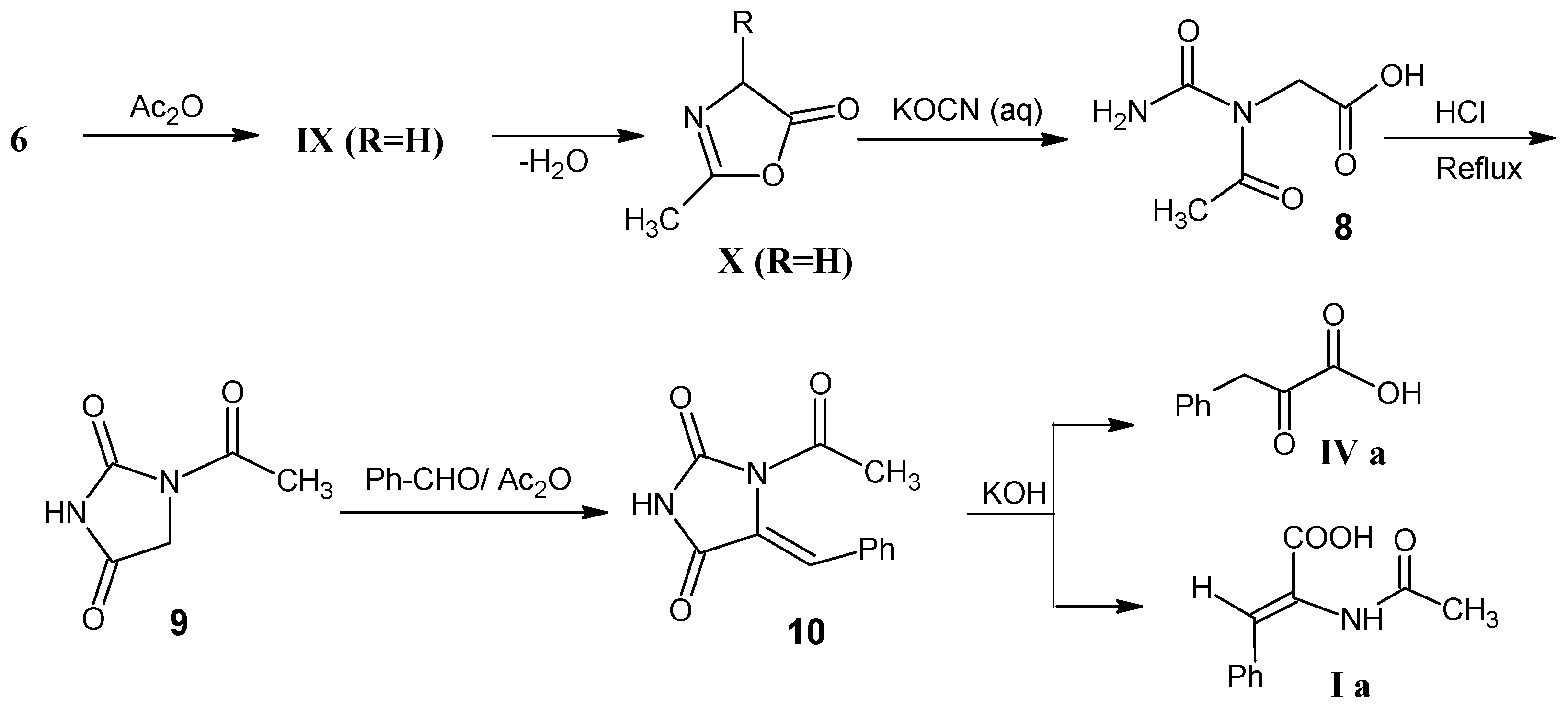
3.1. Catalytic Reduction of 2,3-Dehydroamino Acids I

3.2. Reductive Amination of α-Keto Acids IV

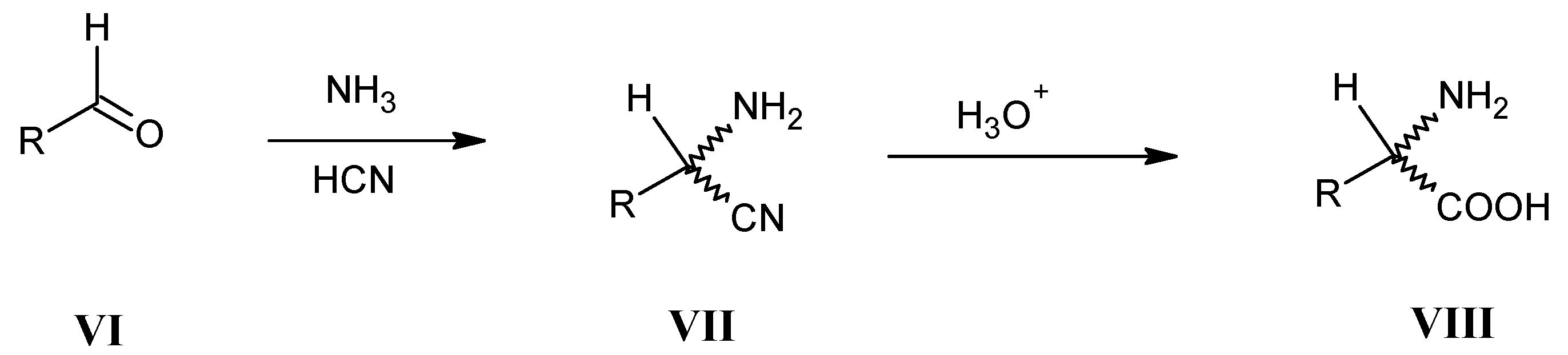
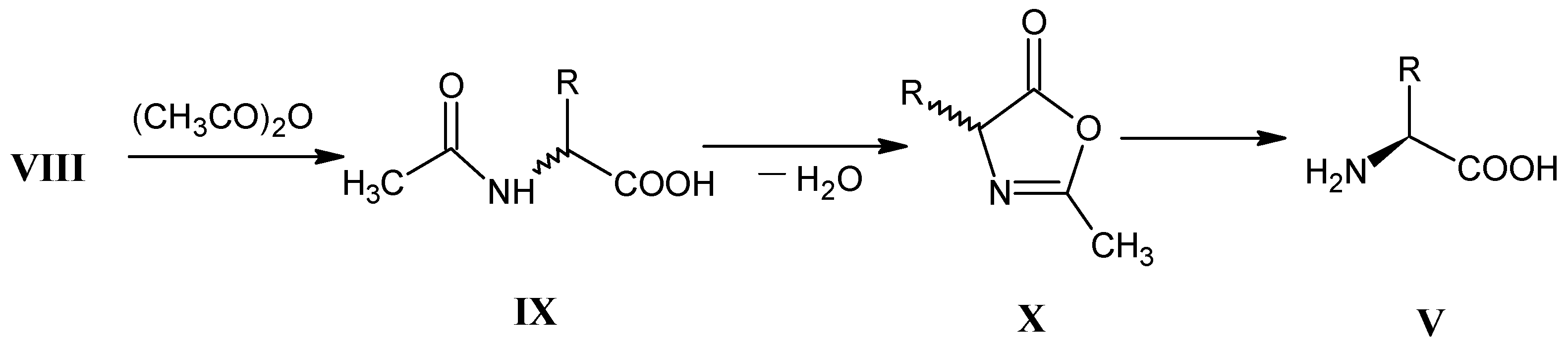
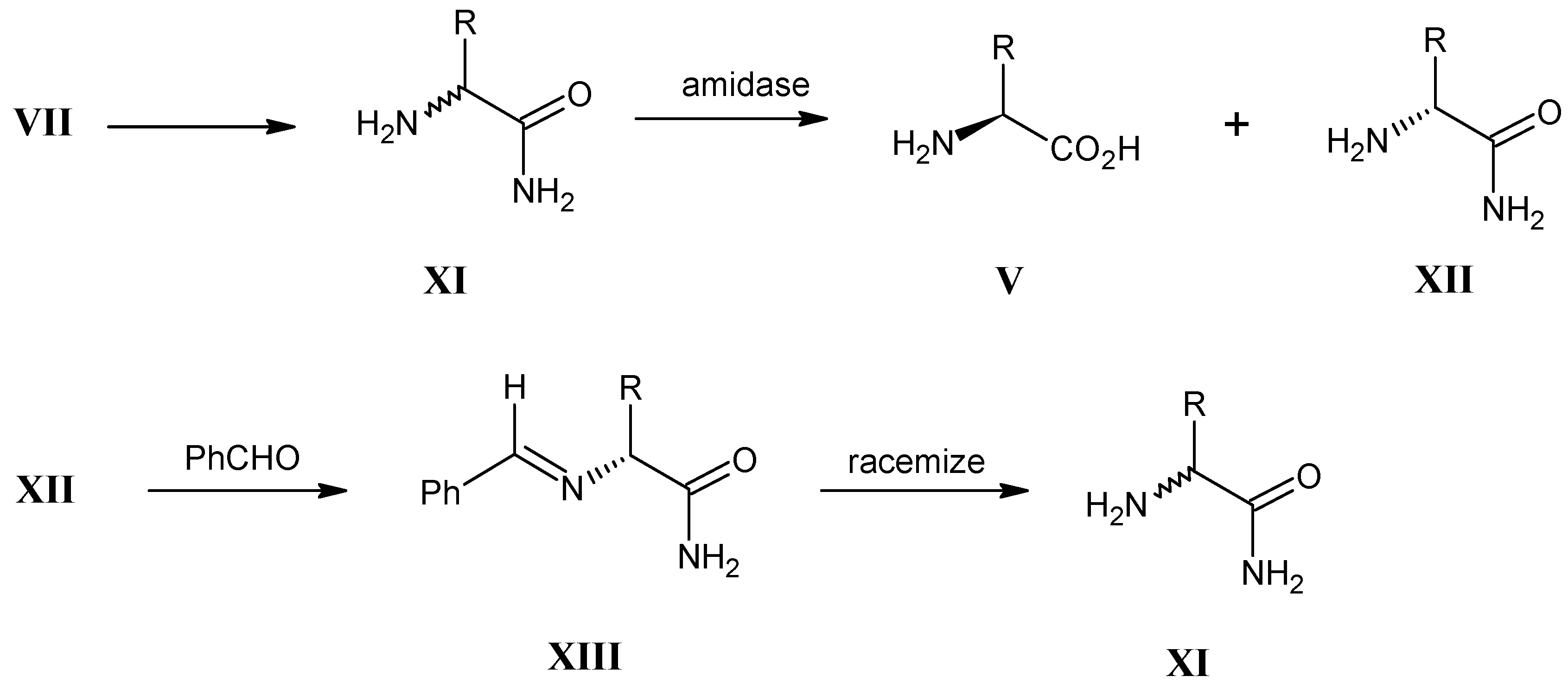
3.3. Hydrolysis of α-Amino Nitriles VII



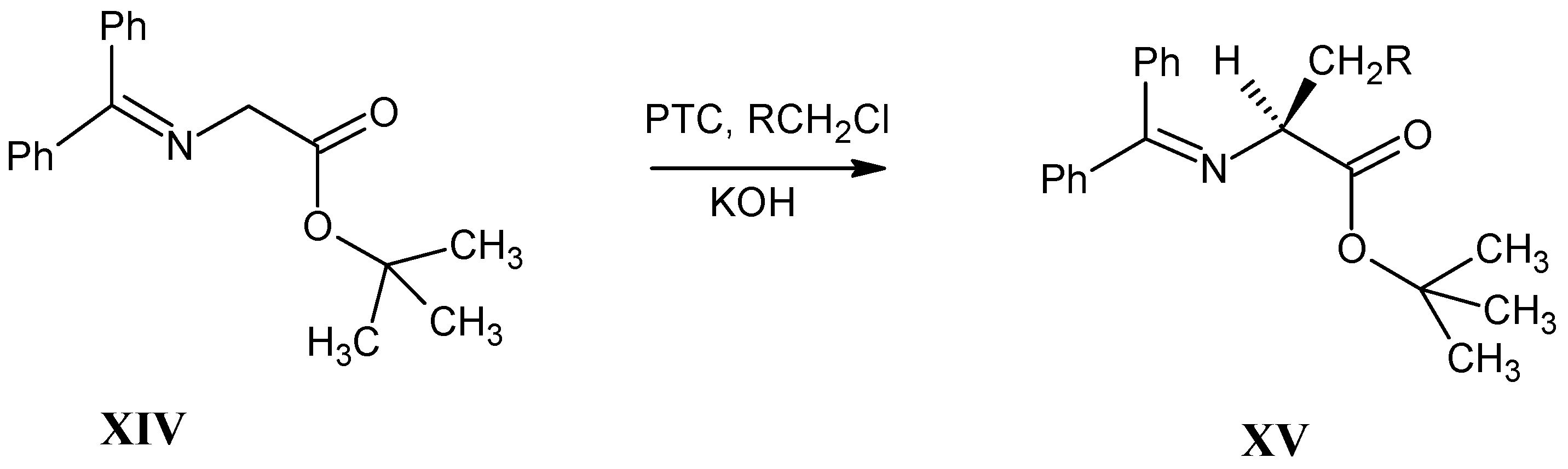
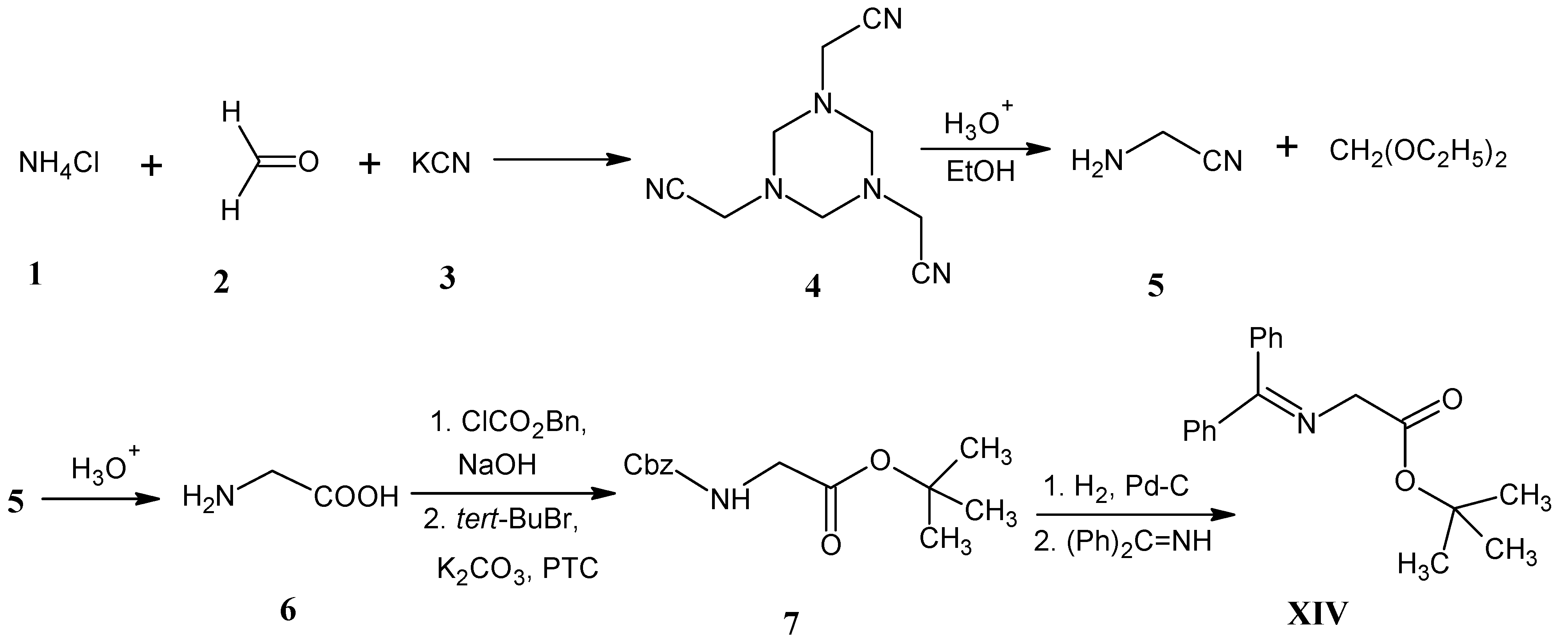
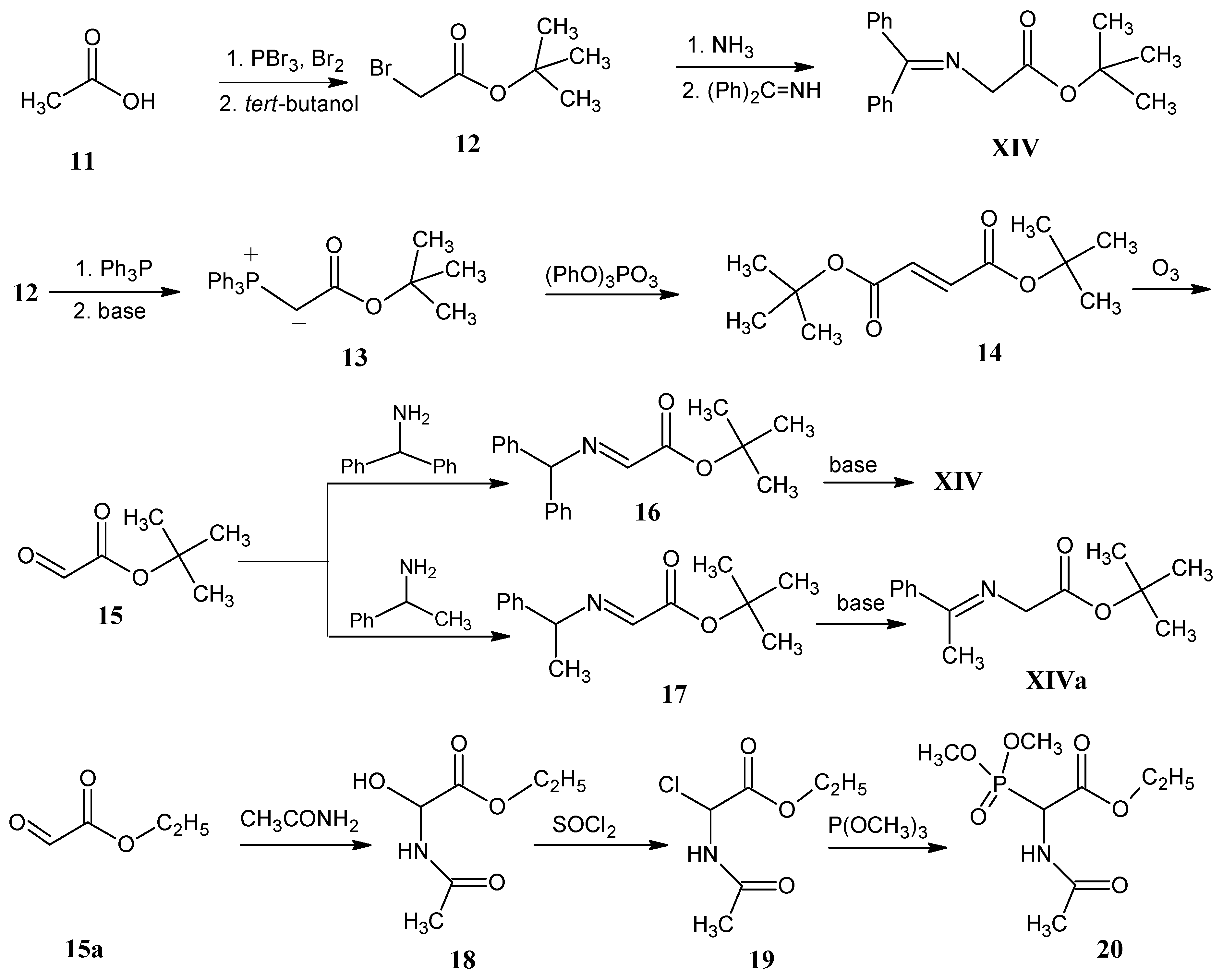
3.4. Alkylation of N-(Diphenylmethylene)glycine tert-Butyl Ester XIV (O’Donnell Method)

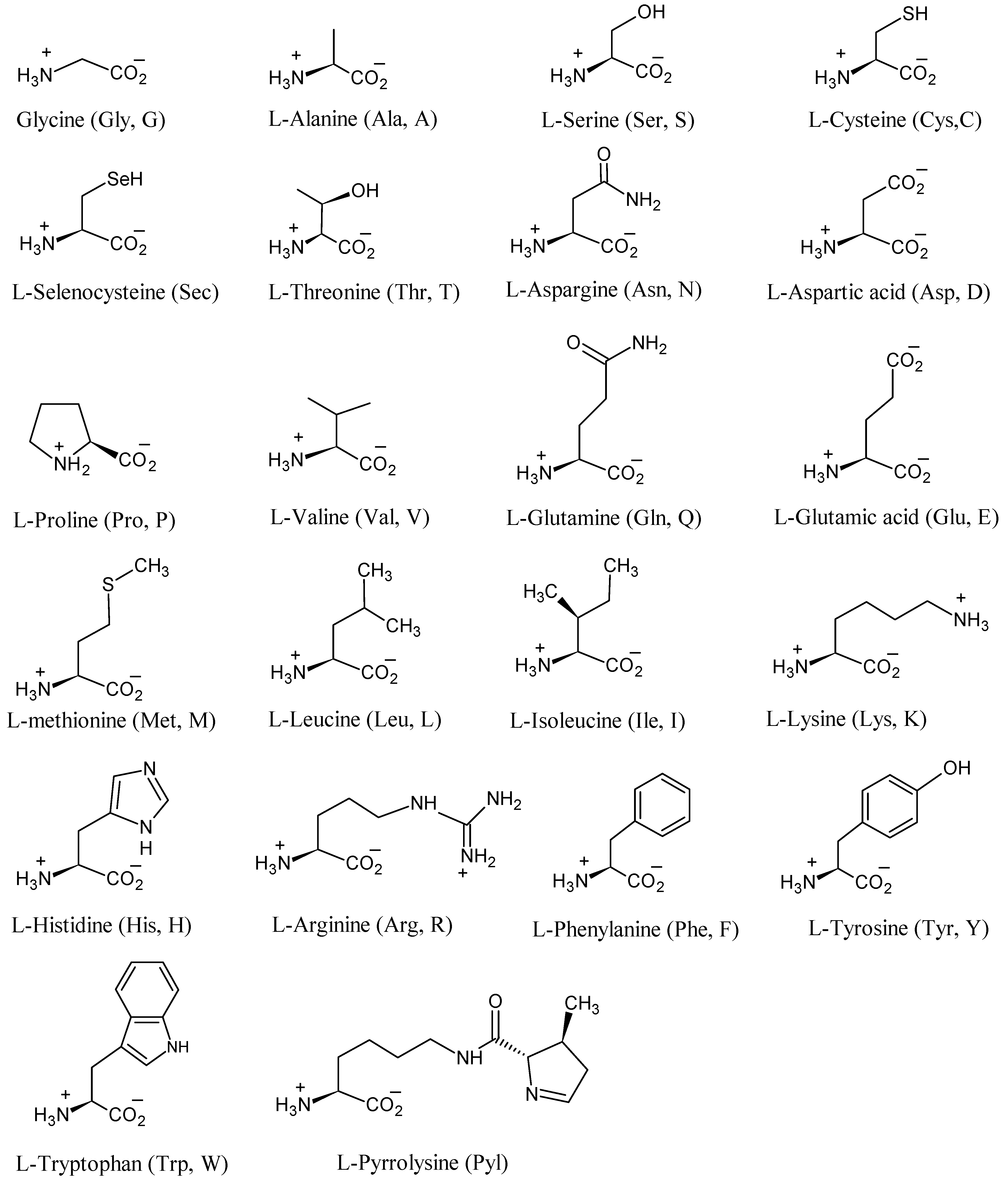
4. Synthesis of 22 Amino Acids
4.1. Glycine



4.2. Alanine

4.3. Serine

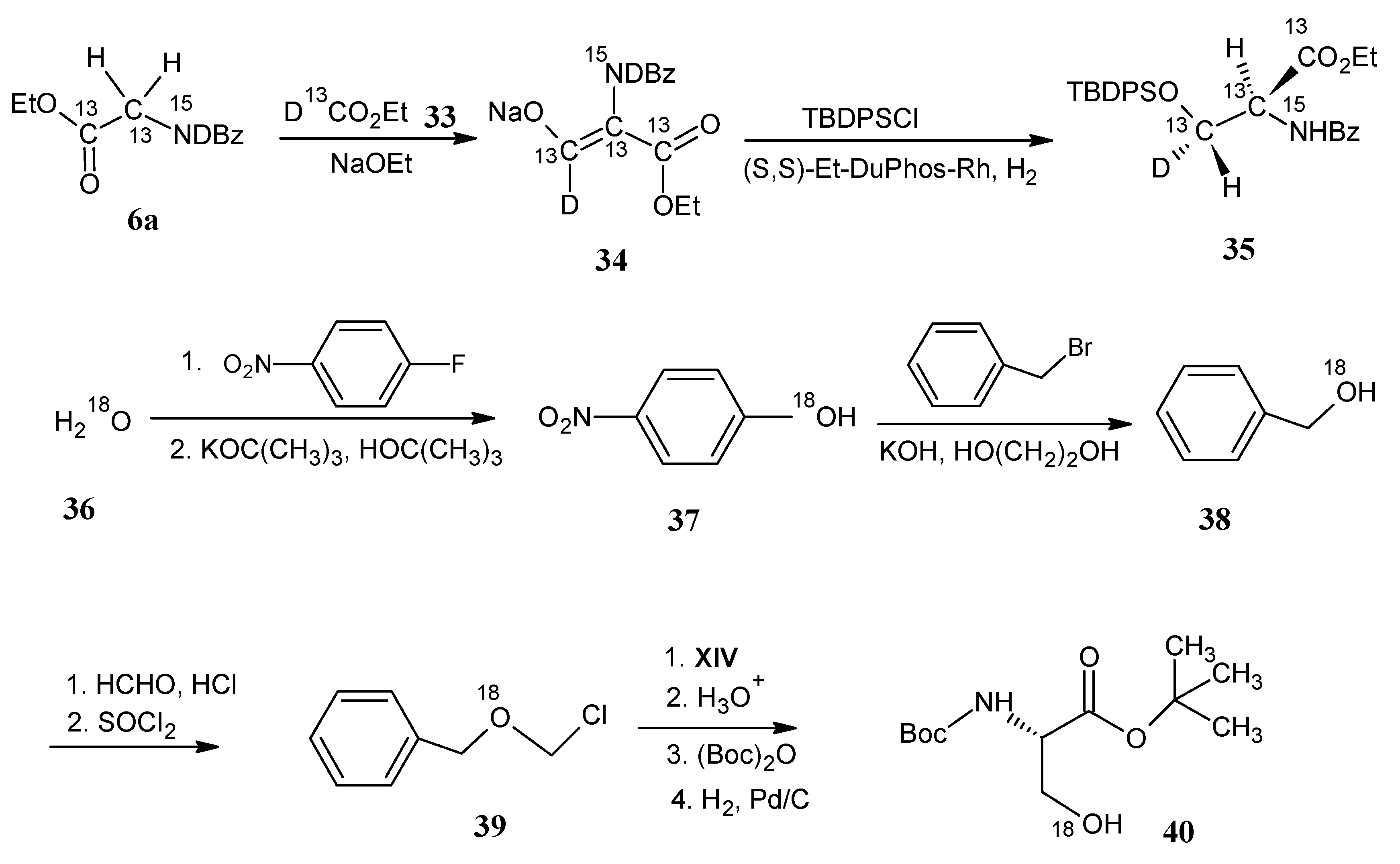
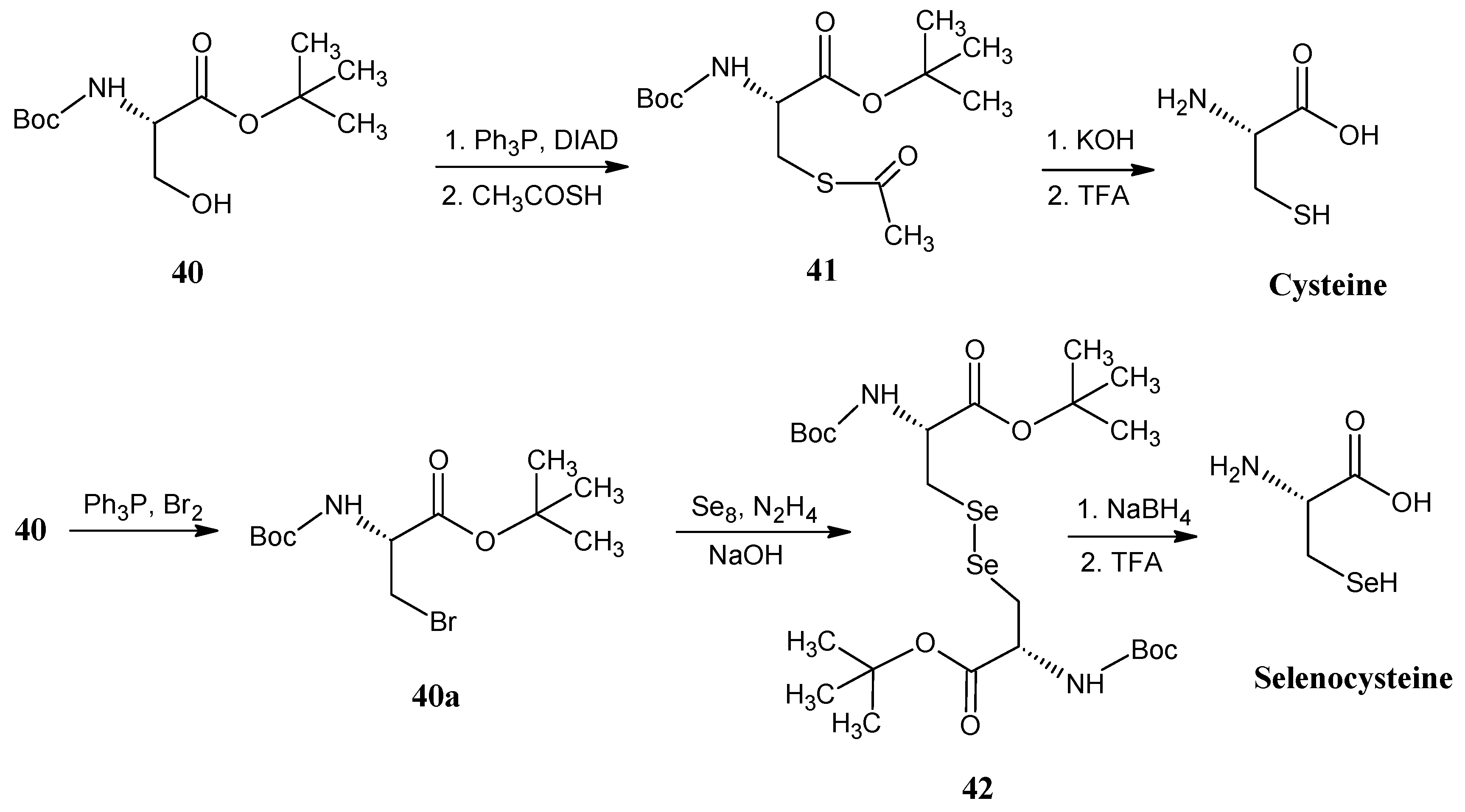
4.4. Cysteine and Selenocysteine

4.5. Threonine

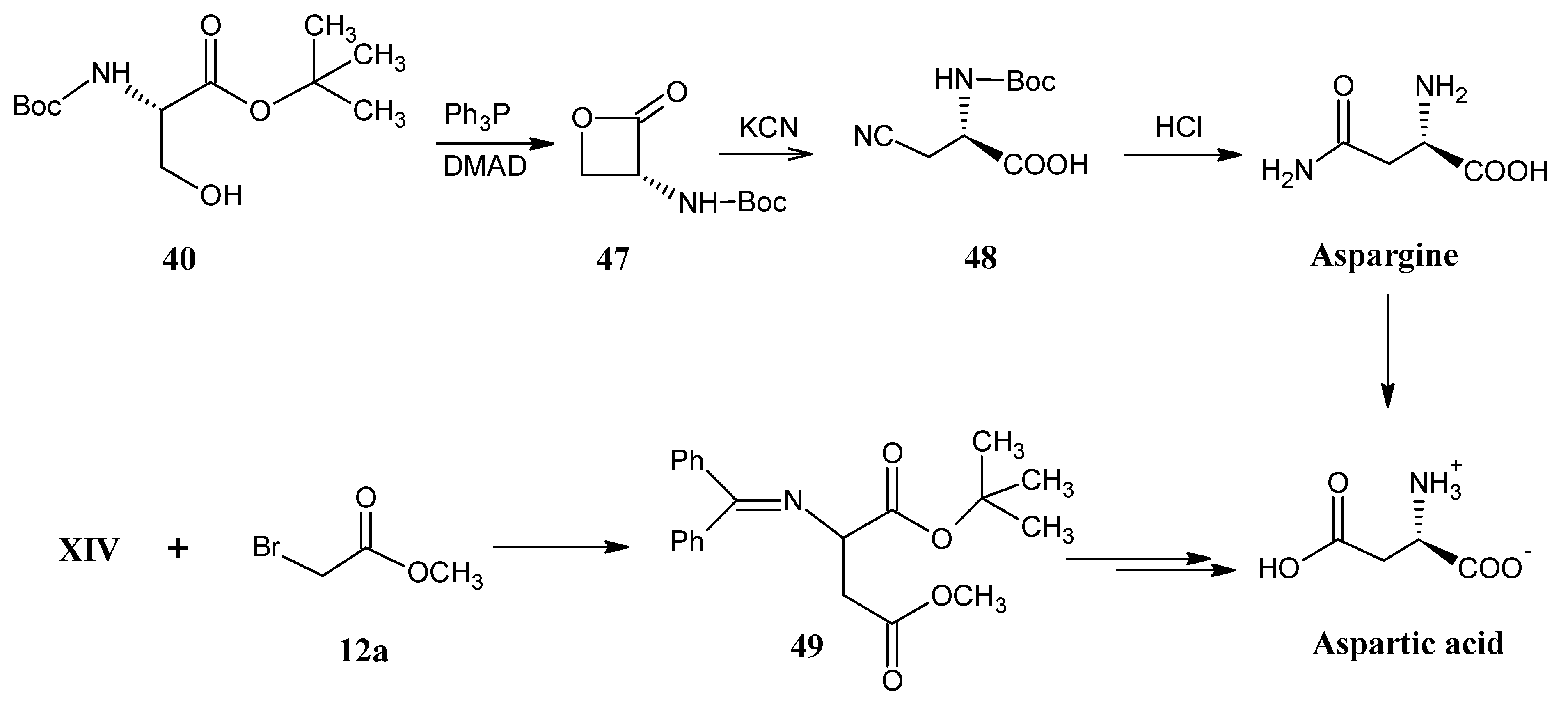
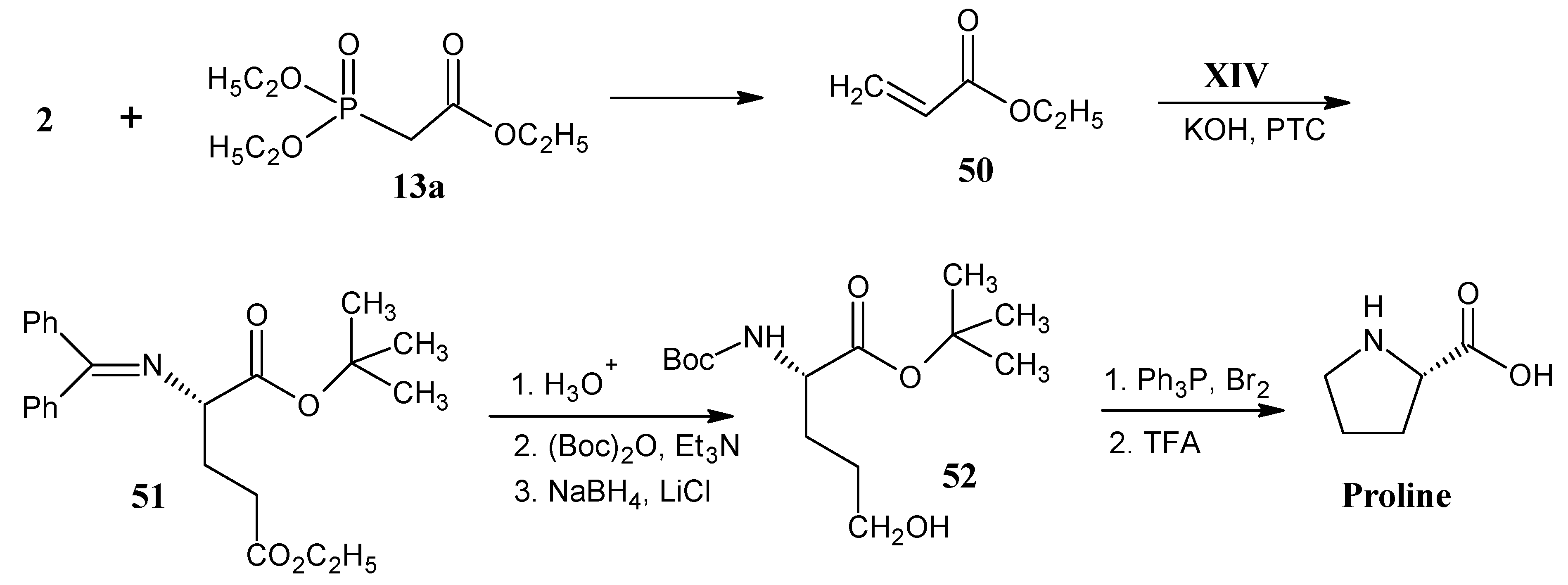
4.6. Asparagine and Aspartic Acid

4.7. Proline

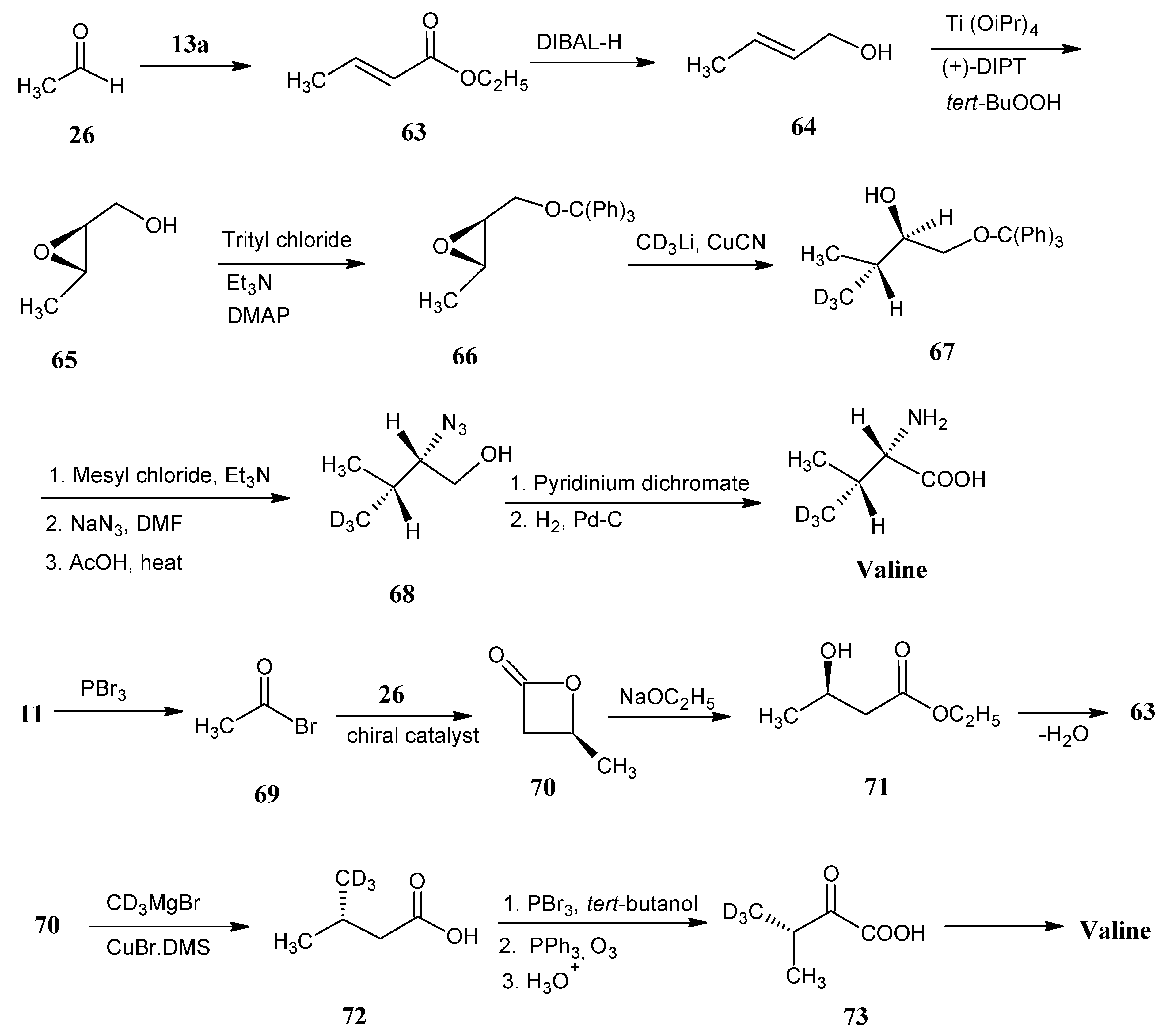
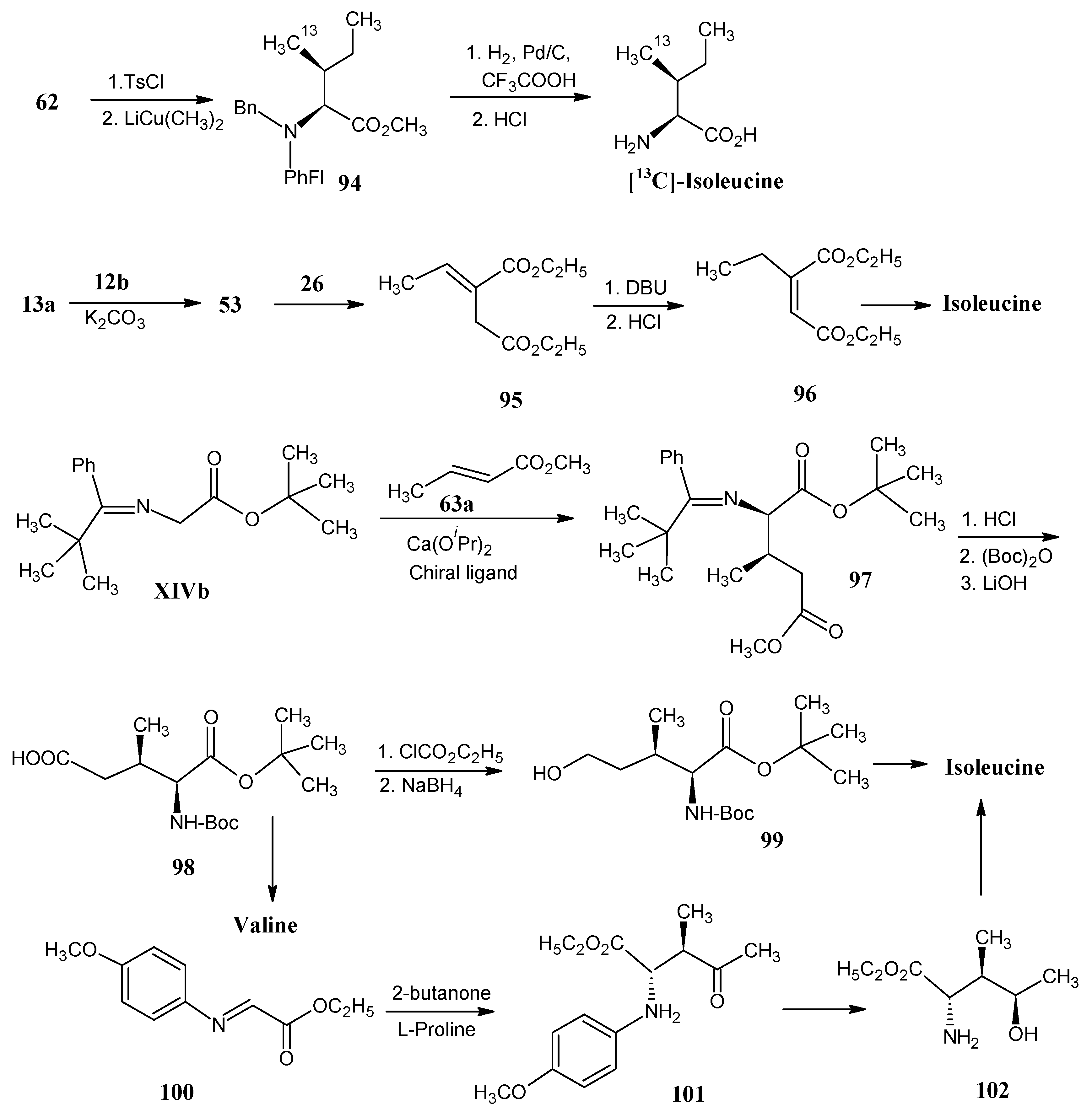
4.8. Valine


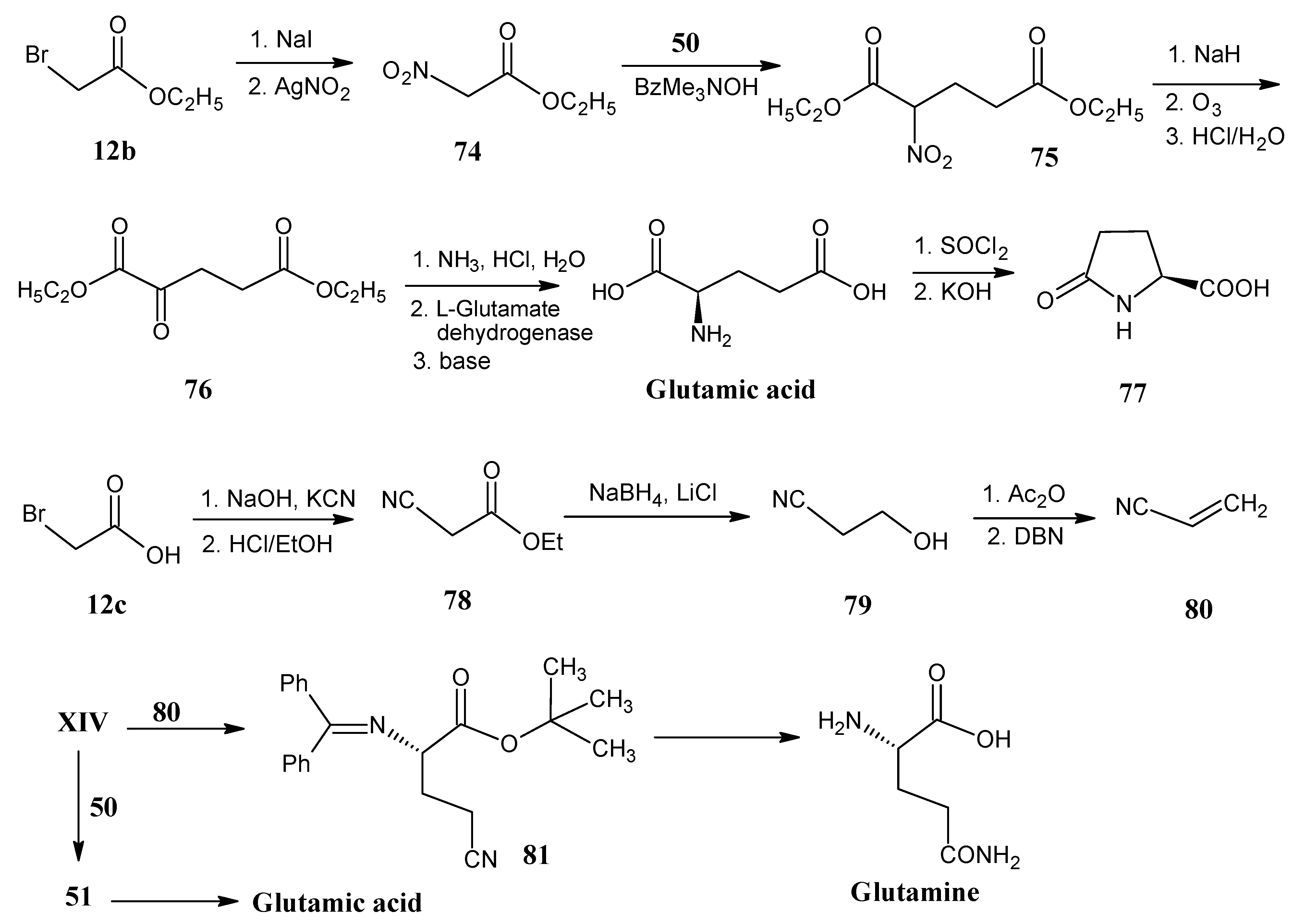
4.9. Glutamine and Glutamic Acid

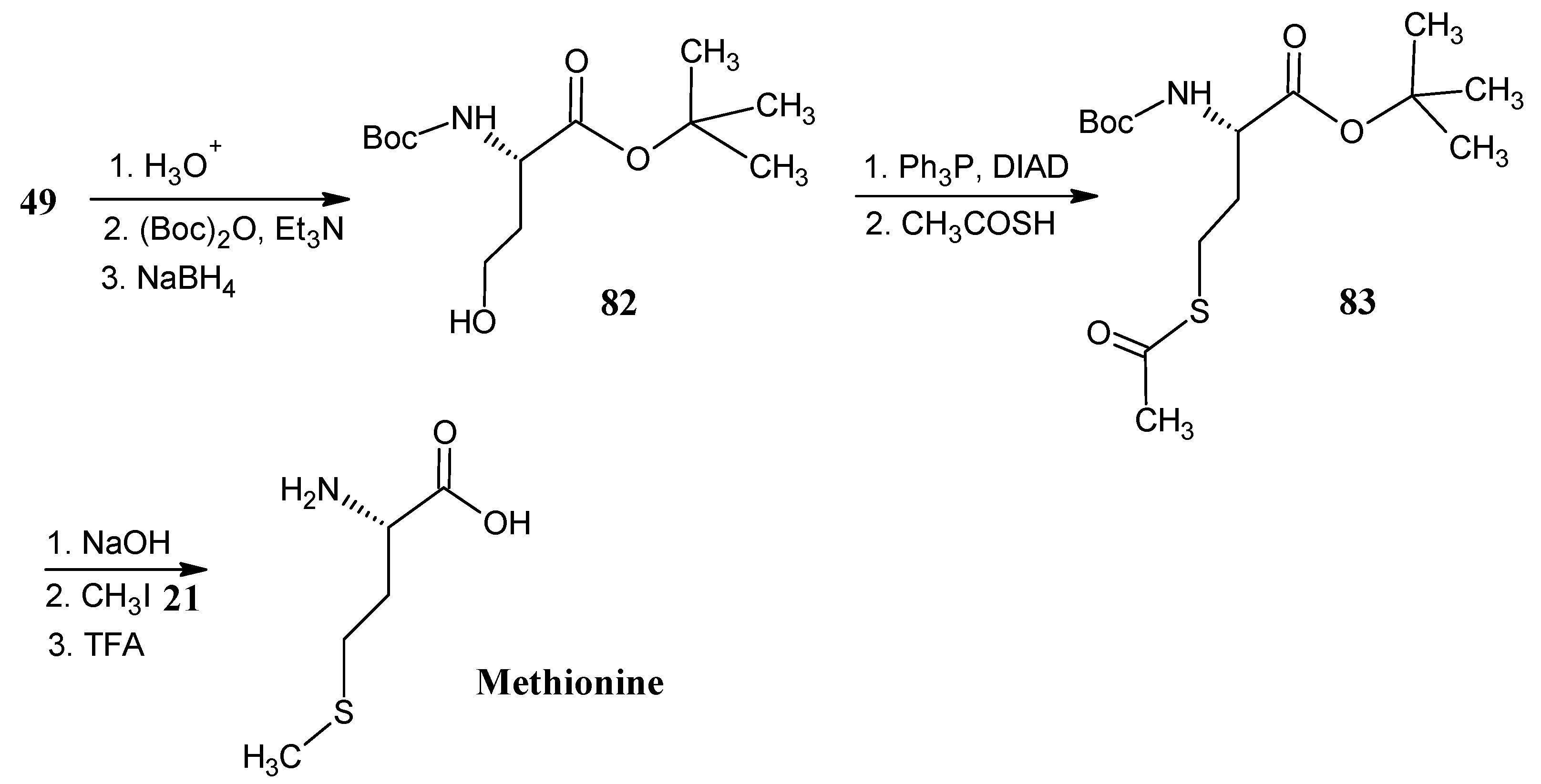
4.10. Methionine

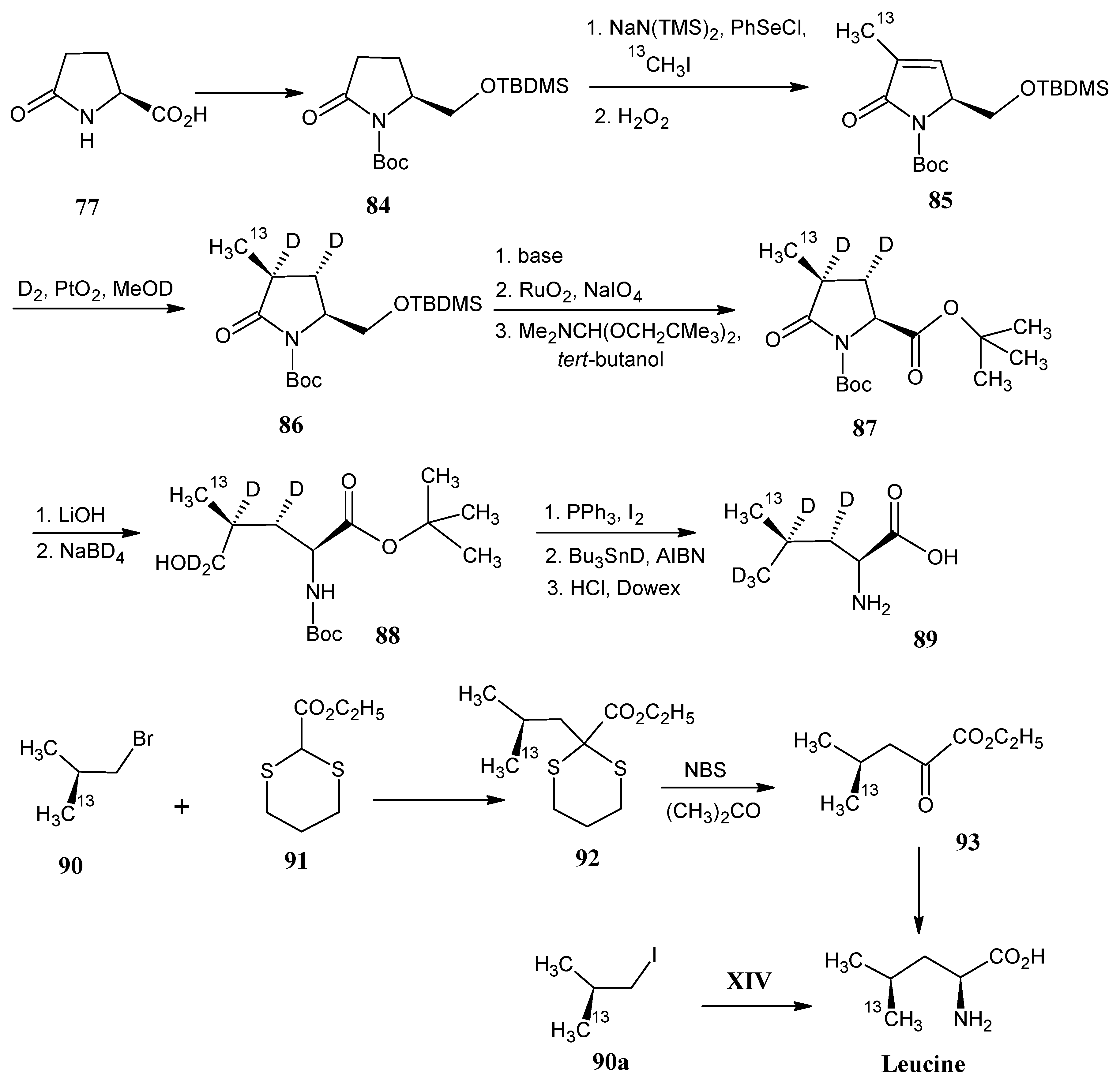
4.11. Leucine

4.12. Isoleucine

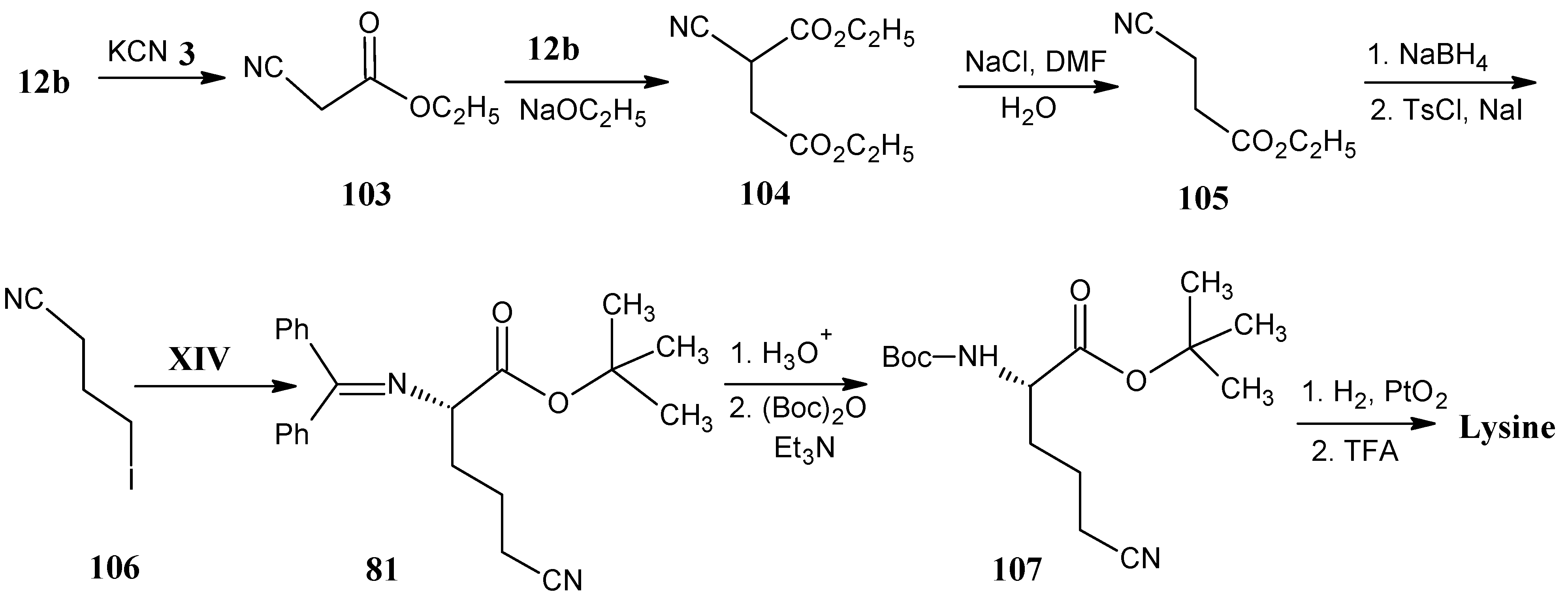
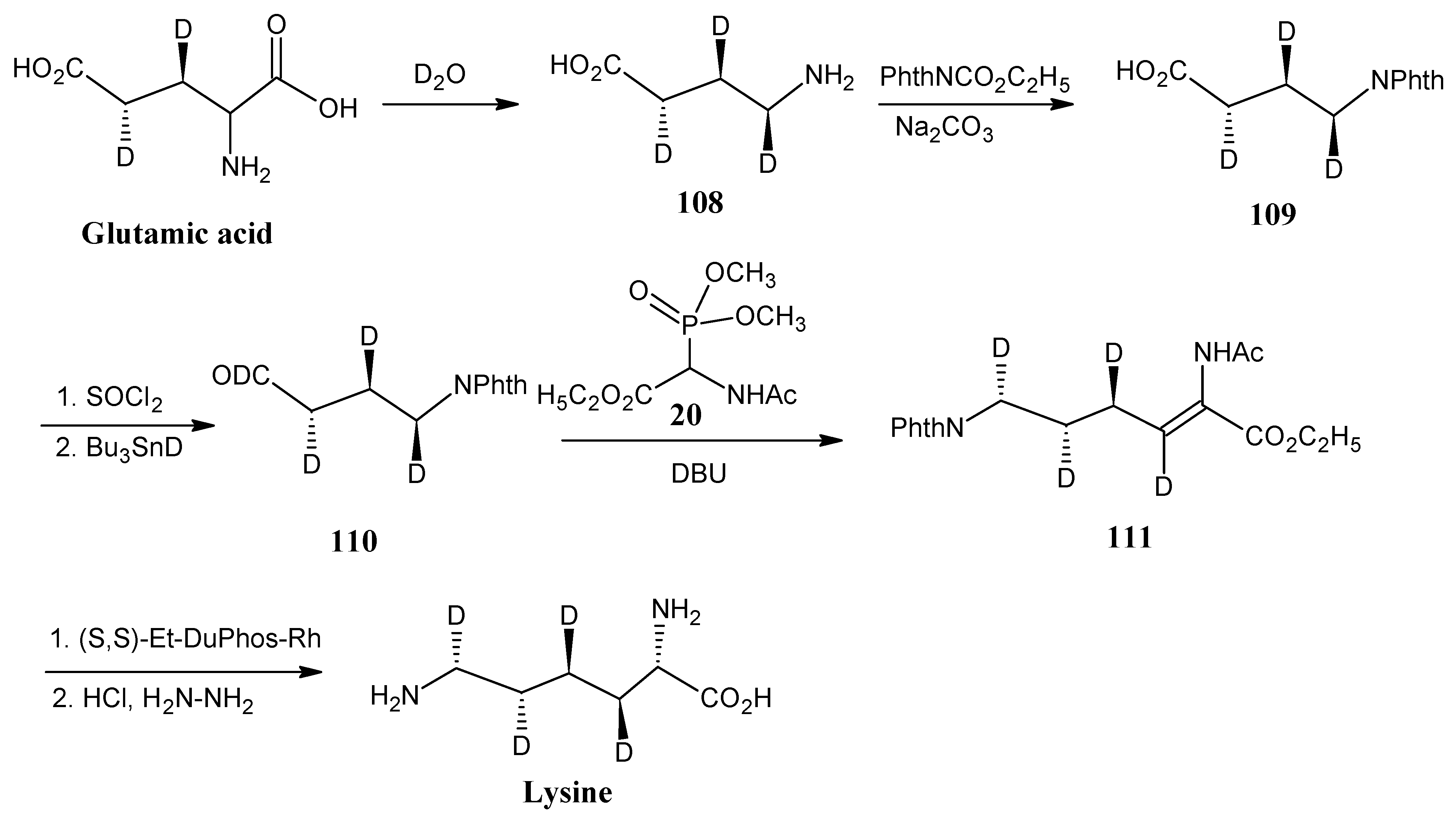
4.13. Lysine


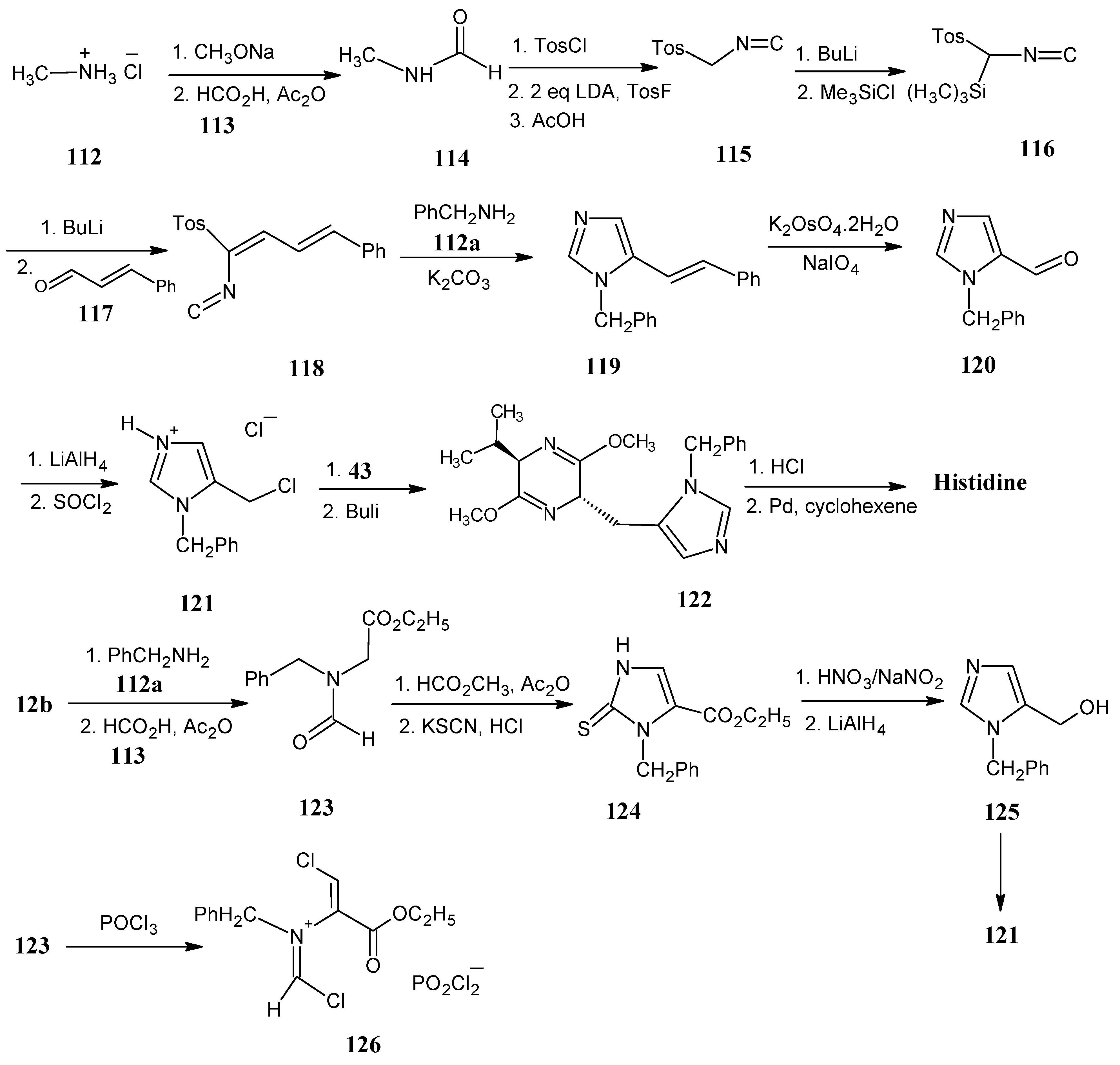
4.14. Histidine

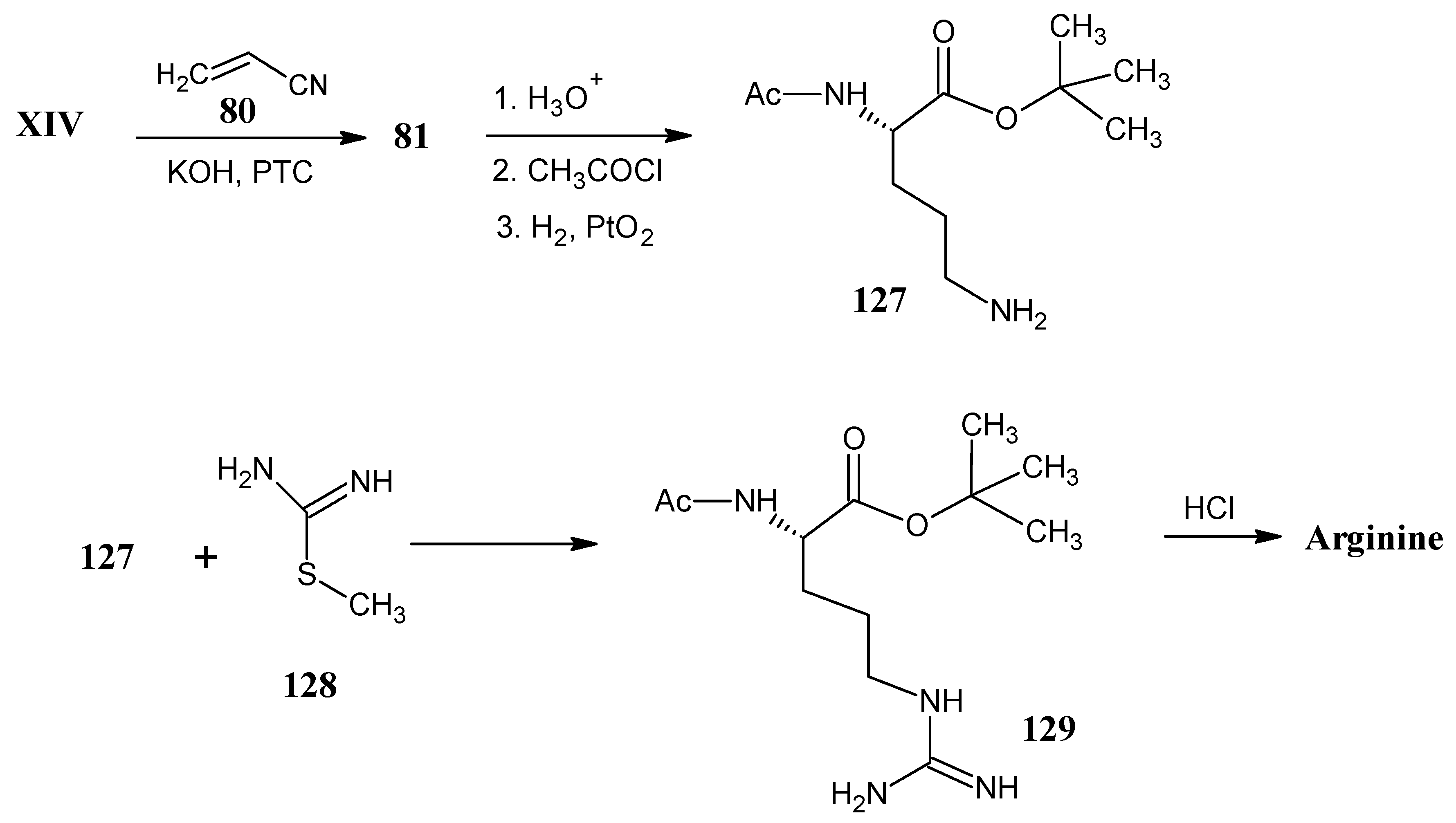
4.15. Arginine

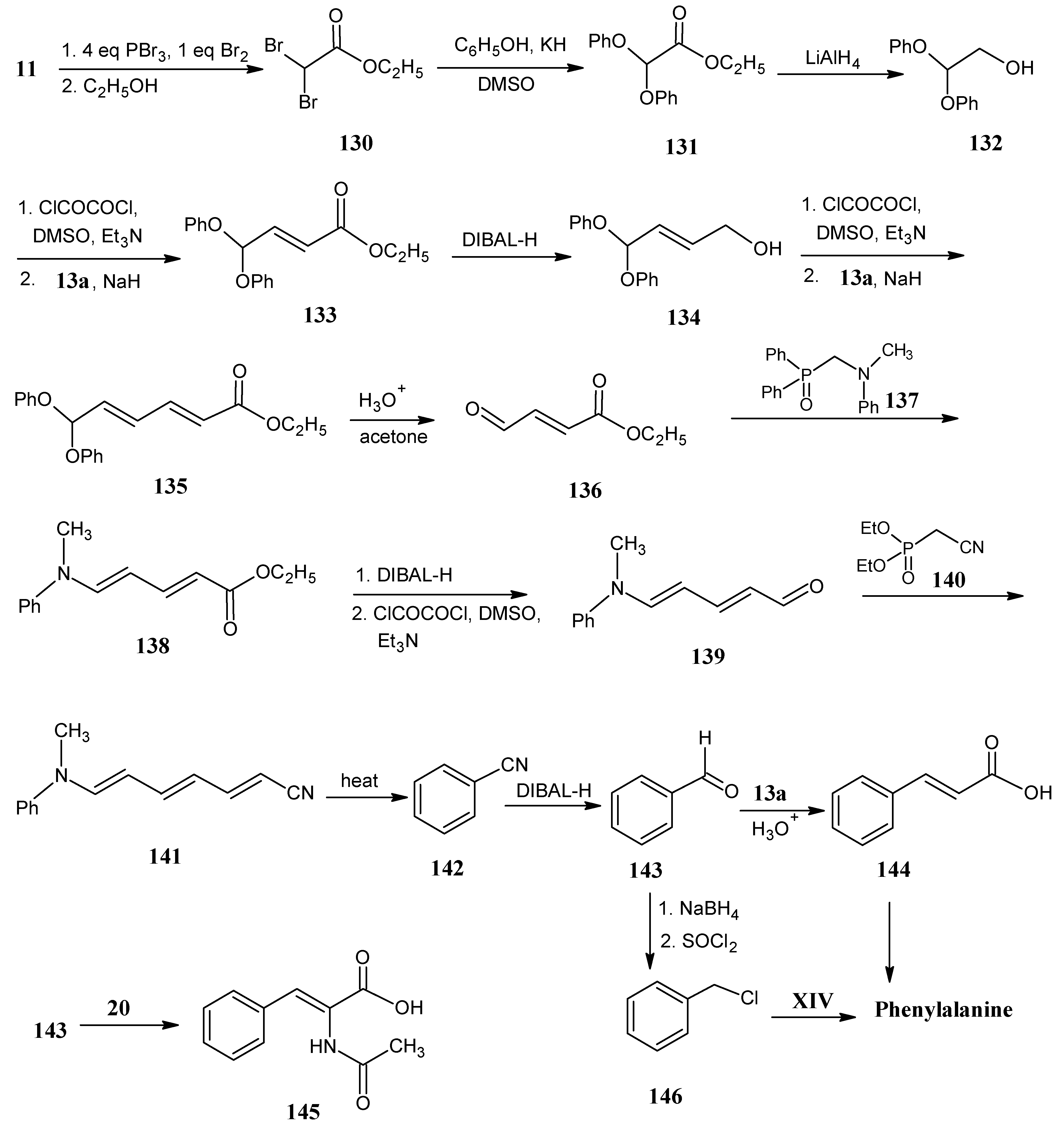
4.16. Phenylalanine

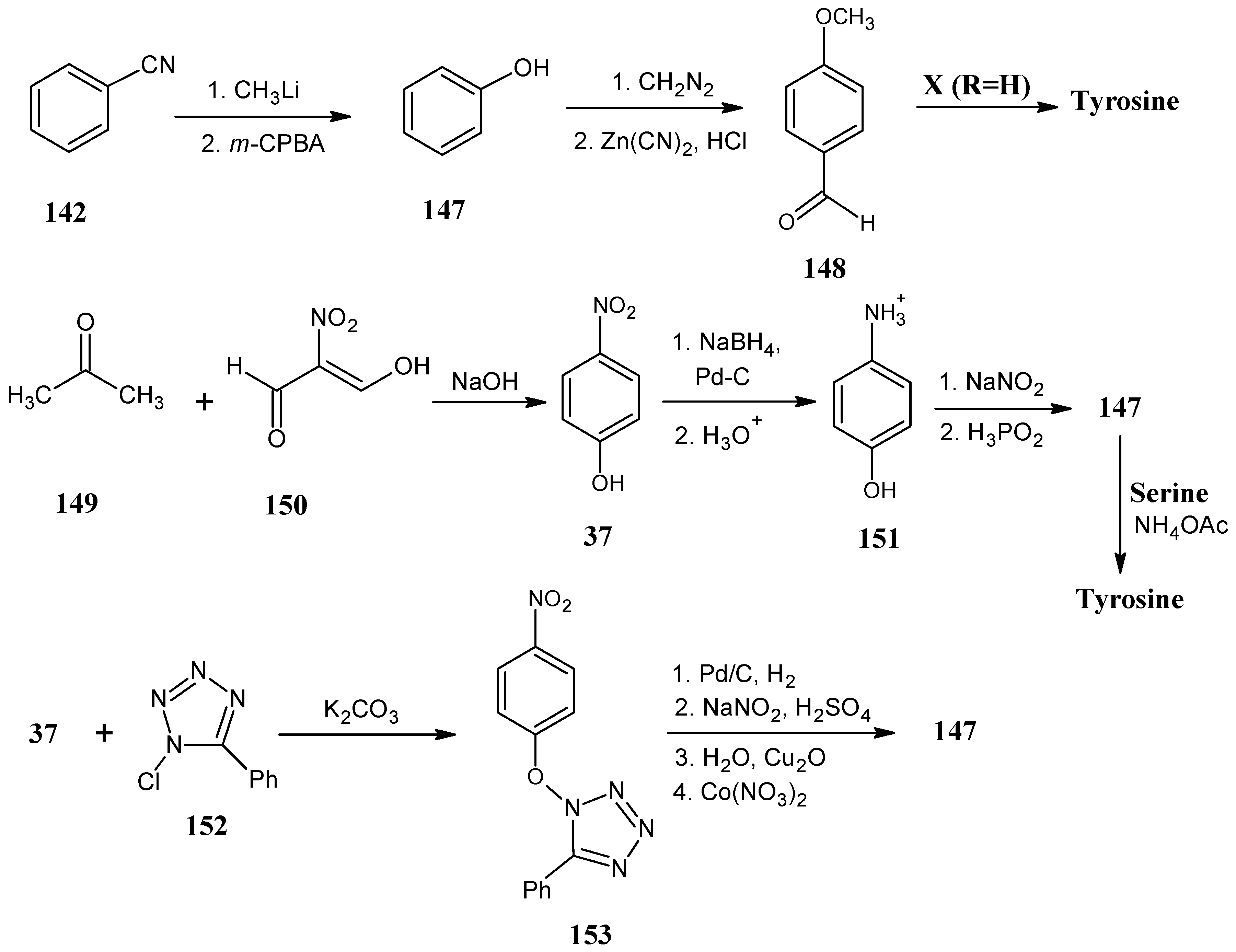
4.17. Tyrosine

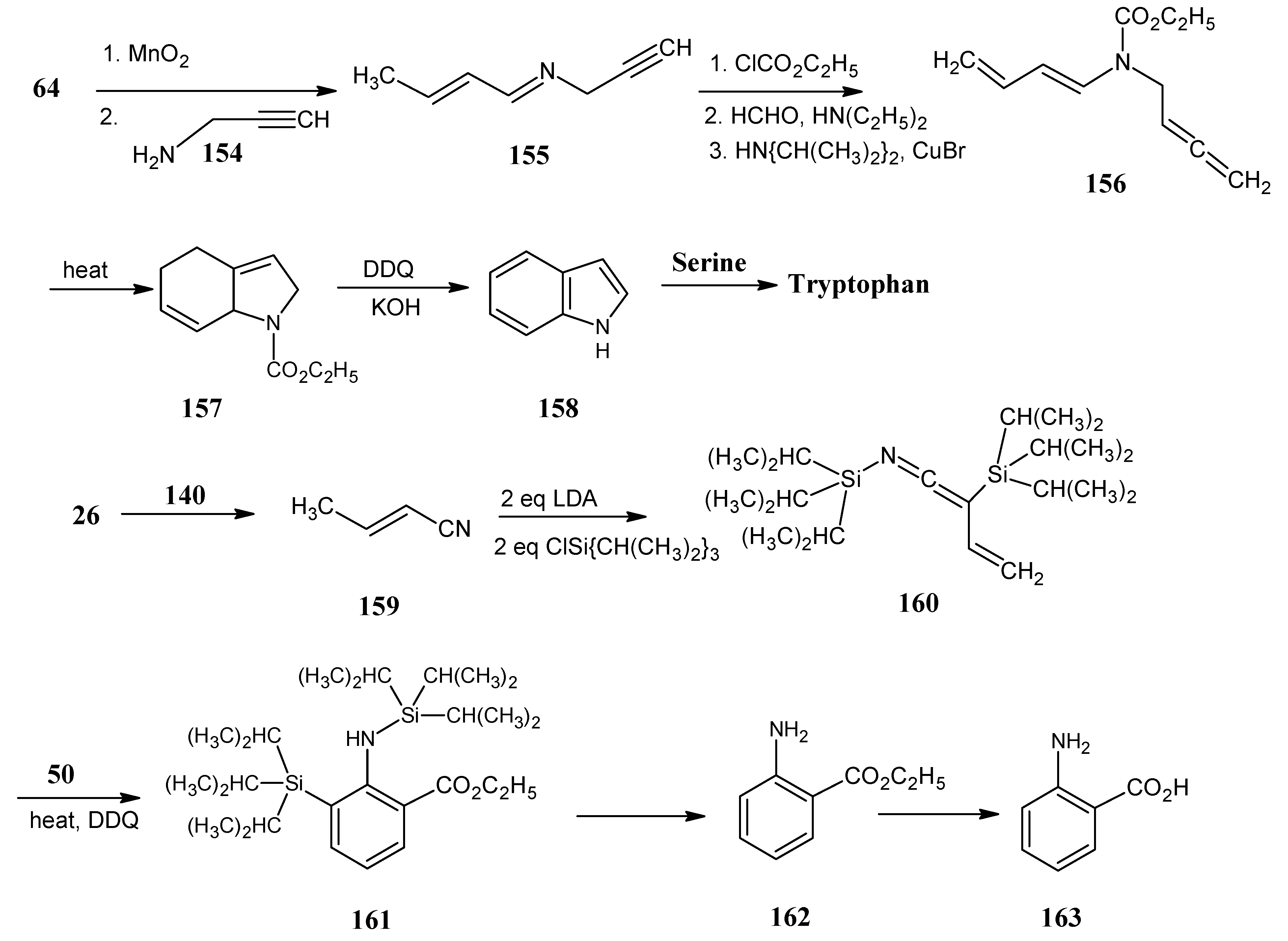
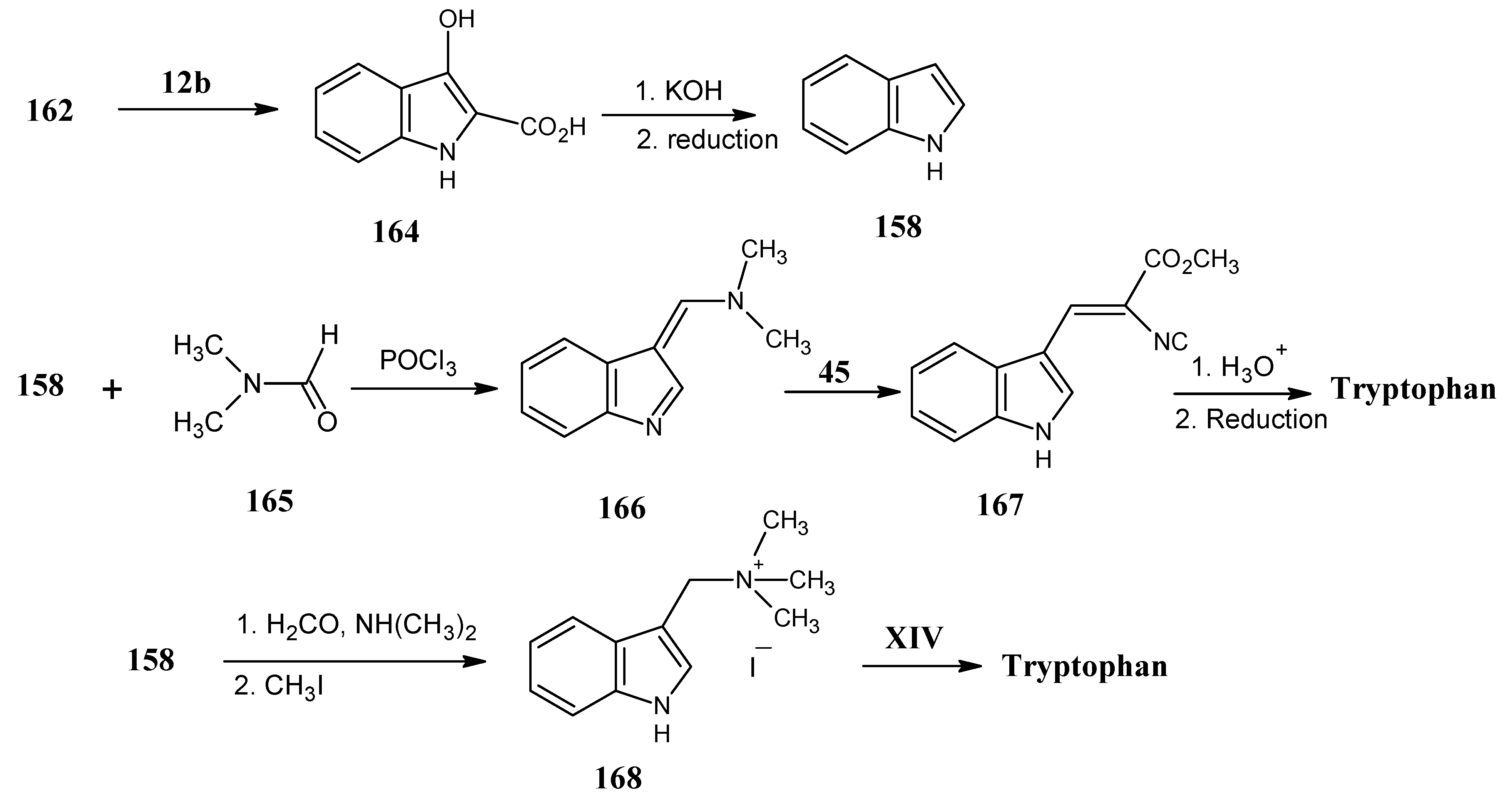
4.18. Tryptophan


4.19. Pyrrolysine

5. Conclusions
Acknowledgments
References
- Stryer, L. Biochemistry, 3rd ed; W.H. Freeman, and Company: New York, NY, USA, 1989. [Google Scholar]
- Blaskovich, M.A. Handbook on Syntheses of Amino Acids. General Routes to Amino Acids; Oxford University Press: New York, NY, USA, 2010. [Google Scholar]
- Hayashi, T.; Hamachi, I. Traceless affinity labeling of endogenous proteins for functional analysis in living cells. Acc. Chem. Res. 2012, 45, 1460–1469. [Google Scholar] [CrossRef]
- Bertini, I.; Felli, I.C.; Gonnelli, L.; Kumar, M.V.; Pierattelli, R. 13C Direct-detection biomolecular NMR Spectroscopy in living cells. Angew. Chem. Int. Ed. 2011, 50, 2339–2341. [Google Scholar]
- Serber, Z.; Straub, W.; Corsini, L.; Nomura, A.M.; Shimba, N.; Craik, C.S.; Ortiz de Montellano, P.; Dötsch, V. Methyl groups as probes for proteins and complexes in in-cell NMR experiments. J. Am. Chem. Soc. 2004, 126, 7119–7125. [Google Scholar]
- Westler, W.M.; Stockman, B.J.; Hosoya, Y.; Miyake, Y.; Kainosho, M.; Markley, J.M. Correlation of carbon-13 and nitrogen-15 chemical shifts in selectively and uniformly labeled proteins by heteronuclear two-dimensional NMR spectroscopy. J. Am. Chem. Soc. 1988, 110, 6256–6258. [Google Scholar] [CrossRef]
- Kainosho, M.; Torizawa, T.; Iwashita, Y.; Terauchi, T.; Ono, A.M.; Güntert, P. Optimal isotope labeling for NMR protein structure determinations. Nature 2006, 440, 52–57. [Google Scholar] [CrossRef]
- Takeda, M.; Ikeya, T.; Güntert, P.; Kainosho, M. Automated structure determination of proteins with the SAIL-FLYA NMR method. Nat. Protoc. 2007, 2, 2896–2902. [Google Scholar]
- Guo, C.; Godoy-Ruiz, R.; Tugarinov, V. High resolution measurement of methyl 13C(m)-13C and 1H(m)-13C(m) residual dipolar couplings in large proteins. J. Am. Chem. Soc. 2010, 132, 13984–13987. [Google Scholar] [CrossRef]
- Otten, R.; Chu, B.; Krewulak, K.D.; Vogel, H.J.; Mulder, F.A.A. Comprehensive and cost-effective NMR Spectroscopy of methyl groups in large proteins. J. Am. Chem. Soc. 2010, 132, 2952–2960. [Google Scholar] [CrossRef]
- Ayala, I.; Hamelin, O.; Amero, C.; Pessey, O.; Plevin, M.J.; Gans, P.; Boisbouvier, J. An optimized isotopic labelling strategy of isoleucine-γ2 methyl groups for solution NMR studies of high molecular weight proteins. Chem. Commun. 2012, 48, 1434–1436. [Google Scholar]
- Pandey, M.K.; Vivekanandan, S.; Ahuja, S.; Pichumani, K.; Im, S.C.; Waskell, L.; Ramamoorthy, A. Determination of 15N chemical shift anisotropy from a membrane-bound protein by NMR Spectroscopy. J. Phys. Chem. B 2012, 116, 7181–7189. [Google Scholar]
- Elavarasi, S.B.; Kumari, A.; Dorai, K. Using the Chemical Shift Anisotropy Tensor of Carbonyl Backbone Nuclei as a Probe of Secondary Structure in Proteins. J. Phys. Chem. A 2010, 114, 5830–5837. [Google Scholar] [CrossRef]
- Zhu, J.; Ye, E.; Terskikh, V.; Wu, G. Solid-state 17O-NMR spectroscopy of large protein-ligand complexes. Angew. Chem. Int. Ed. 2010, 49, 8399–8402. [Google Scholar] [CrossRef]
- Yu, W.; Dawson, P.E.; Zimmermann, J.; Romesberg, F.E. Carbon-deuterium bonds as probes of protein thermal unfolding. J. Phys. Chem. B 2012, 116, 6397–6403. [Google Scholar] [CrossRef]
- Torizawa, T.; Shimizu, M.; Taoka, M.; Miyano, H.; Kainosho, M. Efficient production of isotopically labeled proteins by cell-free synthesis: A practical protocol. J. Biomol. NMR 2004, 30, 311–325. [Google Scholar] [CrossRef]
- Kent, S.B.H. Total chemical synthesis of proteins. Chem. Soc. Rev. 2009, 38, 338–351. [Google Scholar] [CrossRef]
- Metanis, N.; Keinan, E.; Dawson, P.E. Traceless ligation of cysteine peptides using selective deselenization. Angew. Chem. Int. Ed. 2010, 49, 7049–7053. [Google Scholar] [CrossRef]
- Durek, T.; Alewood, P.F. Preformed selenoesters enable rapid native chemical ligation at intractable sites. Angew. Chem. Int. Ed. 2011, 50, 12042–12045. [Google Scholar] [CrossRef]
- Shang, S.; Tan, Z.; Danishefsky, S.J. Application of the logic of cysteine-free native chemical ligation to the synthesis of Human Parathyroid Hormone (hPTH). Proc. Natl. Acad. Sci. USA 2011, 108, 5986–5989. [Google Scholar]
- Dittmann, M.; Sauermann, J.; Seidel, R.; Zimmermann, W.; Engelhard, M. Native chemical ligation of hydrophobic peptides in organic solvents. J. Peptide Sci. 2010, 16, 558–562. [Google Scholar] [CrossRef]
- Bode, J.W.; Fox, R.M.; Baucom, K.D. Chemoselective amide ligations by decarboxylative condensations of N-alkylhydroxylamines and α-ketoacids. Angew. Chem. Int. Ed. 2006, 45, 1248–1252. [Google Scholar] [CrossRef]
- Winkler, F.J.; Kühnl, K.; Medina, R.; Schwarz-Kaske, R.; Schmidt, H.L. Principles and results of stable isotope mnmn7mn7ulabelling of L-α-aminoacids by combined chemical and enzymatic methods. Isot. Environ. Health Stud. 1995, 31, 161–190. [Google Scholar] [CrossRef]
- Reid, C.M.; Sutherland, A. Synthesis of isotopically labeled α-amino Acids. In Amino Acids, Peptides and Proteins in Organic Chemistry: Origins and Synthesis of Amino Acid; Hughes, A.B., Ed.; Wiley-VCH Verlag GmbH and Co. KGaA: Weinheim, Germany, 2009; Volume 1, pp. 473–494. [Google Scholar]
- Hiller, K.; Metallo, C.M.; Kelleher, J.K.; Stephanopoulos, G. Nontargeted elucidation of metabolic pathways using stable-isotope tracers and mass spectrometry. Anal. Chem. 2010, 82, 6621–6628. [Google Scholar]
- Weber, R.J.M.; Southam, A.D.; Sommer, U.; Viant, M.R. Characterization of Isotopic abundance measurements in high resolution FT-ICR and Orbitrap Mass Spectra for improved confidence of metabolite identification. Anal. Chem. 2011, 83, 3737–3743. [Google Scholar] [CrossRef]
- Svatoš, A. Single-cell metabolomics comes of age: New developments in mass spectrometry profiling and imaging. Anal. Chem. 2011, 83, 5037–5044. [Google Scholar] [CrossRef]
- Kozmin, Y.P.; Manoilov, A.V.; Serebryakova, M.V.; Mirgorodskaya, O.A. A direct introduction of 18O isotopes into peptides and proteins for quantitative mass spectroscopy analysis. Russ. J. Bioorg. Chem. 2011, 37, 719–731. [Google Scholar] [CrossRef]
- Poisel, H.; Schmidt, U. Dehydroaminosaüren aus amino saüren. Chem. Ber. 1975, 108, 2547–2553. [Google Scholar] [CrossRef]
- Schmidt, U. α-Mercapto-α-amino acids and dehydro amino acids-syntheses, relationships and interconversions. Pure Appl. Chem. 1977, 49, 163–168. [Google Scholar] [CrossRef]
- Stohlmeyer, M.M.; Tanaka, H.; Wandless, T.J. A stereospecific elimination to form dehydroamino acids: Synthesis of the phomopsin tripeptide side chain. J. Am. Chem. Soc. 1999, 121, 6100–6101. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Bosnich, B. Asymmetric synthesis. Production of optically active amino acids by catalytic hydrogenation. J. Am. Chem. Soc. 1977, 99, 6262–6267. [Google Scholar] [CrossRef]
- Duthaler, R.O. Recent developments in the stereoselective synthesis of α-amino acids. Tetrahedron 1994, 50, 1539–1650. [Google Scholar] [CrossRef]
- Corey, E.J.; Kürti, L. Enantioselective Chemical Synthesis, 1st ed; Direct Book Publishing, LLC: Dallas, TX, USA, 2010. [Google Scholar]
- Winkler, F.J.; Kühnl, K.; Medina, R.; Schwarz Kaske, R.; Schmidt, H.L. Principles and results of stable isotope labelling of L-α-Aminoacids by combined chemical and enzymatic methods. Isot. Environ. Health Stud. 1995, 31, 161–190. [Google Scholar] [CrossRef]
- Sonke, T.; Kaptein, B.; Schoemaker, H.E. Use of enzymes in the synthesis of amino acids. In Amino Acids, Peptides and Proteins in Organic Chemistry: Origins and Synthesis of Amino Acid; Hughes, A.B., Ed.; Wiley-VCH Verlag GmbH and Co. KGaA: Weinheim, Germany, 2009; pp. 79–117. [Google Scholar]
- Chen, Y.; Goldberg, S.L.; Hanson, R.L.; Parker, W.L.; Gill, I.; Tully, T.P.; Montana, M.A.; Goswami, A.; Patel, R.N. Enzymatic preparation of an (S)-amino acid from a racemic amino acid. Org. Process Res. Dev. 2011, 15, 241–248. [Google Scholar] [CrossRef]
- Kürti, L.; Czakó, B. Strategic Applications of Named Reactions in Organic Synthesis; Elsevier Academic Press: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Vachal, P.; Jacobsen, E.N. Structure-based analysis and optimization of a highly enantioselective catalyst for the Strecker Reaction. J. Am. Chem. Soc. 2002, 124, 10012–10014. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in dynamic kinetic resolution. Tetrahedron 2011, 67, 3769–3802. [Google Scholar] [CrossRef]
- Liu, P.; Yang, X.; Birman, V.B.; Houk, K.N. Origin of enantioselectivity in benzotetramisole-catalyzed dynamic kinetic resolution of azlactones. Org. Lett. 2012, 14, 3288–3291. [Google Scholar] [CrossRef]
- O’Donnell, M.J.; Polt, R.L.J. A mild and efficient route to Schiff base derivatives of amino acids. J. Org. Chem. 1982, 47, 2663–2666. [Google Scholar] [CrossRef]
- Park, H.G.; Jeong, B.S. Cinchona-catalyzed nucleophilic alpha-substitution of carbonyl derivatives. In Cinchona Alkaloids in Synthesis and Catalysis Ligands, Immobilization and Organocatalysi; Choong Eui, S., Ed.; Wiley-VCH Verlag GmbH and Co. KGaA: Weinheim, Germany, 2009; Chapter 6. [Google Scholar]
- Yeboah, E.M.O.; Yeboah, S.O.; Singh, G.S. Recent applications of cinchona alkaloids and their derivatives as catalysts in metal-free asymmetric synthesis. Tetrahedron 2011, 67, 1725–1762. [Google Scholar] [CrossRef]
- Marcelli, T.; Hiemstra, H. Cinchona alkaloids in asymmetric organocatalysis. Synthesis 2010, 1229–1279. [Google Scholar] [CrossRef]
- Dawadi, P.B.S.; Schulten, E.A.M.; Lugtenburg, J. Synthesis of [3-13C]-, [4-13C]- and [11-13C]-porphobilinogen. J. Labelled Compd. Radiopharm. 2009, 52, 341–349. [Google Scholar] [CrossRef]
- Siebum, A.H.G.; Woo, W.S.; Lugtenburg, J. Preparation and characterization of [5-13C]-(2S, 4R)-leucine and [4-13C]-(2S, 3S)-valine- Establishing synthetic schemes to prepare any site-directed isotopomer of L-leucine, L-isoleucine and L-valine. Eur. J. Org. Chem. 2003, 4664–4678. [Google Scholar]
- Patching, S.G. Efficient syntheses of 13C- and 14C-labelled 5-benzyl and 5-indolylmethyl L-hydantoins. J. Labelled Compd. Radiopharm. 2011, 54, 110–114. [Google Scholar]
- Raap, J.; Nieuwenhuis, S.; Creemers, A.; Hexspoor, S.; Kragl, U.; Lugtenburg, J. Synthesis of isotopically labelled L-phenylalanine and L-tyrosine. Eur. J. Org. Chem. 1999, 2609–2621. [Google Scholar]
- Ingersoll, A.W.; Babcock, S.H. Hippuric acid. Org. Synth. 1943, 12, 40. [Google Scholar]
- Nieuwenhuis, S.A.M.; Mul, C.; Van Belle, N.J.; Lugtenburg, J.; Raap, J. Synthesis of stereospecifically β-deuterated L-tyrosine. In Photosynthesis, From Light to Biosphere; Mathis, P., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995; Volume II, pp. 313–316. [Google Scholar]
- Heinen, W.; Rosenmoller, C.H.; Wenzel, C.B.; De Groot, H.J.M.; Lugtenburg, J.; van Duin, M. 13C-NMR study of the grafting of maleic anhydride onto polyethane, polypropene and ethene-propene copolymers. Macromolecules 1996, 29, 115–1157. [Google Scholar]
- Hyett, D.J.; Mink, D.; Brvntermann, G.B.; Zeegens, H.M.G. WO 2004/078702 A2, 2004.
- Schmidt, U.; Lieberknecht, A.; Wild, J. Amino acids and peptides; XLIII1. Dehydroamino acids; XVIII2. Synthesis of dehydroamino acids and amino Acids from N-acyl-2-(dialkyloxyphosphinyl)-glycin esters; II. Synthesis 1984, 53-61. [Google Scholar]
- Schmidt, U.; Griesser, H.; Leitenberger, V.; Lieberknecht, A.; Mangold, R.; Meyer, R.; Riedl, B. Diastereoselective formation of (Z)-didehydroamino acid esters. Synthesis 1992, 487–490. [Google Scholar]
- O’Donnell, M.J. The enantioselective synthesis of α-amino acids by phase-transfer catalysis with achiral Schiff base esters. Acc. Chem. Res. 2004, 37, 506–517. [Google Scholar] [CrossRef]
- Reddy, L.R.; Gupta, A.P.; Liu, Y. Asymmetric synthesis of α-amino acids by reduction of N-tert-butanesulfinyl ketimine esters. J. Org. Chem. 2011, 76, 3409–3415. [Google Scholar] [CrossRef]
- Takahiro Soeta, T.; Kojima, Y.; Yutaka Ukaji, Y.; Inomata, K. Borinic acid catalyzed α-addition to isocyanide with aldehyde and water. Tetrahedron Lett. 2011, 52, 2557–2559. [Google Scholar] [CrossRef]
- Corson, B.B.; Dodge, R.A.; Harris, S.A.; Hazen, R.K. Ethyl benzoylformate. Org. Synth. 1941, 1, 241–243. [Google Scholar]
- Roth, P.; Hädener, A.; Tamm, C. Further studies on the biosynthesis of tabtoxin (wildfire toxin): Incorporation of [2,3-13C2]-pyruvate into the β-lactam moiety. Helv. Chim. Acta 1990, 73, 476–482. [Google Scholar] [CrossRef]
- Larsen, B.D.; Eggert, H.; Harrit, N.; Holm, A. Photolysis of 1,2,3-thiadiazole. Formation of thiirene by secondary photolysis of thioketene. Acta Chem. Scand. 1992, 46, 482–486. [Google Scholar] [CrossRef]
- Lee, C.H.; Westling, M.; Livinghouse, T.; Williams, A.C. Acylnitrilium Ion Initiated Heteroannulations in Alkaloid Synthesis. An efficient, stereocontrolled, total synthesis of the Orchidaceae alkaloid (±)-dendrobine. J. Am. Chem. Soc. 1992, 114, 4089–4095. [Google Scholar] [CrossRef]
- Ugi, I.; Werner, B.; Dömling, A. The chemistry of isocyanides, their multi-component reactions and their libraries. Molecules 2003, 8, 53–66. [Google Scholar]
- Xiao, X.; Xie, Y.; Su, C.; Liu, M.; Shi, Y. Organocatalytic asymmetric biomimetic transamination: From α-keto esters to optically active α-amino acid derivatives. J. Am. Chem. Soc. 2011, 133, 12914–12917. [Google Scholar] [CrossRef]
- Chen, Y.; Goldberg, S.L.; Hanson, R.L.; Parker, W.L.; Gill, I.; Tully, T.P.; Montana, M.A.; Goswami, A.; Patel, R.N. Enzymatic preparation of an (S)-amino acid from a racemic amino acid. Org. Process. Res. Dev. 2011, 15, 241–248. [Google Scholar] [CrossRef]
- Kelly, N.M.; O’Neill, B.C.; Probert, J.; Reid, G.; Stephen, R.; Wang, T.; Willis, C.L.; Winton, P. Chemo-enzymatic syntheses of isotopically labelled L-amino acids. Tetrahedron Lett. 1994, 35, 6533–6536. [Google Scholar]
- Terauchi, T.; Kobayashi, K.; Okuma, K.; Oba, M.; Nishiyama, K.; Kainosho, M. Stereoselective synthesis of triply isotope-labeled Ser, Cys, and Ala: Amino Acids for stereoarray isotope labeling technology. Org. Lett. 2008, 10, 2785–2787. [Google Scholar]
- Karstens, W.F.J.; Berger, H.J.F.F.; Van Haren, E.R.; Lugtenburg, J.; Raap, J. Enantioselective synthesis of isotopically labelled L-α-amino acids preparation of 13C-, 18O-and 2H-labelled L-serines and L-threonines. J. Label. Compd. Radiopharm. 1995, 36, 1077–1096. [Google Scholar] [CrossRef]
- Siebum, A.H.G.; Woo, W.S.; Raap, J.; Lugtenburg, J. Access to any site-directed isotopomer of methionine, selenomethionine, cysteine, and selenocysteine- Use of simple, efficient modular synthetic reaction schemes for isotope ncorporation. Eur. J. Org. Chem. 2004, 2905–2913. [Google Scholar]
- Kim, H.Y.; Oh, K. Highly diastereo- and enantioselective aldol reaction of methyl α-isocyanoacetate: A cooperative catalysis approach. Org. Lett. 2011, 13, 1306–1309. [Google Scholar] [CrossRef]
- Lodwig, S.N.; Unkefer, C.J. Stereoselective synthesis of stable isotope labeled L-α-amino acids: Synthesis of L-[4-13C] and L-[3,4-13C2] Aspartic Acid. J. Label. Compd. Radiopharm. 1992, 31, 95–102. [Google Scholar] [CrossRef]
- Lee, K.M.; Ramalingam, K.; Son, J.K.; Woodard, R.W. A highly efficient and large-scale synthesis of (2S,3S)-[2,3-2H2]- and (2S,3R)-[3-2H]-aspartic acids via an immobilized aspartase-containing microbial cell system. J. Org. Chem. 1989, 54, 3195–3198. [Google Scholar] [CrossRef]
- Cappon, J.J.; Baart, J.; van der Walk, G.A.M.; Raap, J.; Lugtenburg, J. Chemo-enzymatic synthesis of specifically stable-isotope labeled L-glutamic acid and 2-oxoglutaric acid. Recl. Trav. Chim. Pays-Bas. 1991, 110, 158–166. [Google Scholar]
- Siebum, A.H.G.; Tsang, R.K.F.; Steen, R.; Raap, J.; Lugtenburg, J. Synthesis of (ϵ-13C-,ϵ-15N)-Enriched L-Lysine- Establishing schemes for the preparation of all possible 13C and 15N isotopomers of L-lysine, L-ornithine, and L-proline. Eur. J. Org. Chem. 2004, 4391–4396. [Google Scholar]
- Humphrey, J.M.; Hart, J.A.; Chamberlin, A.R. Efficient syntheses of diastereoselectively labeled (3S)-[4-13C]-L-valine, and regioselectively labeled [3-13CH3]-L-isoleucine hydrochlorides. Bioorg. Med. Chem. Lett. 1995, 5, 1315–1320. [Google Scholar] [CrossRef]
- Van den Berg, E.M.M.; Van Liemt, W.B.S.; Heemskerk, B.; Lugtenburg, J. Synthesis of indoles and L-tryptophans specifically 2H- or 13C-labelled in the six-membered ring. Recl. Trav. Chim. Pays-Bas. 1989, 108, 304–313. [Google Scholar]
- Shattuck, J.C.; Meinwald, J. The preparation of L-(2S, 3S)-4,4,4-[2H3]-valine. Tetrahedron Lett. 1997, 38, 8461–8464. [Google Scholar] [CrossRef]
- Nelson, S.G.; Wan, Z.; Stan, M.A. SN2 Ring opening of β-lactones:An Alternative to catalytic asymmetric conjugate additions. J. Org. Chem. 2002, 67, 4680–4683. [Google Scholar] [CrossRef]
- Cappon, J.J.; Van der Walle, G.A.M.; Verdegem, P.J.E.; Raap, J.; Lugtenburg, J. Synthesis of specifically stable-isotope-labeled L-proline via L-glutamic acid. Recl. Trav. Chim. Pays-Bas. 1992, 111, 517–523. [Google Scholar]
- Oba, M.; Miyakawa, A.; Nishiyama, K.; Terauchi, T.; Kainosho, M. Stereodivergent synthesis of (2S,3S,4R,5R)- and (2S,3S,4R,5S)-[3,4,5-D3]-proline depending on the substituent of the γ-lactam ring. J. Org. Chem. 1999, 64, 9275–9278. [Google Scholar] [CrossRef]
- Van den Berg, E.M.M.; Richardson, E.E.; Lugtenburg, J.; Jenneskens, L.W. Convenient syntheses for selectively isotopically labelled acrylonitriles. Synth. Commun. 1987, 17, 1189–1198. [Google Scholar] [CrossRef]
- Oba, M.; Kobayashi, M.; Oikawa, F.; Nishiyama, K. Synthesis of 13C/D doubly labeled L-Leucines: Probes for conformational analysis of the leucine side-chain. J. Org. Chem. 2001, 66, 5919–5922. [Google Scholar] [CrossRef]
- Ohfune, Y.; Tomita, M. Total synthesis of (−)-domoic acid. A revision of the original structure. J. Am. Chem. Soc. 1982, 104, 3511–3513. [Google Scholar] [CrossRef]
- Fletcher, M.D.; Harding, J.R.; Hughes, R.A.; Kelly, N.M.; Schmalz, H.; Sutherland, A.; Willis, C.L. Three approaches to the synthesis of L-leucine selectively labelled with carbon-13 or deuterium in either diastereotopic methyl group. J. Chem. Soc. Perkin Trans. 1 2000, 43–51. [Google Scholar]
- Kobayashi, S.; Tsubogo, T.; Saito, S.; Yamashita, Y. Calcium-catalyzed diastereo- and enantioselective 1,4-addition of glycine derivatives to α,β-unsaturated esters. Org. Lett. 2008, 10, 807–809. [Google Scholar] [CrossRef]
- Wong, M.L.; Guzei, I.A.; Kiessling, L.L. An asymmetric synthesis of L-pyrrolysine. Org. Lett. 2012, 14, 1378–1381. [Google Scholar] [CrossRef]
- De Lamo Marin, S.; Catala, C.; Kumar, S.R.; Valleix, A.; Wagner, A.; Mioskowski, C. A practical and efficient total synthesis of potent insulinotropic (2S,3R,4S)-4-hydroxyisoleucine through a chiral N-protected γ-keto-α-aminoester. Eur. J. Org. Chem. 2010, 3985–3989. [Google Scholar]
- Raap, J.; Wolthuis, W.N.E.; Hehenkamp, J.J.J.; Lugtenburg, J. Enantioselective syntheses of isotopically labeled α-amino acids. Preparation of specifically13C-labelled L-lysines. Amino Acids 1995, 8, 171–186. [Google Scholar]
- Terauchi, T.; Kamikawai, T.; Vinogradov, M.G.; Starodubtseva, E.V.; Takeda, M.; Kainosho, M. Synthesis of stereoarray isotope labeled (SAIL) lysine via the “Head-to-Tail” conversion of SAIL glutamic Acid. Org. Lett. 2011, 13, 161–163. [Google Scholar]
- Cappon, J.J.; Witters, K.D.; Baart, J.; Verdegem, P.J.E.; Hoek, A.C.; Luiten, R.J.H.; Raap, J.; Lugtenburg, J. Synthesis of L-histidine specifically labelled with stable isotopes. Recl. Trav. Chim. Pays-Bas. 1994, 113, 318–328. [Google Scholar]
- Talab, S.; Lugtenburg, J. Unpublished results, Leiden University: The Netherlands, 2009.
- Soede-Huijbregts, C.; Van Laren, M.; Hulsbergen, F.B.; Raap, J.; Lugtenburg, J. Improved specific synthesis of [1′-15N]- and [3′-15N]L-histidine. J. Label. Compd. Radiopharm. 2001, 44, 831–841. [Google Scholar] [CrossRef]
- O’Donovan, D.H.; Roza, I. A concise synthesis of asymmetrical N,N′-disubstituted guanidines. Tetrahedron Lett. 2011, 52, 4117–4119. [Google Scholar] [CrossRef]
- Walker, T.E.; Matheny, C.; Storm, C.B.; Hayden, H. An efficient chemomicrobiological synthesis of stable isotope-labeled L-tyrosine and L-phenylalanine. J. Org. Chem. 1986, 51, 1175–1179. [Google Scholar] [CrossRef]
- Truce, W. The Gatterman synthesis of aldehydes. In Organic Reactions; Wiley: New York, NY, USA, 1957; Chapter 2. [Google Scholar]
- Winkel, C.; Aarts, M.W.M.M.; Van der Heide, F.R.; Buitenhuis, E.G.; Lugtenburg, J. Synthesis and NMR spectroscopy of stable isotope-labelled phenols and L-tyrosines. Recl. Trav. Chim. Pays-Bas. 1989, 108, 139–146. [Google Scholar]
- Nishiyama, K.; Oba, M.; Ueno, R.; Morita, A.; Nakamura, Y.; Kainosho, M. Synthesis of phenylalanines regiospecifically labelled with deuterium in the aromatic ring. J. Label. Compd. Radiopharm. 1994, 34, 831–837. [Google Scholar] [CrossRef]
- Nieuwenhuis, S.A.M.; Hulsebosch, R.J.; Raap, J.; Gast, P.; Lugtenburg, J.; Hoff, A.J. Structure of the YD tyrosine radical in photosystem II. Determination of the orientation of the phenoxyl ring by enantioselective deuteration of the methylene group. J. Am. Chem. Soc. 1998, 120, 829–830. [Google Scholar]
- Van den Berg, E.M.M.; Van Liemt, W.B.S.; Heemskerk, B.; Lugtenburg, J. Synthesis of indoles and L-tryptophans specifically 2H- or 13C-labelled in the six-membered ring. Recl. Trav. Chim. Pays-Bas. 1989, 108, 304–313. [Google Scholar]
- Van den Berg, E.M.M.; Baldew, A.U.; De Goede, A.T.J.W.; Raap, J.; Lugtenburg, J. Synthesis of three isotopomers of L-tryptophan via a combination of organic synthesis and biotechnology. Recl. Trav. Chim. Pays-Bas. 1988, 107, 73–81. [Google Scholar]
- Raap, J.; Winkel, C.; De Wit, A.H.M.; Van Houten, A.H.H.; Hoff, A.J.; Lugtenburg, J. Mass spectrometric determination of isotopically labeled tyrosines and tryptophans in photosynthetic reaction centers of Rhodobacter sphaeroides R-26. Anal. Biochem. 1990, 19, 19–15. [Google Scholar]
- Van Liemt, W.B.S.; Van Henegouwen, W.G.B.; Van Rijn, A.; Lugtenburg, J. Synthesis and spectroscopic characterisation of 13C-labelled anthranilic acid. Recl. Trav. Chim. Pays-Bas. 1996, 115, 431–437. [Google Scholar]
- Vorländer, D.; Von Schilling, R. Darstellung von indoxylsäureestern. Justus Liebigs Ann. Chem. 1898, 301, 349–352. [Google Scholar] [CrossRef]
- Vorländer, D.; Apelt, O. Darstellung von indol aus indoxyl. Ber. Dtsch. Chem. Ges. 1904, 37, 1134–1135. [Google Scholar] [CrossRef]
- Jessing, M.; Baran, P.S. Oxidative coupling of indoles with 3-oxindoles. Heterocycles 2010, 82, 1739–1745. [Google Scholar] [CrossRef]
- Noriya, T.; Yoneda, N. Synthesis of methyl 2-isocyano-3-[3(1H)-indolyl] acrylate and related compounds from 3-(aminomethylene)-3H-indoles. Chem. Pharm. Bull. 1982, 30, 158–166. [Google Scholar] [CrossRef]
- Kaur, H.; Heapy, A.M.; Brimble, M.A. The synthesis of dehydrotryptophan and dehydrotryptophan-containing peptides. Org. Biomol. Chem. 2011, 9, 5897–5907. [Google Scholar] [CrossRef]
- Vogel, A.I. Vogel's Textbook of Practical Organic Chemistry, 5th ed; Longman: London, UK, 1989. [Google Scholar]
- Kainosho, M.; Tekauchi, T. Aromatic amino acids labeled with stable isotope methods for incorporating the same into target protein and method for analyzing protein structure using NMR. JSTA Patent Application 20100056799.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dawadi, P.B.S.; Lugtenburg, J. Access to Any Site Directed Stable Isotope (2H, 13C, 15N, 17O and 18O) in Genetically Encoded Amino Acids. Molecules 2013, 18, 482-519. https://doi.org/10.3390/molecules18010482
Dawadi PBS, Lugtenburg J. Access to Any Site Directed Stable Isotope (2H, 13C, 15N, 17O and 18O) in Genetically Encoded Amino Acids. Molecules. 2013; 18(1):482-519. https://doi.org/10.3390/molecules18010482
Chicago/Turabian StyleDawadi, Prativa B. S., and Johan Lugtenburg. 2013. "Access to Any Site Directed Stable Isotope (2H, 13C, 15N, 17O and 18O) in Genetically Encoded Amino Acids" Molecules 18, no. 1: 482-519. https://doi.org/10.3390/molecules18010482